
Quantifying full-length circular RNAs in cancer
- Ken Hung-On Yu1,2,7,
- Christina Huan Shi2,7,
- Bo Wang1,
- Savio Ho-Chit Chow2,3,
- Grace Tin-Yun Chung1,
- Raymond Wai-Ming Lung1,
- Ke-En Tan4,
- Yat-Yuen Lim4,
- Anna Chi-Man Tsang1,
- Kwok-Wai Lo1 and
- Kevin Y. Yip2,5,6
- 1Department of Anatomical and Cellular Pathology, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong;
- 2Department of Computer Science and Engineering, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong;
- 3School of Life Sciences, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong;
- 4Institute of Biological Sciences, Faculty of Science, University of Malaya, Kuala Lumpur 50603, Malaysia;
- 5Hong Kong Bioinformatics Centre, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong;
- 6Hong Kong Institute of Diabetes and Obesity, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong
-
↵7 These authors contributed equally to this work.
Abstract
Circular RNAs (circRNAs) are abundantly expressed in cancer. Their resistance to exonucleases enables them to have potentially stable interactions with different types of biomolecules. Alternative splicing can create different circRNA isoforms that have different sequences and unequal interaction potentials. The study of circRNA function thus requires knowledge of complete circRNA sequences. Here we describe psirc, a method that can identify full-length circRNA isoforms and quantify their expression levels from RNA sequencing data. We confirm the effectiveness and computational efficiency of psirc using both simulated and actual experimental data. Applying psirc on transcriptome profiles from nasopharyngeal carcinoma and normal nasopharynx samples, we discover and validate circRNA isoforms differentially expressed between the two groups. Compared with the assumed circular isoforms derived from linear transcript annotations, some of the alternatively spliced circular isoforms have 100 times higher expression and contain substantially fewer microRNA response elements, showing the importance of quantifying full-length circRNA isoforms.
Circular RNAs (circRNAs) are a class of single-stranded RNAs with the 5′ and 3′ ends covalently linked (Jeck and Sharpless 2014; Chen 2016; Li et al. 2018b). Although they have been known for a long time (Hsu and Coca-Prados 1979; Nigro et al. 1991), research on circRNAs has only been reinvigorated in recent years by the discoveries that some circRNAs are highly abundant (Danan et al. 2012; Salzman et al. 2012) and conserved across species (Rybak-Wolf et al. 2015) and have regulatory potentials by functioning as microRNA (miRNA) sponges (Hansen et al. 2013; Memczak et al. 2013). Several additional sequence- or structure-specific functions of circRNAs have also been proposed (Chen 2016, 2020; Greene et al. 2017; Li et al. 2018b; Liu et al. 2019).
A number of circRNAs are highly expressed in cancer or are differentially expressed between cancer and normal tissues (Kristensen et al. 2018). Some of them have been shown to play oncogenic (Guarnerio et al. 2016) or tumor-suppressive (Li et al. 2015) roles. Because circRNAs are relatively stable compared with their linear counterparts owing to their resistance to RNA exonuclease (Memczak et al. 2013), they can potentially be used as diagnostic biomarkers of cancer (Qu et al. 2015; Vo et al. 2019).
Currently, the standard way of detecting circRNAs genome-wide is RNA sequencing (RNA-seq). Because circRNAs are not polyadenylated, protocols without poly(A) enrichment are used, such as those that involve ribosomal RNA (rRNA) depletion. The resulting data contain a mixture of sequencing reads from both linear and circular transcripts. A usual way to enrich for circRNA reads is to apply RNase R treatment, which preferentially digests linear transcripts (Suzuki et al. 2006; Memczak et al. 2013).
Regardless of the RNA-seq protocol, a common step in identifying circRNAs from the sequencing data is to look for back-splicing junctions (BSJs), that is, junctions that connect the 3′ end of a downstream exon to the 5′ end of an upstream exon, which indicate potential circularization events. Several computational methods have been proposed for identifying circRNAs from RNA-seq data using this idea (Wang et al. 2010; Memczak et al. 2013; Hoffmann et al. 2014; Westholm et al. 2014; Zhang et al. 2014, 2016, 2020; Gao et al. 2015, 2018; Szabo et al. 2015; Cheng et al. 2016; Chuang et al. 2016; Izuogu et al. 2016; Song et al. 2016; Li et al. 2017, 2018a; Metge et al. 2017; Wu et al. 2019; Zheng et al. 2019). Major differences among these methods include their read alignment strategy, signals used to detect BSJs, dependency on annotations of linear transcript isoforms, and ability to distinguish circRNAs from other events that may also create unexpected junctions, such as lariats, fusion genes, and tandem duplications. These methods have been extensively compared using benchmark data sets (Hansen et al. 2016; Zeng et al. 2017; Hansen 2018). The identified circRNAs and their expression information are cataloged in several databases (Glažar et al. 2014; Chen et al. 2016; Zhang et al. 2016; Dong et al. 2018; Xia et al. 2018; Vo et al. 2019; Wu et al. 2020).
Similar to linear transcripts, circular transcripts can also be alternatively spliced to create different isoforms with the same BSJ (Gao et al. 2016; Zhang et al. 2016). Four major types of alternative splice site selection—namely, cassette exon, intron retention, alternative 5′ splice site, and alternative 3′ splice site—can all be found in circRNAs (Zhang et al. 2016). Because the function of a circRNA depends on its exact sequence, for example, in determining the presence of miRNA response elements (MREs) and binding sites of RNA-binding proteins, it is critical to resolve full-length circRNA sequences and quantify their expression levels (Gao and Zhao 2018).
Most existing circRNA detection methods can only identify BSJs but cannot determine full-length circular transcripts. Some other methods can either infer full-length circRNA isoforms or quantify their expression levels, but not both. For example, given a BSJ formed by two exons of a linear transcript, Sailfish-cir (Li et al. 2017) assumes these two exons and all other exons between them are present in the circular transcript and performs quantification of it. The only existing methods that can both identify and quantify full-length circRNA transcripts from standard RNA-seq data are CIRI-full (Zheng et al. 2019) and CircAST (Wu et al. 2019). CIRI-full detects mostly short transcripts such as those shorter than the total length of the two sequencing reads produced by paired-end sequencing from a fragment (Zheng et al. 2019), whereas longer transcripts could be missed. CircAST works best with RNase R–treated data, in which most linear transcripts have been depleted, but may not work well in normal RNA-seq data that contain a large proportion of reads from linear transcripts.
In this paper, we propose pseudoalignment identification of circRNAs (psirc), the first complete pipeline that can detect full-length circRNA transcript isoforms of all lengths and quantify their expression levels directly from standard RNA-seq data. As shown in some previous studies (Li et al. 2018a; Asghari et al. 2020), avoiding full sequence alignments by making use of k-mer matching can reduce the running time, whereas the alignment details are not crucial for circRNA identification and quantification, which motivated our use of pseudoalignment in the current study.
Results
The psirc method
The overall procedure of psirc consists of three steps, namely, (1) identifying BSJs, (2) determining potential full-length transcript isoforms, and (3) quantifying the expression levels of full-length transcript isoforms (Methods) (Fig. 1).
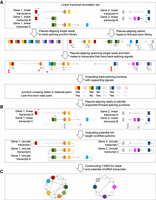
The psirc method. In the first step (A), sequencing reads are pseudoaligned to potential BSJs in single-end mode and to the first and last exons of each transcript in paired-end mode. For each read that is pseudoaligned to a BSJ (by means of rotating the end of the downstream exon to the beginning of the upstream exon), if its mate read is not pseudoaligned to the same transcript or not pseudoaligned in the opposite orientation, both of them will not be considered to support the BSJs (arrows in light gray with a red cross next to them). In the second step (B), the potential circRNA transcript isoforms are determined as those with all forward-splicing and backward-splicing junctions supported by sequencing reads. In the third step (C), a T-DBG is constructed for all linear and circular transcript isoforms for estimating their expression levels by another round of pseudoalignment.
In the first step (Fig. 1A), two types of signals are used to identify BSJs, namely, junction-crossing reads and last–first exon read pairs. A junction-crossing read is a sequencing read split-pseudoaligned to cover the 3′ most positions of a downstream exon and the 5′ most positions of an upstream exon. A last–first exon read pair is a read pair, respectively, pseudoaligned to the first and last exons of an annotated linear transcript in an outward-facing manner. In the second step (Fig. 1B), all RNA-seq reads are pseudoaligned to the linear transcripts that contain both the defining exons of any BSJ. The potential full-length circRNA transcript isoforms are then generated as those with all the involved forward- and backward-splicing junctions supported by sequencing reads using a graph searching algorithm. Because the BSJs identified in the first step of psirc can be defined by any pairs of exons, psirc allows the detection of circRNA isoforms that contain exon combinations not identical to any annotated linear isoforms. Finally, in the third step (Fig. 1C), a transcript de Bruijn graph (T-DBG) (Bray et al. 2016) is constructed for all linear and potential circular transcript isoforms. The pseudoalignments of sequencing reads are then used to quantify the expression level of each linear and circular transcript isoform by likelihood maximization, with boundary effects taken care of by adjusting effective transcript lengths (Methods).
In all three steps, full alignments of sequencing reads are avoided by using kallisto (Bray et al. 2016) to perform pseudoalignments, which makes psirc highly efficient in terms of both running time and memory consumption.
Effective BSJ detection with low time and memory requirements
We first verified the ability of psirc in identifying BSJs using three data sets, involving human fetal samples and two human cell lines (Supplemental Table S1). We used psirc and four other methods to identify BSJs from each sample. These methods were (1) CIRI2 (Gao et al. 2018) and (2) CIRCexplorer2 (Zhang et al. 2016), two methods that consistently ranked top in benchmarking studies (Hansen et al. 2016; Zeng et al. 2017; Hansen 2018), and (3) CircMarker (Li et al. 2018a) and (4) CircMiner (Asghari et al. 2020), two methods that aim at achieving high speed efficiency by using k-mer matching and pseudoalignment to avoid expensive sequence alignments. From the results (Supplemental Figs. S1–S3; Supplemental Results), in terms of identifying BSJs, psirc requires much less computational resources but still achieves comparable sensitivity and precision as the best of the other four methods.
Accurate quantification of full-length circRNA isoforms
We next tested the ability of psirc in quantifying expression levels. First, we used the data set of human fetal samples mentioned above, which also included the expression levels of some BSJs independently measured by RT-qPCR (Szabo et al. 2015). We applied psirc to deduce the expression level of each full-length circRNA isoform using the RNA-seq data, based on which we computed the expression level of each BSJ by aggregating the expression levels of all the full-length circRNA isoforms that involved this junction. We then compared these deduced BSJ expression levels with those measured by RT-qPCR. For benchmarking purposes, we also deduced BSJ expression levels directly from the RNA-seq data using the other four methods.
For all five methods, the deduced BSJ expression levels were correlated with the RT-qPCR results whether the read counts were normalized (Supplemental Fig. S4A) or not (Supplemental Fig. S4B). Among the five methods, the correlation values were slightly stronger for psirc than the other four methods.
In the above comparisons, each method was evaluated based on the BSJs identified by it, which could be different from the BSJs identified by the other methods. To compare the quantification capability of the different methods on the same ground, we designed a novel benchmarking procedure. First, we divided the whole BSJ quantification process into three components, namely, BSJ calling (B), full-length circRNA isoform inference (F), and expression level quantification (Q). The quantification component was further divided into two types, namely, quantification of BSJs without inferring full-length isoforms (Qb) and quantification of BSJs by quantifying and aggregating full-length circRNA isoform expression levels (Qf). Some of the methods provided all three components, whereas the others provided only some of them (Supplemental Table S2). We then established full pipelines by mixing and matching components of different methods, such that the quantification performance of different methods could be fairly compared based on the same set of BSJs or full-length transcript isoforms. For this part of analysis, we included CircAST and Sailfish-cir because, although these methods could not identify BSJs by themselves, the BSJs identified by other methods could be supplied to them as input in the pipelines. In addition, we included two new methods of the CIRI series, CIRI-full (Zheng et al. 2019) and CIRI-quant (Zhang et al. 2020), to provide supplemental or alternative components for CIRI2.
Using the benchmarking procedure, we first compared the quantification performance of five pipelines based on the BSJs identified by CIRI2 (Fig. 2A–E). Among the four pipelines that computed BSJ expression levels by aggregating expression levels of full-length isoforms (i.e., those with the Qf component), three of them achieved stronger correlations than the pipeline that computed BSJ expression levels directly from BSJs (i.e., the one with the Qb component). The only exception was the pipeline involving CIRI-full, which was unable to infer any full-length transcript isoform for many BSJs because it was designed to identify short isoforms. Among all five pipelines, the one with expression quantification performed by psirc achieved the strongest correlation of −0.832.
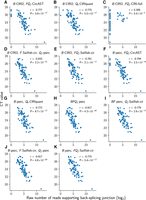
Comparison of different pipelines based on their determined read counts that support each BSJ from the human fetal samples. Each point corresponds to one primer pair in one sample. Each panel (A–K) corresponds to a different pipeline by combining the three components from different methods. If the same method was used for multiple components, they are written together. For example, “B:psirc, FQf:Sailfish-cir” means the BSJs were identified by psirc, whereas both the full-length transcript isoforms and their expression levels were deduced by Sailfish-cir. All pipelines involving full-length quantification (Qf) aggregated the expression levels of all transcripts that involved a BSJ into the expression level of the junction.
Similarly, when the BSJs were identified by psirc (Fig. 2F–K), the five pipelines involving the inference of full-length transcripts achieved stronger correlations than the pipeline that quantified the BSJs directly. Among these five pipelines, when the full-length isoform inference method was fixed, using psirc for quantification achieved stronger correlations than Sailfish-cir (−0.817 for “BFQf :psirc” vs. −0.779 for “BF:psirc, Qf :Sailfish-cir”; −0.817 for “B:psirc, F:Sailfish-cir, Qf:psirc” vs. −0.775 for “B:psirc, FQf :Sailfish-cir”), although the differences do not reach statistical significance. The quantification results of psirc also correlated more with the RT-qPCR results than CircAST (−0.817 for “BFQf :psirc” vs. −0.794 for “B:psirc, FQf:CircAST”).
All these conclusions still hold when the deduced BSJ expression levels were normalized (Supplemental Fig. S5). Overall, these results show that the full-length circRNA isoform inference and quantification of psirc enabled it to deduce BSJ expression levels that were correlated with RT-qPCR results.
Next, we set forth to evaluate psirc's accuracy in quantifying the expression levels of full-length circRNA isoforms. Because large-scale experimental data of full-length circRNA expression levels based on short-read RNA-seq data alone are not available, we first performed this evaluation using two sets of simulated data (Supplemental Methods). We benchmarked the results of psirc against those produced by Sailfish-cir on the basis of its good performance in quantifying BSJs in the evaluations above. To directly compare the quantification component of the two methods, we supplied the simulated full-length transcript isoform sequences as the common input to both methods.
In the first set of simulated data, we followed the original approach of Li et al. (2017) to testing Sailfish-cir to simulate 11 groups of genes. Each group contained 500 genes with one linear isoform and one circular isoform having independent expression levels, leading to 1000 isoforms per group. The 11 groups differed by the degree of overlap between the linear and circular sequences, ranging from 0% to 100% (Supplemental Fig. S6). When the overlap ratio was low, both psirc and Sailfish-cir were able to estimate expression levels accurately, with the correlation between the estimated and actual expression levels of the 1000 isoforms in each group as high as 0.99 (Fig. 3A,B). However, when the overlap ratio was 100%, the correlation value of Sailfish-cir dropped to below 0.93, whereas that of psirc remained higher than 0.99. To check whether psirc's high correlation was owing to biases caused by improper data normalization, we plotted transcript expression levels against their lengths for the group with 100% sequence overlap (Fig. 3C) but did not observe any obvious correlation between expression level and gene length that could have caused biases. We further plotted the estimated and actual read counts of this group of isoforms (Fig. 3D) and found that Sailfish-cir tended to overestimate read counts of linear transcripts and underestimate those of circular transcripts, which could not be shown in the previous BSJ quantification results that involved only circular transcripts. In contrast, psirc was able to quantify both linear and circular transcripts accurately.
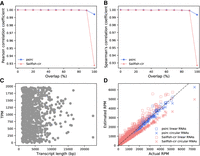
Quantification performance of psirc and Sailfish-cir on the first simulated data set. Pearson (A) and Spearman's (B) correlation coefficients were computed between the estimated and actual expression levels for the 1000 transcript isoforms in each of the 11 groups. For the group with 100% sequence overlap between linear and circular transcript isoforms, the scatter plots show the actual expression levels in transcripts per million (TPM) against transcript lengths (C) and the read counts per million reads aligned (RPM) estimated by the two methods against the actual read count of each transcript isoform (D).
To test whether psirc can handle more complex gene structures, we produced a second set of simulated data. In this set of data, there were 10 genes each in 10 groups. Each gene in the ith group had i linear transcript isoforms and the same number of circular isoforms with exactly the same sequences as the linear counterparts (i.e., 100% sequence overlap) but independent expression levels. For each group, we produced 10 different sets of data by performing 10 independent random sampling of the transcripts in this group. For all 10 groups, psirc outperformed Sailfish-cir by a clear margin (Fig. 4A,B). In general, the performance of both methods dropped as the number of transcript isoforms per gene increased. Yet, the correlation between psirc's estimated expression levels and the actual expression levels remained higher than 0.9 even when there were 10 linear and 10 circular isoforms per gene. When we plotted the estimated and actual read counts of the isoforms (Fig. 4C–I), again we observed that the quantification results of psirc were closer to the actual values than were the results of Sailfish-cir in all cases, even the genes chosen to be included in the plots were already the ones that Sailfish-cir performed the best in each group.
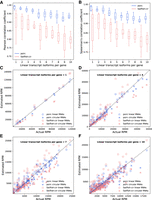
Quantification performance of psirc and Sailfish-cir on the second simulated data set. Pearson (A) and Spearman's (B) correlation coefficients were computed between the estimated and actual expression levels. Each box plot shows the distribution of correlations from the 10 sets of random transcripts, with each correlation coefficient computed based on all the transcripts from the 10 genes in that set. (C–F) The estimated and actual read counts per million reads aligned are shown for each transcript isoform for the gene that Sailfish-cir achieved the strongest Pearson correlation in each group, when each gene had one (C), four (D), seven (E), or 10 (F) linear isoforms per gene.
Finally, we used a data set with both short-read and long-read RNA-seq data to evaluate the full-length circRNA isoforms identified by psirc. This data set contained three biological replicates of RNase R–treated HEK293 cells with short-read data produced (Supplemental Figs. S1–S3; Supplemental Table S1) and three technical replicates from each of two biological replicates of HEK293 cells with long-read data produced (L1–L3, L4–L6). We used psirc and CircAST to identify and quantify full-length circRNAs from the short-read data; isoCirc (Xin et al. 2021), from the long-read data.
From the results (Fig. 5), psirc and isoCirc identified comparable numbers of full-length circRNAs from each sample despite the different types of data they worked on, whereas CircAST identified only around one-fifth on average (Fig. 5A). In general, the circRNAs identified by isoCirc overlapped more with those identified by psirc than those by CircAST, in terms of both the absolute numbers (Fig. 5A) and relative ratios (Fig. 5B). Focusing on the commonly identified circRNAs, we further compared the correlation of their expression levels in pairs of method–sample combinations (Fig. 5C). Again, the correlations between isoCirc and psirc (0.42–0.49) were higher than the correlations between isoCirc and CircAST (0.20–0.32).
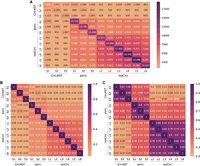
Evaluation of full-length circRNA identification and quantification by comparing with results obtained from long-read sequencing data. (A) The absolute numbers of full-length circRNAs identified by different method–sample combinations and their overlaps. (B) Codetection ratios of the identified full-length circRNAs, defined as the intersection size divided by the union size of the circRNAs identified by each pair of method–sample combinations. (C) Pearson's correlation of the expression levels of the full-length circRNAs commonly detected by two method–sample combinations.
Discovery of cancer-related circRNAs in nasopharyngeal carcinoma
With the effectiveness and efficiency of psirc verified by the series of tests above, we applied it to identify BSJs and full-length circRNA transcript isoforms and deduced their expression levels from rRNA-depleted RNA-seq data of 11 nasopharyngeal carcinoma (NPC) cell lines, xenografts, and patient tumor specimens and four normal nasopharynx (NP) cell lines (Supplemental Table S3). Sequencing reads were aligned to both the human and Epstein–Barr virus (EBV) genomes at the same time, allowing for a joint detection and quantification of both human and EBV full-length circRNAs.
We detected 8401–28,809 BSJs from the different samples, from which 8350–32,959 full-length circRNA isoforms were inferred. Focusing on only the frequently expressed cases, defined as those expressed in at least 70% of NPC or NP samples, 3145–5862 BSJs and 2247–4637 full-length isoforms were identified from the samples (Table 1; Supplemental Files S1–S4).
Numbers of human and EBV BSJs and full-length circular isoforms identified by psirc from the NPC and NP samples
Taking all the NPC and NP samples together, 6723 unique frequently expressed BSJs were identified from the human cellular genome (plus seven from the EBV genome). Looking up these BSJs from three circRNA databases, namely, CIRCpedia v2 (Dong et al. 2018), CSCD (Xia et al. 2018), and MiOncoCirc v0.1 (Vo et al. 2019), we found that 5786 of them (86.2%) were contained in at least one of these databases, whereas the remaining 930 (13.8%) were novel (Fig. 6A). Because of our definition of frequently expressed BSJs, each of these novel BSJs was called in at least three, and usually even more, samples (Fig. 6B), suggesting that these novel BSJs are frequently expressed in NP and NPC in a tissue-specific or cancer type–specific manner. We also checked the average number of supporting reads for each of these novel BSJs among the samples from which it was called, and found that around half of these novel BSJs had an average of five or more supporting reads across those samples (Fig. 6C), further supporting the reliability of these BSJ calls.
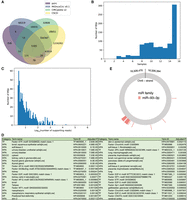
Analyses of the results produced by psirc from the NPC and NP samples. (A) A Venn diagram comparing the frequently expressed BSJs identified from the NPC and NP samples with those in three circRNA databases. (B,C) Histograms of the frequently expressed novel BSJs not contained by any of the three databases, in terms of the number of NPC and NP samples from which they were called (B) and their average number of supporting reads among the samples from which they were called (C). (D) Enriched functional terms (adjusted P < 0.01) of the down-regulated genes based on the full-length circRNA isoform analysis. (E) MREs on the differentially expressed ATXN1 circRNA.
To see how the expression levels of linear and circular isoforms are related to each other, from each sample we extracted all pairs of linear and circular isoforms with identical sequences with at least one of them expressed. These pairs reveal a general positive correlation between the linear and circular expression levels, but some variations do exist (Supplemental Fig. S7). This is consistent with observations from previous studies based on specific examples or BSJs (Chen 2020).
We then performed differential expression analysis and obtained 222 BSJs and 319 full-length circRNA isoforms, coming from 177 and 271 genes, respectively, with relatively strong differential expression between the NPC and NP samples (Wilcoxon Q-value ≤ 0.2) (Supplemental Files S2, S4). Among these genes, 41 from the BSJ list and 57 from the full-length list were up-regulated in NPC, 19 of which were common. The numbers of down-regulated genes were larger, with 136 from the BSJ list and 214 from the full-length list, 89 of which were common. This trend of reduced circRNA expression in NPC is consistent with similar observations in prostate cancer and colorectal cancer (Bachmayr-Heyda et al. 2015; Chen et al. 2019). The differences between the BSJ and full-length lists show that some differentially expressed full-length circRNA isoforms would be missed if only the BSJs were quantified. Next, we performed a functional enrichment analysis of the differentially expressed genes on the full-length list using g:Profiler (Raudvere et al. 2019). For the up-regulated genes, there were no significantly enriched functional terms. For the down-regulated genes, a fairly large number of functional terms were significantly enriched (Fig. 6D), including the Human Protein Atlas (Uhlén et al. 2015) terms “tonsil; squamous epithelial cells” (adjusted P = 4.98 × 10−5), “thyroid gland” (adjusted P = 8.30 × 10−5), and “nasopharynx” (adjusted P = 1.38 × 10−3).
Among the full-length circRNA isoforms with a differential expression P-value < 0.05, we found 24 of them with at least 10 MREs of a single miRNA family (Methods) (Supplemental Table S4). Among them, a circRNA generated by back-splicing of an exon of the ATXN1 gene was predicted to harbor 29 MREs of the miRNA family miR-93-3p (Fig. 6E).
To further explore the significance of quantifying full-length circRNA isoforms, we compared the MREs of each frequently expressed circRNA isoform involving exon skipping with the corresponding isoforms derived from all annotated linear isoforms that contain the two exons defining the BSJ. We call the latter the “default isoforms” and the former the “alternative isoform.” In total, from 252 frequently expressed alternative isoforms, we derived 276 default isoforms. On average, each of these alternative isoforms harbors 45.6 fewer MREs than their default isoforms. In one extreme example, an alternative isoform harbors 10 fewer MREs from the same miRNA family than its default isoforms. Importantly, 31.2% of these alternative isoforms had an expression level over 100 times higher than the corresponding default isoforms. These results show that if full-length circRNA isoforms are not inferred and quantified but rather default isoforms are assumed based on BSJs and annotated linear isoforms alone, functional studies of circRNA could be seriously misinformed.
Finally, we experimentally verified some of the differentially expressed circRNAs between the NPC and NP groups. We started with a verification of the BSJs using RT-PCR (Fig. 7A). The results are highly consistent with the TPM values of these BSJs determined by psirc based on the supporting reads (Fig. 7B). For example, the BSJ from NETO2 was mostly expressed in the NP group but not the NPC group according to both RT-PCR and psirc. In contrast, the BSJs from NTRK2 and the EBV-encoded RPMS1 were mostly expressed in the NPC group but not the NP group according to both methods.
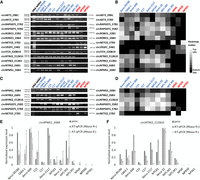
Experimental validations of the computational results of psirc. (A) Validation of differentially expressed BSJs in the RNase R–treated NP and NPC RNA samples using RT-PCR. Each row corresponds to a BSJ and each column corresponds to a sample, with the NPC samples labeled in blue and the NP samples labeled in red. (B) Expression levels of the same BSJs determined by psirc. The expression value of each BSJ was computed by summing up the TPM values of all transcripts that involved this BSJ. (C) Validation of differentially expressed full-length transcript isoforms using RT-PCR. (D) TPM values of the same full-length isoforms inferred by psirc. (E,F) RT-qPCR results of two short full-length transcript isoforms, circRPMS1_E6B4 (E) and circNTRK2_E12B10 (F), with or without RNase R treatment. Each RT-qPCR experiment was repeated three times with independent RT preparations, with the standard deviation of each triplicate indicated by the error bars. In each of these two panels, normalization was performed by dividing each value by the largest value among all samples.
Next, we designed primers specifically for some differentially expressed full-length transcript isoforms. The RT-PCR results (Fig. 7C) again show high consistency with the corresponding TPM levels deduced by psirc in distinguishing between the two sample groups (Fig. 7D). This trend was further confirmed by the RT-PCR results based on different reverse transcriptases for an NTRK2 BSJ and a full-length transcript isoform of it (Supplemental Fig. S8A).
To get a more quantitative evaluation of the deduced expression levels, we further performed RT-qPCR on two short full-length isoforms, selected according to the detection limit of RT-qPCR. The results (Fig. 7E,F) show that the two isoforms had almost no expression in the NP group based on both the psirc and RT-qPCR results. When considering only the NPC samples, the two sets of results correlated positively (Pearson correlation = 0.75/0.48 for circRPMS1_E6B4 with/without RNase R treatment, and 0.71/0.82 for circNTRK2_E12B10 with/without RNase R treatment), with some differences between them likely caused by a combination of technical and biological reasons such as cell passages. We further tested the circRPMS1_E6B4 case using a BaseScope RNA in situ hybridization assay, a sensitive non-RT-based experiment, and observed the same differential expression between the NPC and NP groups (Supplemental Fig. S8B).
Discussion
In this study, we have developed psirc, the first complete pipeline that can identify both BSJs and full-length circRNA transcript isoforms of all lengths and quantify their expression levels. We have shown the effectiveness and computational efficiency of psirc using simulated data and RNA-seq data from human cell lines and fetal tissue samples. At the BSJ level, psirc achieved comparable sensitivity and precision as the best of CIRCexplorer2, CIRI2, and CircMarker while requiring substantially less running time and memory. At the full-length isoform level, psirc detected a lot more isoforms than CIRI-full and produced more accurate expression level quantification than Sailfish-cir, especially in terms of the relative expression levels of linear and circular transcripts. The full-length quantification results of psirc were more consistent with the isoCirc results obtained from long-read sequencing data than the results of CircAST.
The efficiency of psirc is owing to its use of pseudoalignment by kallisto, which avoids time-consuming full alignments of sequencing reads but is still able to accurately determine the transcript isoforms from which each sequencing read could come by using the T-DBG. In contrast, the k-mer-based method of Sailfish-cir can assign reads to wrong transcript isoforms by ignoring the order of k-mers.
The good performance of psirc in identifying and quantifying full-length circRNA isoforms is owing to a number of factors. First, the design of psirc permits the detection of circRNAs of different lengths, including both short and long ones. Second, psirc can detect many possible isoforms by considering exon combinations based on read-supported BSJs and forward-splicing junctions. Third, psirc quantifies linear and circular transcript isoforms together, allowing it to accurately determine the relative expression levels of the linear and circular isoforms.
We have found that for some differentially expressed circRNA isoforms between NPC and NP, their expression levels were much higher than default isoforms produced by “circularizing” annotated linear isoforms that contain the two exons defining the BSJ. The highly expressed alternative isoforms could have far fewer MREs than the default isoforms owing to exon skipping. Being able to identify full-length circRNA isoforms and quantify their expression levels thus enables much more accurate study of potential circRNA functions such as their interactions with miRNAs and RNA-binding proteins. In general, psirc can help identify potentially interesting miRNA–circRNA interactions by providing information about the expression levels of full-length circRNA isoforms and miRNAs that may interact with them for further validations and detailed studies with additional experiments.
A recent study has shown that circRNAs can form 16- to 26-bp duplex structures, which act as inhibitors of double-stranded RNA-activated protein kinase (EIF2AK2 [also known as PKR]), and these circRNAs are degraded by RNase L for activating EIF2AK2 during early innate immune response (Liu et al. 2019). The structures of circRNAs and their corresponding functional mechanisms are still under investigation, but the full-length sequences of circRNAs likely play some roles in determining the possible structures.
By considering the read supports of individual forward-splicing junctions and BSJs, psirc is able to infer circular transcript isoforms that contain an exon combination different from any annotated linear isoforms. However, psirc still relies on an input set of linear transcript isoforms to define exon boundaries. As a result, it cannot detect nonexonic circRNAs such as intron–exon circRNAs (elciRNAs) or circRNAs that involve cryptic 5′ or 3′ splice sites not at the boundaries of annotated exons. This limitation can be potentially overcome by first performing a linear transcript assembly on the RNA-seq data and augmenting the transcript annotation set with the novel transcripts identified, at the expense of extra computational resources.
Methods
Details of the psirc method
Identification of BSJs
In the first step of psirc (Fig. 1A), to detect junction-crossing reads, we construct a library of all possible BSJ sequences according to the transcripts defined in a gene annotation set. In the default setting of psirc, GENCODE (v29) (Harrow et al. 2012) is used. For each annotated linear transcript, we consider each pair of exons in turn to construct a sequence that contains the x nucleotides at the 3′ end of the 3′ exon in the pair followed by the x nucleotides at the 5′ end of the 5′ exon, and add the sequence to the library. The value of x is determined according to the size of k-mers (length-k subsequences) used in pseudoalignment as a trade-off between the sensitivity and specificity of BSJ detection. In the default setting of psirc with a k-mer size of 31, x is set to 24 such that each of the two exons has at least 7 bp of the k-mer pseudoaligned to. All the BSJs with pseudoaligned sequencing reads are then collected.
Similarly, we also constructed sequence libraries to gain additional support for these BSJs and for detecting the last–first exon read pairs (Supplemental Methods).
In all the above processes, pseudoalignment is performed using kallisto, which determines the set of all sequences that could have produced the read. This is performed by comparing k-mers in the read with the k-mers in the library sequences. This pseudoalignment step in psirc is very fast because (1) the total length of the library sequences is small compared with the whole genome or transcriptome, (2) kallisto uses a hash table to efficiently map each k-mer to the sequences that contain it (called its “k-compatibility class”), and (3) kallisto uses a T-DBG to determine the k-mers that do not need to be checked when they belong to the same k-compatibility class and appear on the same nonbranching path in the graph. Although the pseudoalignment results do not contain full alignments between the reads and the library sequences at the per-nucleotide resolution, they do contain the aligned location(s) of each read, which is sufficient for our purpose of identifying BSJs. By default, we allow each read to be pseudoaligned to, at most, 10 different locations. The version of kallisto used in psirc is a forked version that we modified from the main trunk, which can support multithreading and produce a SAM file as output.
Identification of potential full-length circular transcript isoforms
In the second step of psirc (Fig. 1B), for each linear transcript isoform with a BSJ detected in the first step, we identify a set of potential full-length circular transcript isoforms as follows. Suppose the original linear transcript isoform in the annotation file involves exons E1,E2,…,En, and in the first step of psirc, a BSJ was detected between exons Ei and Ej, where 1 ≤ i ≤ j ≤ n. A directed graph is constructed with each exon Ei, Ei + 1,…, Ej forming a node. For each pair of nodes Ea and Eb where i ≤ a < b ≤ j, if the forward-splicing junction from Ea to Eb is supported by sequencing reads, an edge will be drawn from the former to the latter. In addition, edges are also added for exons that are adjacent in the original linear transcript isoform, that is, from Ea to Ea + 1 for 1 ≤ a < n. A depth-first search is then performed to identify all noncyclic paths from Ei to Ej, and the nodes on each of these paths will form a potential full-length circular transcript isoform. Finally, if i = j, a potential full-length circular transcript isoform involving this exon alone will also be added to the list.
A concrete example and some additional filtering steps are described in Supplemental Methods.
Expression quantification of full-length transcript isoforms
In the third step of psirc (Fig. 1C), the expression level of each linear and circular transcript isoform is estimated based on likelihood maximization. The overall workflow involves indexing, pseudoalignment, and quantification (Supplemental Fig. S9).
In the indexing stage, all the potential linear and circular transcript isoforms, including the ones originally in the annotation file of linear transcript isoforms and the ones identified in the second step of psirc, are used to construct a colored T-DBG. In the pseudoalignment stage, all sequencing reads are pseudoaligned to the transcriptome defined by the T-DBG containing both linear and circular transcript isoforms. In the quantification stage, the expression level of each transcript isoform, defined as the number of transcript copies, is quantified by maximizing the data likelihood according to a probabilistic model (Bray et al. 2016) under the assumption that reads are correctly pseudoaligned.
More details and additional steps for bias correction and quality checking are described in the Supplemental Methods.
Data sets for testing psirc's performance
We obtained four RNA-seq data sets for testing psirc's performance and compared it with other circRNA detection methods (Supplemental Table S1). The first data set contained rRNA-depleted RNA-seq data of human fetal samples (NCBI Gene Expression Omnibus [GEO; https://www.ncbi.nlm.nih.gov/geo/] accession GSE64283) (Szabo et al. 2015). Among the 35 samples in this data set, 11 of them had RT-qPCR measurements of the expression of BSJs from eight genes, with 79 measurements in total. We used only these 11 samples in our analyses and downloaded the corresponding RNA-seq data from the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) (Leinonen et al. 2011). The CT values were obtained from column 5 of the additional file 15 of Szabo et al. (2015). The second and third sets of data contained rRNA-depleted RNA-seq and RNase R–treated RNA-seq data of HeLa and Hs68 cells (Jeck et al. 2013; Mercer et al. 2015). Replicate samples were combined by pooling the data directly. The fourth data set contained short-read RNA-seq data produced from three biological replicates of HEK293 cells, with long-read RNA-seq data produced from three technical replicates from each of two biological replicates of HEK293 cells (GEO accession GSE141693) (Xin et al. 2021). In the original data set, long-read data were also produced from 12 human tissues, and the full-length circRNAs reported by isoCirc were those identified from at least two of the 18 samples. In our analyses, we took the subset of these circRNAs from the six HEK293 samples directly from the original investigators’ results.
Comparisons with other circRNA-calling methods
We compared psirc with a number of existing methods that identified and/or quantified circRNAs from short-read RNA-seq data, including CircAST (Wu et al. 2019; https://github.com/xiaofengsong/CircAST, git commit c2f36ad4), CIRCexplorer2 (v2.3.8) (Zhang et al. 2016), CircMarker (Li et al. 2018a; https://github.com/lxwgcool/CircMarker, git commit 06aa680a), CircMiner (v0.4.5) (Asghari et al. 2020), CIRI2 (v2.0.6) (Gao et al. 2018), CIRI-full (v2.0, with CIRLAS v1.2 and CIRI-vis v1.4) (Zheng et al. 2019), CIRI-quant (v1.1) (Zhang et al. 2020), and Sailfish-cir (v0.11, with sailfish v0.9.2) (Li et al. 2017).
We categorized these methods based on their abilities to identify BSJs, infer full-length transcript isoforms, and quantity
the expression of them (Supplemental Table S2). For methods that infer and quantify full-length transcript isoforms, BSJ read counts were computed as 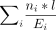 , where ni is the read count of transcript i, l is the read length, Ei is the effective length of transcript i, and the summation is over all transcripts that involve the BSJ. In our analyses, we considered both raw read counts and
normalized read counts, defined as raw read counts per million aligned reads.
, where ni is the read count of transcript i, l is the read length, Ei is the effective length of transcript i, and the summation is over all transcripts that involve the BSJ. In our analyses, we considered both raw read counts and
normalized read counts, defined as raw read counts per million aligned reads.
Additional details of the experimental configurations are described in the Supplemental Methods.
Testing psirc's ability to identify BSJs
We applied CIRCexplorer2, CircMarker, CircMiner, CIRI2, and psirc to identify BSJs from the two data sets as explained in the Results. For CIRCexplorer2, we failed to run it in its default setting on the sample Hs68 C1. Therefore, we instead downloaded the CIRCexplorer2 results of HeLa and Hs68 from the Supplemental Materials (“Presentation2.zip”) of Hansen (2018) and used these results in the comparisons directly.
For the analyses involving the HeLa and Hs68 data, for each BSJ identified, we classified it into either enriched, unaffected, depleted, or abolished, according to the numbers of reads that support this junction in the rRNA-depleted data and RNase R–treated data. Following methods of previous studies (Hansen et al. 2016; Zeng et al. 2017; Hansen 2018), unnormalized read counts were used to define these classes. For each of the two samples, because the total number of read pairs after pooling the data form replicates is highly consistent with or without the RNase R treatment (Supplemental Table S1), using unnormalized read counts should not create any strong systematic bias. For a particular BSJ, suppose the number of supporting reads in the rRNA-depleted data is d, the number of supporting reads in the RNase R–treated data is t, and α is an enrichment factor (Hansen et al. 2016; Hansen 2018); the definitions of the four classes are as follows:
-
Enriched: t ≥ d × α;
-
Unaffected: d × α > t ≥ d;
-
Depleted: d > t > 0;
-
Abolished: t = 0.
We used the enrichment factor α = 1.5 for HeLa and α = 5 for Hs68. The value for Hs68 was taken directly from previous studies (Hansen et al. 2016; Hansen 2018), whereas the value for HeLa was the total number of identified BSJ reads in the RNase R–treated samples divided by that in the untreated samples.
Although the enriched cases were likely true positives and the abolished cases were likely false positives, the unaffected and depleted cases were more ambiguous. We therefore considered four different measures of precision in order to provide a comprehensive evaluation of the performance of the four methods. In all four definitions, precision was defined as TP/(TP + FP), where TP stands for true positives and FP stands for false positives. The four precision measures differed by their definitions of TP and FP:
-
Precision 1—TP involved the enriched cases only, and FP involved the abolished cases only;
-
Precision 2—TP involved the enriched and unaffected cases, and FP involved the abolished cases only;
-
Precision 3—TP involved the enriched cases only, and FP involved the depleted and abolished cases;
-
Precision 4—TP involved the enriched and unaffected cases, and FP involved the depleted and abolished cases.
We quantified the computational costs by elapsed time, CPU time, and RAM usage. Elapsed time was defined as the duration of the physical running time, from the time that a method started to the time that it completed. CPU time was the total amount of time used by the CPU on all the threads of the method. These two time measurements differed mainly in two aspects, namely, (1) elapsed time could be shortened by multithreading, but CPU time was the total of all threads, and (2) elapsed time included all the overheads such as disk I/O, but CPU time just included the time spent on CPU cycles. RAM usage was defined as the peak memory usage during the whole execution process.
When gathering the running time and RAM usage information, all the methods were run on a machine with 64 Intel Xeon E5-4610 v2 cores@2.30 GHz with 520 GB of RAM. At any time, only one method was run on one sample without any other user processes running in the background.
Comparing with RT-qPCR measurements
Expression values deduced from RNA-seq data were log2-transformed after addition of a small constant (c) to handle the zero-expression cases. The value of c was set to one and 0.01 for raw and normalized BSJ read counts, respectively. These values were chosen because they were close to the smallest nonzero values observed.
Production and processing of RNA-seq data from the NPC and NP samples
Four immortalized normal nasopharyngeal epithelial cell lines (NP69, NP361, NP460, and NPC550), two EBV-positive NPC cell lines (C666-1 and NPC43), seven patient-derived xenografts (Xeno-666, Xeno-2117, Xeno-1915, Xeno-99186, C15, C17, and Xeno-32) (Huang et al. 1989; Bernheim et al. 1993; Cheung et al. 1999; Tsao et al. 2002; Li et al. 2006; Tsang et al. 2012; Chung et al. 2013; Lin et al. 2018), and two patient tumor specimens (NPC-M1 and NPC-M2) were used in this study. NPC tumor specimens were from patients admitted to Prince of Wales Hospital, The Chinese University of Hong Kong. Patient consents were obtained according to institutional clinical research approval (IRB) at The Chinese University of Hong Kong, Hong Kong Special Administrative Region. Total RNA extracted from the NP and NPC samples were subjected to rRNA depletion by a Ribo-Zero kit (Illumina), followed by TruSeq stranded total RNA library construction and sequencing on the Illumina HiSeq 2000 system according to the manufacturer's protocols (Chung et al. 2013).
Comparing the frequently expressed BSJs identified from the NPC and NP samples with those in circRNA databases
The frequently expressed cellular BSJs identified from the NPC and NP samples were combined and deduplicated, resulting in a set of 6723 unique BSJs from the human cellular genome. These BSJs were compared with those from three circRNA databases. For CIRCpedia v2, a total of 183,943 unique BSJs from all human cell lines were downloaded from https://www.picb.ac.cn/rnomics/circpedia/. For CSCD (accessed on June 9, 2020), the union of all common, normal, and cancer-specific BSJs was taken, all downloaded from http://gb.whu.edu.cn/CSCD/, resulting in a set of 1,393,002 unique BSJs. For MiOncoCirc v0.1, BSJs from all cancer samples were downloaded from https://mioncocirc.github.io/download/, with a total of 232,665 unique BSJs. All genomic positions in the four sets were based on the human reference genome GRCh38.
Functional enrichment analysis
We used g:Profiler to perform functional enrichment analysis of the differentially expressed circRNAs obtained from the full-length quantification results of psirc. We ran g:Profiler using its default setting, which included the following functional categories: Gene Ontology subontologies (molecular function, cellular component, and biological process), biological pathways (KEGG, Reactome, and WikiPathways), regulatory motifs in DNA (TRANSFAC and miRTarBase), protein databases (Human Protein Atlas and CORUM), and human phenotype ontology (HP).
Prediction of MREs
Information about high-confidence human and EBV miRNAs and their families was downloaded from miRBase (release 22) (Kozomara et al. 2019). In total, 897 human miRNAs from 736 families and 44 EBV miRNAs from 44 families were involved. Different miRNAs in the same family share the same seed. One EBV miRNA (ebv-miR-BART4-3p) was found to have the same seed as a human miRNA (hsa-miR-499a-3p). Therefore, altogether 736 + 44-1 = 779 miRNA families were considered. MREs on potential circRNA sequences were identified using TargetScan (v7.0) (Agarwal et al. 2015) with default parameter values. All bases on the sequences were not masked, which allowed MREs to appear anywhere on the sequences. To allow for detection of MREs that overlap the BSJs, 10 bases from the 5′ end of each sequence were copied and pasted to the 3′ end. Redundant MREs that appeared completely within the 10 bases were removed to avoid double counting.
Experimental validations
The candidate BSJs and full-length isoforms of selected differentially expressed cellular and viral circRNAs of NPC and NP samples identified by psirc were subjected to conventional RT-PCR analysis and RT-qPCR analysis. The predicted BSJs of cancer gene–derived circRNAs either predominantly overexpressed (e.g., circAKT3, circNTRK2) or down-regulated (circNETO2, circVEGFC) in multiple NPC tumor samples were selected for validation by conventional RT-PCR with primers flanking the junction. For validation of full-length isoforms predicted by psirc, conventional RT-PCR analysis with an inverse primer pair in the same exon for selected overexpressed EBV-encoded (circRPMS1) and cellular (circNTRK2, cir-cRAPGEF5) circRNAs, as well as down-regulated circRNAs (circNETO2), was performed in a panel of NPC and NP samples. For the RT-PCR experiments, the total RNA of the samples was extracted with TRIzol reagent (Invitrogen), treated with 10 units of RNase R (BioVision) for 30 min at 37°C and purified with a miRNeasy kit (Qiagen). The RNA samples were then subjected to cDNA synthesis with SuperScript III reverse transcriptase (Invitrogen) and PCR amplification with a KAPA2G fast HotStart PCR kit (Roche) according to the manufacturer's protocol. PCR products were run on a 2% agarose gel. The primers involved in the validations of BSJs and full-length transcripts are listed in Supplemental Tables S5 and Table S6, respectively. The predicted BSJs in the amplified RT-PCR products of the samples were confirmed by Sanger sequencing (Supplemental Fig. S10).
For the additional experimental validations using different reverse transcriptases, 2 μg of total RNA was treated with 10 units of RNase R (BioVision M1228-500) for 30 min at 37°C and purified with a miRNeasy kit (Qiagen 217004). First-strand cDNA was generated with M-MLV (Invitrogen 18080044) or AMV (Promega M5101) reverse transcriptase. The cDNA products were then amplified by PCR with a KAPA2G fast HotStart PCR kit (Roche KK5519) as described above. The PCR products were run on a 1.5% agarose gel.
Two relatively small-sized circRNAs (circRPMS1_E6B4, 399 bp; circNTRK2_E12B10, 237 bp) were further validated by RT-qPCR analysis and DNA sequencing. cDNA samples were subjected to quantitative PCR analysis using the TaqMan universal PCR master mix (Applied Biosystems) on a LightCycler 480 instrument II as previously described (Ungerleider et al. 2018). The primers involved are listed in Supplemental Table S7. The RT-qPCR results were analyzed by delta-delta Ct method and normalized by GAPDH expression.
Following the method of Huang et al. (2019), BaseScope RNA in situ hybridization assays were performed using the BaseScope reagent kit v2-RED according to the manufacturer's instructions (Advanced Cell Diagnostics). Briefly, the cell or tissue sections were baked for 1 h at 60°C, deparaffinized, and treated with pretreat solution for 10 min at room temperature. Target retrieval was performed for 15 min at 100°C, followed by protease treatment for 15 min at 40°C. The circRPMS1_E6B4 BaseScope probe (BaseScope probe-BA-V-EBV-BART-E4-2zz-st, Advanced Cell Diagnostics) was then hybridized for 2 h at 40°C followed by BaseScope amplification and fast red chromogenic detection.
Software availability
The source code of psirc is available at GitHub (https://github.com/Christina-hshi/psirc.git) and is also provided as a Supplemental Code file.
Data access
The RNA-seq data generated in this study have been submitted to NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE165181.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This project was supported by the Hong Kong Research Grants Council Theme-based Research Scheme T12-401/13-R. Y.-Y.L. was supported by the Fundamental Research Grant Scheme (FRGS/1/2017/SKK08/UM/02/11). K.-W.L. and C.-M.T. were supported by the Hong Kong Research Grants Council Area of Excellence AoE/M-401/20, Collaborative Research Fund C4001-18GF, and General Research Fund 14113620. K.Y.Y. was supported by Hong Kong Research Grants Council Collaborative Research Funds C4045-18WF, C4054-16G, C4057-18EF, and C7044-19GF and General Research Funds 14107420, 14170217, and 14203119; the Hong Kong Epigenomics Project (EpiHK); and the Chinese University of Hong Kong Young Researcher Award and Outstanding Fellowship.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.275348.121.
- Received February 4, 2021.
- Accepted October 12, 2021.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








