
DENT-seq for genome-wide strand-specific identification of DNA single-strand break sites with single-nucleotide resolution
- 1MIT Department of Biological Engineering, Cambridge, Massachusetts 02139, USA;
- 2Broad Institute of MIT and Harvard, Cambridge, Massachusetts 02142, USA;
- 3Koch Institute for Integrative Cancer Research at MIT, Cambridge, Massachusetts 02142, USA
-
↵4 These authors contributed equally to this work.
Abstract
DNA single-strand breaks (SSBs), or “nicks,” are the most common form of DNA damage. Oxidative stress, endogenous enzyme activities, and other processes cause tens of thousands of nicks per cell per day. Accumulation of nicks, caused by high rates of occurrence or defects in repair enzymes, has been implicated in multiple diseases. However, improved methods for nick analysis are needed to characterize the mechanisms of these processes and learn how the location and number of nicks affect cells, disease progression, and health outcomes. In addition to natural processes, including DNA repair, leading genome editing technologies rely on nuclease activity, including nick generation, at specific target sites. There is currently a pressing need for methods to study off-target nicking activity genome-wide to evaluate the side effects of emerging genome editing tools on cells and organisms. Here, we developed a new method, DENT-seq, for efficient strand-specific profiling of nicks in complex DNA samples with single-nucleotide resolution and low false-positive rates. DENT-seq produces a single deep sequence data set enriched for reads near nick sites and establishes a readily detectable mutational signal that allows for determination of the nick site and strand with single-base resolution at penetrance as low as one strand per thousand. We apply DENT-seq to profile the off-target activity of the Nb.BsmI nicking endonuclease and an engineered spCas9 nickase. DENT-seq will be useful in exploring the activity of engineered nucleases in genome editing and other biotechnological applications as well as spontaneous and therapeutic-associated strand breaks.
Cells continuously accrue DNA damage as a result of exposure to environmental stressors. The single-strand break (SSB), or “nick,” is estimated to occur at rates of tens of thousands per cell per day, making it the most frequent type of damage to DNA (Caldecott 2008). Reactive oxygen species such as hydrogen peroxide–derived free radicals can nick DNA directly by reacting with its sugar-phosphate backbone or indirectly by altering nucleobases, which are then subject to repair pathways that entail nicked intermediates (Hegde et al. 2008). Endogenous processes including DNA replication and topoisomerase activity also generate nicks (Pommier et al. 2003).
In addition to the spontaneous accumulation of breaks, enzymes able to make engineered site-specific DNA breaks are fundamental to recombinant DNA technology and important genome editing technologies. RNA-guided nucleases, such as CRISPR associated protein 9 (Cas9) and its engineered nickase variants, represent robust tools for targeted genome editing owing to their ability to generate site-specific DNA double-strand breaks (DSBs) and nicks in cells (Garneau et al. 2010; Gasiunas et al. 2012; Jinek et al. 2013; Mali et al. 2013). Cas9 shows off-target activity, however, and the off-target DSBs generated by wild-type Cas9 can lead to significant unintended mutagenesis, toxicity, and cell death (Wu et al. 2014; Zhang et al. 2015). In efforts to make precise edits and minimize off-target DSB toxicity, Cas9's engineered nickase variants are an increasing focus in genome editing applications (Davis and Maizels 2011, 2014; Jinek et al. 2012; Ran et al. 2013; Komor et al. 2016; Gaudelli et al. 2017; Satomura et al. 2017; Anzalone et al. 2019).
For example, two guide RNAs can be designed to flank a target site and generate nicks on opposite DNA strands that together comprise an on-target DSB with reduced off-target DSBs (Ran et al. 2013). CRISPR base editors are composed of Cas9 nickase enzymes fused to either a cytidine deaminase or an adenosine deaminase enzyme to generate targeted C→T or A→G mutations, respectively (Komor et al. 2016; Gaudelli et al. 2017). The nick is incorporated on the DNA strand opposite the desired mutation to increase editing efficiency by stimulating mismatch repair at the site before base excision repair can replace the deaminated nucleotide (Komor et al. 2016; Gaudelli et al. 2017). Additionally, it has been shown that insertions can be made at nicks via homology directed repair with a designed donor DNA without nonhomologous end joining (NHEJ) and the potential for undesired insertion/deletion (indel) mutations (Davis and Maizels 2011). Creating these nicks with Cas9 nickase can also expand the targeting scope for editing, because desired edits can reside outside of the guide RNA target sequence and farther from PAM sites (Davis and Maizels 2014; Satomura et al. 2017). Furthermore, repair at nicks shows significantly higher editing precision than repair at DSBs, measured as the ratio of desired editing to unintended mutagenesis such as indels formed from NHEJ (Davis and Maizels 2011). Finally, the recently reported prime editing strategy can generate specific base substitutions, insertions, and deletions by initiating DNA synthesis at a precisely located nick using an extended guide RNA as the template for reverse transcription (Anzalone et al. 2019).
Although nicks are less toxic and pose reduced potential for unwanted indel generation than DSBs, the off-target activity of Cas9 nickase remains a concern. Large numbers of nicks can still interrupt cellular functions and lead to cell death (Zhou and Doetsch 1993; Kuzminov 2001; Fan and Zong 2014), and it has been shown that nicking base editors can cause off-target modifications (Kim et al. 2019). Furthermore, homology directed repair at off-target nicks can lead to loss of heterozygosity, a form of genetic instability commonly observed in tumor cells (Davis and Maizels 2014). The potential for unintended, potentially oncogenic modifications in even a rare subset of cells is a significant concern for clinical applications of genome editing. Genome editing technologies that rely on Cas9 nickases are not commonly tested for off-target nicking activity directly, because high-resolution nick detection technologies have not been available. Rather, off-target activity is predicted by proxy using analysis of off-target DSB sites of the nickases’ wild-type Cas9 counterparts (Komor et al. 2016; Gaudelli et al. 2017; Anzalone et al. 2019).
The study of off-target nicks in genome editing has been limited by a lack of available methods for direct, highly resolved determination of nicks. The alkaline comet assay reveals the overall extent of nicks and other alkali-labile sites (such as abasic sites) when compared with the results of a neutral comet assay, but does not provide information about the locations of breaks in genomes (Collins 2004). Ligation-mediated PCR has been used to generate and amplify DNA fragments at nicks for applications of genome sequencing, protein footprinting, and analysis of DNA damage. This approach could be applied to nick detection, although the dependence of published protocols on site-specific primers renders it suitable for targeted analysis of specific loci rather than genome-wide applications (Pfeifer et al. 1989). The SSB-seq method was developed to map nicks to the genome through nick translation with digoxigenin-modified nucleotides followed by anti-digoxigenin immunoprecipitation, although it does not achieve single-nucleotide resolution (Baranello et al. 2014). SSiNGLe is a recently reported method for nick detection with high resolution, but because it relies on detection via terminal transferase extension, its libraries are not specific to nick sites but represent both double- and single-strand breaks (Cao et al. 2019). Further, owing to the poly(A) tailing at nicks by terminal transferase, the true single-strand break site cannot be determined when the nick is flanked by at least one A residue—nearly half the sites in a random DNA sequence. GLOE-seq detects nicks with high resolution by ligation of adapters to free 3′ hydroxy (-OH) ends of DNA but, like SSiNGLe, is not specific to nicks and captures DSBs as well (Sriramachandran et al. 2020). Finally, Nick-seq is able to identify nicks with single-nucleotide resolution without capturing DSBs but has not yet been applied to gigabase-scale genomes (Cao et al. 2020). Descriptions of these methods and the DENT-seq method reported here can be found in Supplemental Table S1.
Here, we built on SSB-seq (Baranello et al. 2014) to create degenerate and enrichment nick translations followed by sequencing (DENT-seq), a method capable of specifically identifying DNA nicks with single-nucleotide resolution in human genomic DNA. This method relies on an engineered mutational signal arising from nick translation with degenerate nucleotides (Hill et al. 1998) and produces a sequence library enriched for single-strand break sites. Utilizing the mutational signal in conjunction with the enrichment of reads near nicks allows us to achieve single-nucleotide resolution and strand-specific detection of nicks with low false-positive rates. We applied DENT-seq to assess the off-target activities of the nicking endonuclease Nb.BsmI and the D10A nickase variant of spCas9. DENT-seq has many potential applications that have been underserved by existing methods for nick detection.
Results
Degenerate nucleotides provide a mutational signal demarking the locations of nicks
DENT-seq takes advantage of synthetic noncanonical deoxynucleotides capable of base-pairing with more than one of the four naturally occurring deoxynucleotides (Fig. 1). The bases 6H,8H-3,4-dihydropyrimido[4,5-c][1,2]oxazin-7-one (P) and N6-methoxy-2,6-diaminopurine (K) act as a universal pyrimidine and purine, respectively (Fig. 1A; Hill et al. 1998). dPTP and dKTP can be incorporated into a DNA molecule at the site of a single-strand break via nick translation (Fig. 1B) and generate readily detectable transition mutations when amplified by PCR and sequenced (Fig. 1C). Additionally, further extension of this nick translation product with biotin-tagged nucleotides and streptavidin pulldown enrich reads with mutational signal at nick sites (Fig. 1D).
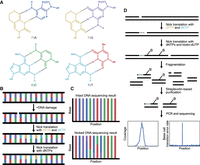
DENT-seq overview and workflow. (A) Chemical structures of the two degenerate nucleotides, dPTP and dKTP, in both of their tautomeric forms enabling their respective activities as universal pyrimidine and purine. Black dotted lines represent hydrogen bonds between nucleotides. (B) During nick translation with only the two degenerate nucleotides, P residues (gold) are inserted across from A (blue) and G (purple) residues while K residues (cyan) are inserted across from C (green) and T (red) residues. (C) Sequencing DNA fragments without nicks or that had nick translation occur with only regular dNTPs will not show a mutational signal (top). Sequencing DNA fragments that underwent nick translation with dPTP and dKTP will show a mutational signal that extends a few bases 3′ of the nick's original location (bottom). (D) Workflow used to perform DENT-seq. Nick translations are performed consecutively with dPTP plus dKTP and then with regular dNTPs plus biotinylated dUTP. Of note, nick translation will move a nick from its original location but will not actually repair the nick. DNA is fragmented and streptavidin-based purification enriches for fragments around the original (pre-nick translation) sites of nicks. After PCR and sequencing, sequence coverage and mutational information allow for sensitive single-nucleotide-resolved and strand-specific identification of nick sites.
To characterize the mutational signatures that arise from P and K, we annealed custom oligonucleotides to obtain four DNA molecules, each with a nick present just 5′ and just 3′ of each of the four canonical deoxynucleotides (Fig. 2A). After PCR with Taq DNA polymerase and sequencing, we found evidence of P and K incorporation extending 4–8 bases downstream from the original nick site, with the ratio of C:T at sites where P was incorporated being ∼40:60 and the ratio of G:A at sites where K was incorporated being ∼15:85 in agreement with a previous study (Fig. 2B; Hill et al. 1998). PCR with KAPA HiFi DNA polymerase also results in 4–8 downstream bases displaying evidence of dPTP and dKTP incorporation, although with different distributions of C:T and G:A at such sites (Fig. 2C). Whereas PCR with Taq tends to result in higher signal at nicks, PCR with KAPA HiFi tends to result in lower background noise. Overall, although both polymerases are suitable for use in DENT-seq, KAPA HiFi is a better choice for complex samples like large genomes. We considered other degenerate nucleotides for use in DENT-seq, such as inosine (I) and ribavirin (R). I is recognized as G by DNA polymerases during PCR (Alseth et al. 2014), however, and as such would not allow for true single-nucleotide resolution of nicks flanked by G residues. We tested R in place of K as a universal purine (Sala et al. 1996) but found K to produce superior results (Supplemental Fig. S1).
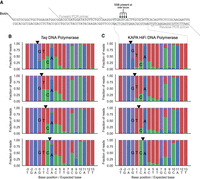
Effect of degenerate nucleotides during PCR. (A) Schematic of the oligonucleotides used to test the mutational outcome when dPTP and dKTP are incorporated into DNA and amplified by PCR. Four different oligonucleotides were generated, each with a nick directly 5′ of a different one of the four native dNTPs. The strand with the nick also contained a 5′ biotin-TEG modification to allow for purification of just that strand before PCR. PCR primers were designed to specifically amplify the region of the oligonucleotides containing the nicks and were tailed with 5′ sequences compatible with secondary PCR using barcoded P5 and P7 sequencing primers. (B,C) Sequencing result of the four oligonucleotides following consecutive nick translations with dPTP plus dKTP and then with standard dNTPs, purification, and PCR with Taq DNA polymerase (B) or KAPA HiFi DNA polymerase (C). The black triangles represent the location of the nick for each sample.
Biotinylated nick translation products can be enriched to focus sequencing effort at nick sites and enable confident strand determination
Nick translation with unmodified and biotin-tagged nucleotides after incorporation of the degenerate nucleotides allows for selective pulldown of DNA molecules containing P and K residues via a streptavidin-based purification. By using a low dNTP concentration (5000 times lower than typical PCR protocols), we limit incorporation of biotin-tagged nucleotides to within a few hundred bases of the original nick site. Increased sequence coverage around nick sites reduces the total amount of sequencing needed, provides independent evidence of nicks that can be analyzed in conjunction with the mutational signal, and reduces the number of bases that need to be tested for mutational signal (Fig. 1D). To test library enrichment, we digested a plasmid containing two recognition sites with the nicking endonuclease Nb.BsmI (Fig. 3A). After performing DENT-seq on this sample, we observed up to 1000-fold higher coverage of sequence reads at sites around the nicks (Fig. 3B). These coverage peaks can be identified by MACS2, a peak caller originally designed for use with ChIP-seq data (Zhang et al. 2008), and the mutational signal from P and K incorporation reveals the exact location of nicks within the peaks (Fig. 3C,D).
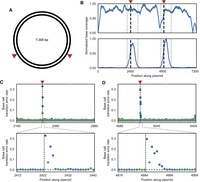
Single-base-pair resolution of nicks in plasmid DNA. (A) Schematic of the plasmid used to test DENT-seq in which red arrows represent the expected locations of nicks after treatment with the nicking endonuclease Nb.BsmI (both are expected on the reference strand). (B) Sequence coverage normalized to the location with maximum read depth for untreated plasmid (top) and plasmid with two nicks introduced by Nb.BsmI treatment (bottom) after performing DENT-seq. Black dashed lines represent the expected locations of nicks after Nb.BsmI treatment and gray dashed lines represent the bounds of MACS2 peaks called on data from the treated sample. (C,D) Mutational signal for all locations within the called MACS2 peaks (top) and close-up views where there is high mutational signal (bottom). Black dashed lines represent the expected locations of nicks after Nb.BsmI treatment. Data in gray represent the untreated plasmid, whereas data in blue and green represent the Nb.BsmI treated sample where a P or K residue would be inserted after a reference strand nick, respectively.
In enriched samples, we observe mutational signal from P and K in a large majority (∼90%) of unique sequence reads that contain the original nick site. These signals are robust to the enzyme used to incorporate the biotinylated nucleotides (Supplemental Fig. S2) and the enzyme used to conduct PCR (Supplemental Fig. S3). Furthermore, similar levels of enrichment are achieved when using nucleotides that are modified with desthiobiotin (Hirsch et al. 2002; Supplemental Fig. S4). Although we originally developed DENT-seq using a biotin-tagged uracil deoxynucleotide (biotin-dUTP), desthiobiotin-tagged nucleotides are a superior option. Use of a desthiobiotin-tagged adenosine deoxynucleotide (desthiobiotin-dATP) provides better compatibility with DNA polymerases and sequencing library construction protocols than biotin-dUTP. In addition, desthiobiotin-tagged nucleotides can be eluted from streptavidin-coated magnetic beads with free biotin, so that PCR can be carried out without potential interference from the presence of the beads. Peak width and the magnitude of enrichment are highly dependent on the concentration of nucleotides during the nick translation reaction. Concentrations higher than 40 nM per dNTP are not recommended due to an associated increase in background sequence coverage (Supplemental Fig. S5).
Library enrichment also plays a crucial role in determining the strandedness of a nick. As DNA polymerases extend DNA in the 5′→3′ direction and the nick translation steps are carried out consecutively, the biotinylated nucleotides are always 3′ of the degenerate nucleotides and the position with maximum enrichment is expected 3′ of the mutational signal. Sequence data are typically viewed with respect to the reference strand of double-stranded DNA, so a nick that occurred on the reference strand will result in the coverage peak appearing 3′ of the mutational signal. A nick that occurred on the non-reference strand, however, will result in the coverage peak appearing 5′ of the mutational signal because it is on the 3′ side with respect to the non-reference strand (Supplemental Fig. S6). Determining the strandedness of a nick is, in turn, important for single-nucleotide resolution because multiple P and K residues can be incorporated at a nick, resulting in a few consecutive bases with mutational signal that enhances the mutation signal's detectability, but does not unambiguously identify the nick site. Stranded determination unambiguously identifies the nick site, which is immediately 5′ of the mutational signal on the nicked strand.
Finally, this enrichment is essential to DENT-seq's high sensitivity. Not all DNA molecules in a sample will necessarily be nicked at the same locus, but by including this enrichment we can selectively sequence a larger fraction of DNA fragments originating from nicked molecules versus those originating from non-nicked molecules. In this way, the mutational signal arising from dPTP/dKTP can remain high even as the fraction of molecules containing nicks at a given locus decreases. Even so, DENT-seq has finite sensitivity and specificity. To explore these limits in different scenarios, we carried out a series of experiments and analyses.
By serially diluting a nicked plasmid in a non-nicked plasmid, we showed that DENT-seq identifies nicks present in as few as 1 in 1000 DNA molecules (Supplemental Fig. S7). We then performed bootstrap resampling of reads in each of these nick dilution data sets to estimate DENT-seq's false-negative rate. Similarly, we performed bootstrap resampling of a data set obtained by performing DENT-seq on non-nicked DNA to estimate the false-positive rate (false-positive and false-negative rate estimates are provided in Supplemental Table S2). False-negative rate is highly dependent on nick penetrance; whereas we estimate rates as high as 12.18% when nicks are rare at a locus (1 nicked molecule out of 1000 copies at a single locus), we estimate false-negative rates <0.1% for more penetrant nicks (≥1 nicked molecule out of 100). Of note, the false-negative rate for low penetrance nicks is based on a conservative mutation rate cutoff for nick calling and lowering this cutoff could reduce the false-negative rate with little effect on false-positive rate. False-positive rate is highly dependent on the class of DNA being tested. Based on our results, we estimate about one false positive occurring across every four experiments performed on human genomic DNA (i.e., less than one false positive call in 10 billion genomic bases) and lower rates for less complex samples like microbial genomes and synthetic DNAs.
Nicking endonuclease Nb.BsmI has detectable off-target activity
Nb.BsmI has been reported to display off-target activity in reactions with too high an enzyme concentration, that proceed for too long, or that have modified buffer conditions. Utilizing an increased concentration of Nb.BsmI (cf. Fig. 3) causes off-target activity that is detectable by DENT-seq (Fig. 4A). Enrichment of the two on-target sites still occurs, but to a lesser extent than when off-target nicks are absent. Because read coverage is relatively high throughout the plasmid, we did not restrict our nick calls to MACS2 peaks in this instance. In addition to the on-target sites, there are 12 sites with mutational signal above background level (Fig. 4B; Supplemental Table S3). Each of these sites has a single-nucleotide mismatch within the 6-nt consensus Nb.BsmI recognition sequence. Given a total of 57 such sites in the plasmid, observing 12 sites with high mutational signal occurring at such near-cognate Nb.BsmI sites by chance is highly unlikely (P < 10−25, hypergeometric test), suggesting these mutational signals indeed reflect the presence of nicks resulting from true off-target Nb.BsmI activity. Because these off-target nicks are not associated with highly enriched MACS2 peaks, their strandedness cannot be determined from the mutation signal alone. Here, even without the presence of enrichment peaks, the orientation of the off-target Nb.BsmI recognition sequence in the plasmid reference can be used as an alternative method to determine the nicked strand. Although the identified off-targets do not occur in enrichment peaks, the enrichment procedure significantly enhances their detection (Supplemental Fig. S8). This likely results from such off-target nicks having low penetrance and the enrichment selecting for DNA molecules that underwent the nick translation with dPTP and dKTP.
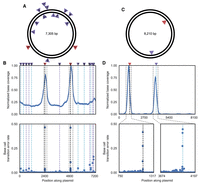
Detection of off-target nicks in plasmid DNA. (A) Schematic of a plasmid used to test DENT-seq, in which red arrows represent on-target activity of Nb.BsmI and purple arrows represent detected off-target activity of Nb.BsmI. (B) Normalized sequence coverage (top) and base transition rate (bottom) for the plasmid in A. Black dashed lines represent on-target locations of Nb.BsmI activity and gray dashed lines represent the bounds of called MACS2 peaks. Purple and cyan dashed lines represent detected off-target locations of Nb.BsmI activity on the reference and non-reference strands, respectively. (C) Schematic of a plasmid used to test DENT-seq in which the red arrow represents on-target activity of D10A spCas9 nickase and the purple arrow represents detected off-target activity (both are on the non-reference strand). (D) Normalized sequence coverage (top) and base transition rate (bottom) for the plasmid in C. The black dashed line represents on-target activity of D10A spCas9 nickase, whereas the cyan dashed line represents off-target activity. The gray dashed lines represent the bounds of called MACS2 peaks.
Cas9 nickase has detectable off-target activity in plasmid DNA
Cas9 off-target activity has been observed at locations with significant, but not full, complementarity to the guide RNA (Kim et al. 2015; Tsai et al. 2015, 2017; Slaymaker et al. 2016; Yan et al. 2017). We were able to observe this phenomenon directly in D10A spCas9 nickase activity via DENT-seq in plasmid DNA. We used a guide RNA with one fully complementary on-target site and one predicted off-target site where proper base-pairing would occur for 15 of the 20 nucleotides of the guide RNA (Fig. 4C). Significant enrichment and a strong mutational signal occurred at both sites, with slightly lower enrichment observed at the off-target site (Fig. 4D; Supplemental Table S4).
Nick detection at genome scale
After demonstrating the capabilities of DENT-seq on plasmid DNA, our next goal was to detect nicks in a genome. We used D10A spCas9 nickase to incorporate nicks in an Escherichia coli genome of 4.62 Mbp by carrying out a reaction with two guide RNAs wherein we predicted nine nicks would occur at fully cognate target sites (Fig. 5A). After performing DENT-seq, all nine nicks were identified with no false positive identifications. Eight nicks, all originating from the same guide RNA, resulted in highly significant sequence coverage peaks (Fig. 5B), and single-nucleotide resolution was achieved using the mutational signal (Fig. 5C; Supplemental Fig. S9A). The remaining nick, originating from the other guide RNA, was located within a MACS2 called peak but with lower enrichment. In fact, this peak could not be distinguished from background peaks based on sequence coverage or MACS2 statistics alone (Fig. 5B). The mutational signal present in this peak allowed the nick to be detected and localized (Fig. 5D). We hypothesize that the difference in sequence coverage between the peaks containing nicks is owing to different guide RNA activity levels that result in differential nick penetrance. Other sequence coverage peaks either lacked mutational signal altogether, contained singleton mutational signal (not at multiple consecutive or near-consecutive nucleotides), or contained mutation types other than the transition mutations that dPTP and dKTP are known to cause (Supplemental Fig. S9B). These signals likely arose from sources other than P and K incorporation, such as misincorporation of canonical dNTPs during PCR or sequencing errors, and as such were straightforwardly filtered out to achieve high specificity. We did not expect to observe off-target activity from either guide RNA in this experiment. One guide showed low on-target activity, and there were no sites in the genome with significant homology to the other, high activity guide that lacked mismatches at the 3′ end of the target sequence and were also adjacent to a Cas9 PAM site.
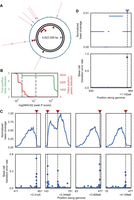
Single-base-pair resolution detection of nicks in genomic DNA. (A) Schematic of an E. coli genome used to test DENT-seq, in which red arrows represent the activity of Cas9 nickase paired with one guide RNA and the purple arrow represents the activity of Cas9 nickase paired with a different guide RNA. Circularized plot data (all colors) show normalized sequence coverage. Green signals represent the locations of called MACS2 peaks with quality score greater than 0.4. Red signals represent locations of such peaks wherein a nick is identified with the exact location of that nick represented by a black dot. (B) Cumulative number of nicks contained within MACS2 peaks (green) and cumulative number of MACS2 peaks containing no nick (red) as a function of P-value threshold. The left gray dashed line represents the P-value where the final nick-containing peak is identified, and the right gray dashed line represents the P-value where the first peak not containing a nick is identified. This panel uses only the coverage signal and does not incorporate mutational data. (C) Normalized sequence coverage (top) and base transition rate (bottom) for four MACS2 peaks that contain nicks resulting from Cas9 nickase in conjunction with one of the guide RNAs used (corresponding to the red arrows in A). The second set of plots from the left represent a single MACS2 peak, which encompassed two nicks owing to their close proximity. (D) Normalized sequence coverage (top) and base transition rate (bottom) for the MACS2 peak that contains the nick resulting from Cas9 nickase in conjunction with the other guide RNA used (corresponding to the purple arrow in A). Coverage is lower for this guide, but there are still nucleotide positions proximal to the Cas9 nickase target site with high mutational signal that enable nick identification.
Off-target Cas9 nickase activity can be detected across the human genome
We performed DENT-seq on human genomic DNA samples treated with D10A spCas9 nickase and guide RNAs targeting either the AAVS1 gene on Chromosome 19 (the same guide used in the plasmid experiment described above) or the VEGFA gene on Chromosome 6. With the AAVS1 guide, this allowed for the identification of six nicks without using a priori knowledge of expected Cas9 on- or off-target activity to inform nick calling. Of these six nicks, the one with the highest enrichment is located at the on-target site (Fig. 6A), and the other five are located at off-target sites each with three or four mismatches to the guide RNA (Fig. 6B). Comparing maximum read depth around the on-target nick site to average read depth across the whole genome reveals ∼28-fold higher sequencing effort would have been required to obtain similar read depth around the on-target site without the streptavidin pulldown. Of note, this calculation underestimates the benefit of the pulldown because it only considers read depth. Mutational signal is also enhanced by the pulldown, particularly for nick sites where the nick is not fully penetrant as reads from the nick translation products are enriched relative to unmodified sequences from non-nicked molecules at the same locus. In particular, nuclease off-target sites often have lower nick penetrance than the on-target site, and some may not have any apparent mutational signal above background if the pulldown was not performed. Nonetheless, in the absence of a priori target information, we only identified the on-target nicking site in the VEGFA nicking experiment (Supplemental Fig. S10). Although prior work using the same VEGFA guide, wild-type spCas9 nuclease, and genome-wide DSB detection did identify off-target DSB sites (Kim et al. 2015; Tsai et al. 2015; Slaymaker et al. 2016; Yan et al. 2017), we wanted to explore off-target nick sites in a minimally biased fashion, and for this reason focused our further analyses on data from experiments using the AAVS1 guide.
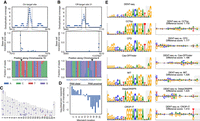
Detection and analysis of D10A spCas9 nickase on- and off-target activity in human genomic DNA. (A) D10A spCas9 nickase single-site on-target activity is represented by the most prominent MACS2 peak, denoted by the gray dashed lines, which also contains strong mutation signal. The reverse complement of the guide sequence is shown in blue with the PAM in green at bottom. (B) One of the five off-target nicks that were identified without using a priori knowledge of expected off-target sites. Gray lines mark the MACS2 peak bounds. At bottom, the genomic sequence where the guide RNA binds is shown in blue with mismatches in red. (C) Ideogram showing the site of on-target activity in red and candidate off-target sites with DENT-seq mutational signal in purple. (D) Comparison of the frequency of mismatch locations in the guide RNA at the sites of detected off-target activity to all potential sites proposed by Cas-OFFinder. (E) Sequence motifs created from the sites with evidence of D10A spCas9 nickase off-target activity and the top hits predicted by multiple Cas9 off-target predictors (left). The motif for the sites identified by DENT-seq is compared to the motifs for each of the software predictions using DiffLogo (right). The bases composing the PAM sequence are shown with a light gray background. Dispersion metrics for the prediction software sequence logos were generated by performing 10,000 bootstrap resamples of the entire list of potential off-target sites proposed by each algorithm and calculating the position probability matrix for the highest rated 52 sites in each resample. Ninety percent confidence intervals for the frequency of each base at each position are displayed under the assumption that the information content at each position remains unchanged.
Nick calling can be enhanced further by considering only prospective off-target locations. To this end, we used Cas-OFFinder (Bae et al. 2014) to identify all 714,765 PAM-adjacent sites in the genome with up to eight mismatches to the AAVS1 guide RNA or with up to two mismatches and a 2-nt DNA or RNA bulge. Analyzing these locations for sequence reads and mutational signal enabled the identification of 53 sites with strong evidence for off-target activity (Fig. 6C; Supplemental Table S5). These off-target sites were generally concordant with previously reported characteristics of wild-type Cas9 off-target DSB activity (Tsai et al. 2015). Mismatches closer to the 5′ end of the guide RNA, distal from the PAM, are better tolerated than mismatches at the 3′ end, proximal to the PAM (Fig. 6D). Further, the PAM sequence 5′-NGG is the most prominent but there is observable off-target nicking activity at the alternative PAM 5′-NAG.
The guide RNA used here was previously tested for off-target activity in the human genome with wild-type spCas9 (Wang et al. 2014; Tan et al. 2015). The methods previously used for DSB analysis with this particular guide were neither unbiased nor genome-wide, but rather relied on PCR and deep sequencing of select locations in the genome predicted as likely off-target sites (99 unique sites were tested between the two studies), which hinders direct comparison to our findings. However, DENT-seq provides evidence of D10A spCas9 nickase activity at five off-target sites where the previous studies described no wild-type spCas9 DSB off-target activity using the same guide RNA. Three of these five sites were detectable as nicks without the use of a priori knowledge of expected off-target sites and showed highly significant peak enrichment as measured by MACS2 (Supplemental Table S5). Furthermore, the previous studies identified eight off-target sites with wild-type spCas9 DSB activity where DENT-seq did not show D10A spCas9 nickase activity, suggesting that wild-type Cas9 and the D10A nickase mutant could have different off-target spectra.
To summarize the off-target activity of the D10A spCas9 nickase variant, we generated a sequence motif from the 52 sequences where nickase activity was identified and base-pairing between the guide RNA and target DNA did not result in a DNA or RNA bulge. To compare the D10A spCas9 nickase off-targets detected by DENT-seq with predicted wild-type spCas9 off-targets, we used several DSB off-target prediction tools. CCTop (Stemmer et al. 2015), CRISPOR (Concordet and Haeussler 2018), DeepCRISPR (Chuai et al. 2018), and CROP-IT (Singh et al. 2015) ranked all genome-wide potential off-targets and we generated sequence motifs for the top 52 sites proposed by each algorithm (for direct comparison with the 52 DENT-seq identified sites). Two motifs were created based on CRISPOR's output as it reports both MIT off-target scores (score calculation is based on data from Hsu et al. 2013) and CFD off-target scores (Fig. 6E, left column; Doench et al. 2016). To compare the motifs created from our results and from each of the prediction tools, we used DiffLogo (Nettling et al. 2015). Each position in the DiffLogo motif summarizes the base magnitude and polarity of difference between the two motifs being compared and enables the calculation of a scalar total difference score between the motifs. We created DiffLogo motifs to perform pairwise comparisons between the DENT-seq identified off-targets and the proposed off-targets from each prediction tool, including the background distribution of all possible off-targets identified by Cas-OFFinder (Fig. 6E, right column). From the DiffLogo scores, the motif generated by the CCTop predictions most closely resembles the motif generated by the DENT-seq results. The biggest discrepancy between these two motifs is at the base just next to the PAM region where T is not as frequent at off-target sites as CCTop predicts. This is in contrast to CROP-IT and, to a lesser extent, DeepCRISPR, where too little emphasis is placed on this position. For the CFD and MIT scores, it appears as though the bases at the 5′ end of the target sequence are being weighed too heavily and the 5′-NAG alternative PAM is nearly ignored.
All of the prediction tools used here are meant to predict wild-type spCas9 DSB off-target activity. We expect there to be some differences in the off-target strand breaks created by wild-type spCas9 and its D10A nickase variant, so our analysis here is not a test of how well these tools perform at the task they were designed for. Rather, it reveals which of the currently available methods for a related task performs best at predicting D10A spCas9 nickase off-target activity. As additional D10A spCas9 nickase off-target sites are characterized using DENT-seq, optimal parameters for each algorithm can be identified for predicting D10A spCas9 nickase activity and the prediction tools could have the option to search for expected wild-type/DSB or D10A nickase off-target activity.
Discussion
We show a method for identifying nicks in human genomic DNA with single-nucleotide resolution and strand specificity using a mutational signature and read enrichment. The technique relies on incorporating degenerate nucleotides and a tag for enrichment at nicks through serial nick translations and reading out a mutational signal by sequencing. Because this results in multiple consecutive transition mutations being identified, true signal can be readily distinguished from PCR misincorporations, sequencing errors, and other sources of noise that occur randomly and are unlikely to be present at consecutive nucleotides. In addition, library enrichment around nick sites reduces the sequencing effort required for nick detection in complex samples like large genomes and enhances specificity by reducing the number of candidate sites. Further, the nicked strand can be identified by comparing the relative locations of the mutational signal and the peak in enriched sequence coverage.
In addition to the significance of site-specific nicks in genome engineering, the accumulation of nicks from increased DNA damage or dysregulated DNA repair activity has many consequences. DNA lesions, including nicks, can block RNA polymerase progression and result in incomplete transcription of the genes that contain such lesions (Zhou and Doetsch 1993). Furthermore, nicked genomic DNA can lead to replication fork collapse during DNA synthesis, resulting in DSBs and indel mutations after error-prone DSB repair (Kuzminov 2001). Excessive damage can activate poly(ADP-ribose) polymerase (PARP), a protein capable of identifying nicks, signaling for their repair, and triggering cell death via necrosis or apoptosis (Fan and Zong 2014). Nicks have also been implicated in heart failure in mouse models through increased expression of inflammatory cytokines (Higo et al. 2017) and are thought to be a major factor affecting the rate of telomere shortening (von Zglinicki et al. 2000; von Zglinicki 2002). Ataxia-oculomotor apraxia 1 (AOA1) (Le Ber et al. 2003) and spinocerebellar ataxia with axonal neuropathy 1 (SCAN1) (Takashima et al. 2002) are neurodegenerative disorders associated with deficient repair of nicks resulting from abortive DNA ligase and topoisomerase 1 activities, respectively. Finally, deficient nick repair is observed in many tumors (Caldecott 2008).
Despite their importance, nicks have been less of a focus for study than DSBs. Previously available nick detection methods offered low resolution, preventing the identification of fine-scale sequence biases and genomic features that help identify processes affecting nick accumulation and repair. Here, we showed the capability of DENT-seq to profile the off-target activity of Cas9 nickase. DENT-seq could also be beneficial in other contexts in which nick detection is relevant, such as analysis of damage caused by reactive oxygen species and other genotoxic agents or the relationships between nick accumulation and neurodegeneration in diseases characterized by deficient nick repair, telomere shortening, and cell death. For such applications, though, it is possible that nicks appearing at extremely low penetrance would not be detected using DENT-seq. Furthermore, as recently reported by Cao et al. (2020), nick detection methods like DENT-seq could be applied to study other types of DNA damage as well by converting the damage into nicks; for example, base modifications could be studied through treatment with a glycosylase and AP endonuclease from the base excision repair pathway before readout by DENT-seq.
We showed the ability of DENT-seq to identify nicks expected to be rare within a sample of DNA molecules by detecting off-target activity of the nicking endonuclease Nb.BsmI and the D10A nickase variant of spCas9. The off-target activities of these and other enzymes are important to quantify as they critically limit applications including therapeutic genome editing. CRISPR-Cas9 nickase is used as an essential component of several advanced genome editing strategies (Davis and Maizels 2011, 2014; Ran et al. 2013; Komor et al. 2016; Gaudelli et al. 2017; Satomura et al. 2017; Anzalone et al. 2019). However, no method has been available to directly test the off-target activity of Cas9 nickase because nicks could not previously be detected in human genomic DNA with single-nucleotide resolution. Our results suggest that prior work to understand the off-target characteristics of wild-type Cas9 may not fully translate to nickases, underscoring the need for express analysis of off-target nicking to model off-target activity and support the development of next-generation nickase enzymes and computational tools that predict activity spectra. Of additional interest, Cas enzymes that nominally generate DSBs may create nicks that require further study by methods able to distinguish double- and single-strand breaks to fully characterize the spectrum of cleavage products. In particular, Cas12a (formerly Cpf1) was recently reported to show nicking activity in addition to its duplex cleaving activity (Murugan et al. 2020). DENT-seq provides a compelling combination of performance characteristics and ease of implementation to tackle the growing need to characterize single-strand breaks in complex DNA substrates across a range of applications.
Methods
Preparation of DNA containing nicks
Biotinylated double-stranded oligonucleotides with one nick each were generated by annealing single-stranded oligonucleotides ordered from Integrated DNA Technologies. Nine oligos were ordered in total: a constant bottom strand (89 nt long) used for each annealing reaction, four 5′ top strands of varying lengths (54, 55, 56, and 57 nt) with 5′ biotin-TEG modifications, and four 3′ top strands of varying lengths (35, 34, 33, and 32 nt) (Fig. 2A). Annealing was performed by creating an equimolar mixture of the bottom strand and a top strand pair, denaturing any base-pairing by heating to 95°C on a thermocycler, and reducing the temperature to 25°C at a rate of 0.1°C per sec.
Nicking endonuclease Nb.BsmI (New England Biolabs) was used to incorporate nicks in one of the plasmids (extracted from bacteria with the Plasmid Plus Midi Kit, Qiagen) used. Two nicks, both on the reference strand, were expected based on sequence. A 10 μL reaction was prepared containing 1× NEBuffer 3.1 (New England Biolabs), 5 units of endonuclease, and 100 ng of plasmid and incubated for 1 h at 65°C. Enzyme inactivation then occurred by incubation for 20 min at 80°C.
EnGen Spy Cas9 nickase (New England Biolabs), a Cas9 nuclease variant containing a D10A mutation in the RuvC nuclease domain, was used in conjunction with guide RNA (Synthego, synthetic cr:tracrRNA kit) to incorporate nicks in the other plasmid used as well as the E. coli genomic DNA (Thermo Fisher Scientific; genomic DNA purified from E. coli type B cells, ATCC 11303 strain) and human genomic DNA (extracted from cells with DNeasy Blood and Tissue Kit, Qiagen). One guide was used with the plasmid, expected to produce one nick on the non-reference strand. Two guides were used with the E. coli genome, expected to produce eight and one nicks, respectively, based on sequence. One guide, targeting the AAVS1 gene on Chromosome 19, was used with the human genome. crRNA and tracrRNA were rehydrated and annealed according to the manufacturer's instructions. For plasmid and E. coli genomes, 30 μL reactions were prepared containing 1× NEBuffer 3.1, 50 pmol of each guide RNA being used, 2 pmol Cas9 nickase, and 300 ng of DNA and incubated for 1 h at 37°C. Enzyme inactivation then occurred by incubation for 5 min at 65°C. For reactions with human genomic DNA, a larger reaction volume and 10 μg of DNA were used.
Incorporation of degenerate and biotinylated nucleotides
The degenerate nucleotides dPTP (TriLink Biotechnologies) and dKTP (Axxora) were incorporated into DNA at nicks via nick translation with Taq DNA polymerase (New England Biolabs). A 10 μL reaction was prepared containing 1× ThermoPol buffer (New England Biolabs), 0.25 units of polymerase, 20 pmol each of dPTP and dKTP, and 10 ng of DNA (or 10 μg of DNA in a larger reaction volume in the case of human genome) and incubated for 15 min at 72°C. Polymerase and excess nucleotides were removed by 1:1 SPRI with Agencourt AMPure XP beads (Beckman Coulter) followed by elution in TE buffer. Ten nanograms of DNA was used because it is a standard input quantity for the downstream library preparation step. For human genomic DNA, 10 μg was used to maintain approximately the same number of genome equivalents used in the E. coli genomic DNA experiment.
Next, nick translation was performed with regular dNTPs (New England Biolabs) and biotin-11-dUTP (Thermo Fisher Scientific). Optimal results were obtained with biotinylated dUTP and dTTP in a 1:5 ratio. The purified DNA was incubated for 30 min at 72°C with 1× ThermoPol buffer, 0.25 units Taq polymerase, and 0.4 pmol of each dNTP (a 1 in 5000 dilution compared to the manufacturer's recommended PCR protocol) plus biotinylated dUTP. Polymerase and excess nucleotides were again removed by 1:1 SPRI and elution in TE buffer. During experiments with the oligonucleotides only (Fig. 2), biotinylated dUTP was not used during the second nick translation (because the oligonucleotides already contained a 5′ biotin-TEG modification). During experiments with human genomic DNA only, desthiobiotin-7-dATP (Jena Bioscience) was used in place of biotinylated dUTP for compatibility with the library preparation protocol.
Targeted pulldown and library construction
Plasmid DNA and bacterial genome were tagmented for 10 min at 55°C in a 20 μL reaction containing 5 mM magnesium chloride, 10 mM tris acetate, and 4 μL TDE1 (tagment DNA enzyme, Illumina). Then 0.1% SDS treatment denatured the enzyme and 1:1 SPRI followed by elution in TE buffer was used to remove SDS. Human genomic DNA was fragmented by a dsDNA fragmentase (New England Biolabs) followed by end preparation and adapter ligation according to the NEBNext Ultra II DNA Library Prep Method for Illumina (New England Biolabs).
Twenty-five micrograms of Dynabeads MyOne Streptavidin C1 (Invitrogen) was washed in 100 μL of bind and wash buffer, 1 M sodium chloride and 0.1% tween-20 in TE buffer. DNA was diluted to 150 μL in bind and wash buffer and incubated on the beads for 30 min at room temperature with occasional gentle manual agitation. Following three washes in 180 μL bind and wash buffer, beads and bound biotinylated DNA were resuspended in 20 μL of TE buffer plus 0.01% tween-20. In experiments with the oligonucleotides only (Fig. 2), treatment with 100 mM sodium hydroxide for 5 min was used after the third wash followed by a final wash and resuspension to disrupt base-pairing and allow for purification of the biotinylated strand (which initially contained the nick) only. In experiments with human genomic DNA only, the final wash was followed by a 20-min incubation in 10 μL of 0.11 mg/mL biotin (Sigma-Aldrich) to elute desthiobiotinylated DNA from the beads.
One microliter of DNA (with beads unless elution was performed) was the substrate for a 10 μL qPCR reaction using either Taq DNA polymerase or KAPA HiFi (KAPA HiFi HotStart PCR kit, Kapa Biosystems) according to the manufacturer's protocol with barcoded P5 and P7 sequencing primers at 0.5 mM and with 1× EvaGreen dye (Biotium) and 1× ROX reference dye (Invitrogen) used to monitor DNA amplification. Reagents and short primer-derived products were removed by 1:1 SPRI followed by elution in TE buffer.
Sequencing
Enriched libraries were quantified using the Qubit dsDNA high sensitivity assay kit (Invitrogen) according to the manufacturer's instructions. DNA was diluted to 1 nM and prepared for paired-end sequencing on a MiniSeq (Illumina) using a 75- or 150-cycle kit according to the manufacturer's instructions with a final loading concentration of 1.1 pM.
Following sequencing and barcode-based demultiplexing, cutadapt (Martin 2011) removed primer sequences present from both the 5′ and 3′ ends of the reads. Bowtie 2 (Langmead and Salzberg 2012) aligned reads to the appropriate reference genome with default parameters except maximum insert length (which we increased from 500 to 800) and discordant read pair mappings were disallowed; mapping rates and other sequencing metrics can be found in Supplemental Table S6. The high read depths reported in Supplemental Table S6 (especially for plasmids) can improve nick detection confidence but are not required for performing DENT-seq; for example, subsampling in SAMtools revealed that the nicks displayed in Figure 3 could be identified had the plasmid been sequenced to as low as ∼0.53× coverage as opposed to the observed ∼13,261× coverage. Experiments on human genomic DNA were mapped to GRCh37 (hg19) to compare results with literature that also mapped to this reference (Wang et al. 2014; Tan et al. 2015). The reads were also mapped to GRCh38 (hg38), the most recent human genome reference build, but this did not alter the findings except for small variations to MACS2 Q-scores (Supplemental Table S7). SAMtools (Li et al. 2009) created two sorted and indexed BAM files: one including all reads and one with duplicate reads removed. Peaks in sequence coverage were identified by MACS2 (version 2.1.1) (Zhang et al. 2008), a peak caller originally created for use with ChIP-seq data. We applied MACS2 with default parameters except genome size, which was set to match each sample and FDR (Q-value) cutoff set to 0.95. (Such a high value was chosen such that all potential peaks would be identified and low-quality peaks could be filtered out in downstream analyses.)
Computational analysis
Further analysis of sequencing data was performed with custom scripts written in Python 3.6.8. In plasmid DNA, on-target nicking by Nb.BsmI and nicking by Cas9 nickase are readily identified by manual inspection because the signals are overwhelming and the number of nicks is small. Each plasmid contains only two MACS2 peaks, these peaks are very highly enriched, and there is very high mutational signal only at the nicked loci. Off-target Nb.BsmI nicking is identified programmatically by examining the transition mutation rate at every locus. All loci with transition mutation rate above a cutoff are considered hits. Each hit can be determined to be either a potential Nb.BsmI off-target or a potential false positive based on the plasmid's reference sequence. The cutoff was set using logic to ensure that hits are called at the two Nb.BsmI on-target sites and at 12 off-target sites with no calls at sites lacking significant homology with the Nb.BsmI target sequence. Cutoff values between 0.0115 and 0.0163 produce the same results on this data set.
We initiated nick identification in genomic DNA by locating all the peaks called by MACS2. In human genomic samples, we excluded MACS2 peaks with Q-scores <0.4 and peaks overlapping an ENCODE blacklist region to address the challenge of accurately mapping short reads in the large, repetitive human reference genome. As peaks with low-quality scores are included here there are many false-positive peak calls, but use of the mutational signal prevents false-positive nick calls in these peaks. Within the MACS2 peaks, all nucleotide positions are assigned transition mutation Q-values based on binomial tests determining the probability of receiving the observed transition mutation rate (C↔T and A↔G, the only mutations expected from dPTP and dKTP incorporation) at each location with the null expectation being that the observed rate is equal to the maximum of either the observed transversion (nontransition) mutation rate at that location or 0.03. Candidate nick sites were determined in this manner where the observed transition mutation rate at that location is likely attributed to dPTP or dKTP incorporation and not to other factors such as misincorporation during PCR, sequencing errors, or mapping errors. These sites were then filtered based on proximity to one another; that is, if a site did not have another site within close proximity (<5 nt) it was removed because dPTP and dKTP incorporation is expected to produce a string of mutations during nick translation under the conditions of our protocol.
At this point, each locus with a predicted nick consists of the range of locations over which there was mutational signal with the nick expected to lay at one end of the range. Comparing the location of the mutational signal range to the location of maximum coverage in the MACS2 peak reveals which end of the range as well as which strand of DNA the causal nick likely occurred on. Because DNA is always synthesized 5′→3′, the location of maximum coverage is expected 3′ relative to the nick. Therefore, if the nick occurs 5′ to the peak's maximum on the reference strand, it is a reference strand nick at the 5′ end of the predicted range. If the nick occurs 3′ to the peak's maximum on the reference strand (which is 5′ on the non-reference strand), it is a non-reference strand nick at the reference strand's 3′ end (non-reference strand's 5′ end) of the predicted range. During this analysis and in all figures, mutation rates are determined from the BAM file with all sequencing reads, and sequence coverage is determined from the BAM with duplicate reads removed. (Using the BAM with all sequencing reads slightly alters figure appearance but does not alter nick prediction.) Coverage was calculated by inferring the full DNA molecules from the sequenced paired-ends and counting the number of times each locus in the reference is represented. When normalized, all coverage values were simply divided by the maximum coverage value (so that the normalized values range from 0 to 1).
Data access
All sequence data generated in this study have been submitted to the NCBI BioProject database (https://ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA643848. A Jupyter notebook containing all custom Python code used can be found on the Blainey Lab GitHub (https://github.com/blaineylab/DENTseq) and in Supplemental Code.
Competing interest statement
The Broad Institute and MIT have filed patent applications related to the work described here. One or more coauthors are inventors on the applications. The title of the patent application is “Single-Stranded Break Detection In Double-Stranded DNA.” The U.S. Provisional Application was filed on April 19, 2018, 62/660,028. The PCT application was filed on April 18, 2019, PCT/US19/28091, nationalized in the United States and Europe. P.C.B. is a consultant to and/or holds equity in companies that may develop or apply genomic or genome editing technologies: 10x Genomics; General Automation Lab Technologies; Celsius Therapeutics; Next Gen Diagnostics, LLC; Cache DNA; and Concerto Biosciences.
Acknowledgments
The authors thank Arnaud Gutierrez for discussions in designing the DENT-seq method, as well as Dr. David Liu and members of his laboratory for discussions about the application of DENT-seq to genome editing. We also thank all members of the Blainey laboratory for discussions and providing materials used in this work. This work was supported by start-up funds from the Broad Institute and a New Innovator Award from the National Institutes of Health (HL141005). P.C.B. was supported by a Burroughs Wellcome Fund Career Awards at the Scientific Interface (CASI) Award. S.E.D. was supported by Massachusetts Institute of Technology Undergraduate Research Opportunities Program (MIT UROP) funding.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.265223.120.
-
Freely available online through the Genome Research Open Access option.
- Received April 27, 2020.
- Accepted November 23, 2020.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








