
Parallel bimodal single-cell sequencing of transcriptome and chromatin accessibility
- Qiao Rui Xing1,2,11,
- Chadi A El Farran1,3,11,
- Ying Ying Zeng1,2,
- Yao Yi1,3,
- Tushar Warrier1,3,
- Pradeep Gautam1,3,
- James J. Collins4,5,6,
- Jian Xu3,7,
- Peter Dröge2,
- Cheng-Gee Koh2,
- Hu Li8,
- Li-Feng Zhang2 and
- Yuin-Han Loh1,3,9,10
- 1Epigenetics and Cell Fates Laboratory, Institute of Molecular and Cell Biology, A*STAR, Singapore 138673, Singapore;
- 2School of Biological Sciences, Nanyang Technological University, Singapore 637551, Singapore;
- 3Department of Biological Sciences, National University of Singapore, Singapore 117558, Singapore;
- 4Institute for Medical Engineering and Science, Department of Biological Engineering, and Synthetic Biology Center, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA;
- 5Broad Institute of MIT and Harvard, Cambridge, Massachusetts 02142, USA;
- 6Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, Massachusetts 02115, USA;
- 7Department of Plant Systems Physiology, Institute for Water and Wetland Research, Radboud University, Heyendaalseweg 135, 6525 AJ, Nijmegen, The Netherlands;
- 8Center for Individualized Medicine, Department of Molecular Pharmacology & Experimental Therapeutics, Mayo Clinic, Rochester, Minnesota 55905, USA;
- 9NUS Graduate School for Integrative Sciences and Engineering, National University of Singapore, Singapore 119077, Singapore;
- 10Department of Physiology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 119228, Singapore
-
↵11 These authors contributed equally to this work.
Abstract
Joint profiling of transcriptome and chromatin accessibility within single cells allows for the deconstruction of the complex relationship between transcriptional states and upstream regulatory programs determining different cell fates. Here, we developed an automated method with high sensitivity, assay for single-cell transcriptome and accessibility regions (ASTAR-seq), for simultaneous measurement of whole-cell transcriptome and chromatin accessibility within the same single cell. To show the utility of ASTAR-seq, we profiled 384 mESCs under naive and primed pluripotent states as well as a two-cell like state, 424 human cells of various lineage origins (BJ, K562, JK1, and Jurkat), and 480 primary cord blood cells undergoing erythroblast differentiation. With the joint profiles, we configured the transcriptional and chromatin accessibility landscapes of discrete cell states, uncovered linked sets of cis-regulatory elements and target genes unique to each state, and constructed interactome and transcription factor (TF)–centered upstream regulatory networks for various cell states.
With the growing interest in understanding cellular heterogeneity, development of single-cell technologies has exploded in recent years. There are a multitude of single-cell techniques measuring genome (Navin et al. 2011; Wang et al. 2012; Gawad et al. 2014), transcriptome (Tang et al. 2009, 2010; Klein et al. 2015; Macosko et al. 2015), protein abundance (Huang et al. 2007; Hughes et al. 2014), cell surface protein (Stoeckius et al. 2017), copy number variation (Zong et al. 2012), DNA methylation (Guo et al. 2013; Lorthongpanich et al. 2013; Smallwood et al. 2014), chromatin accessibility (Buenrostro et al. 2015; Cusanovich et al. 2015; Jin et al. 2015; Pott 2017), immunoprecipitated chromatin (Rotem et al. 2015; Grosselin et al. 2019; Wang et al. 2019), chromatin architecture (Nagano et al. 2013; Flyamer et al. 2017; Ramani et al. 2017), and lineage tracing (Raj et al. 2018) at a single-cell resolution. These single-cell approaches allow for the deconstruction of cell types and cell states from mixed populations and the identification of cell state–specific genomic, transcriptomic, proteomic, and epigenomic programs.
However, unimodal single-cell techniques present limited values in uncovering intricate relationships across modalities, which is of utmost importance to gain comprehensive understanding of cell states. To tackle this, multimodal single-cell methods have been developed for concurrent measurement of genome and transcriptome (Dey et al. 2015; Macaulay et al. 2015) and of transcriptome and DNA methylome within a single-cell (Angermueller et al. 2016; Cheow et al. 2016; Hu et al. 2016). With the technical advancement in the multi-omic fields, single-cell toolkits for joint profiling of transcriptome and chromatin accessibility are being developed to unravel the epigenetic mechanisms regulating gene transcription (Cao et al. 2018; Clark et al. 2018; Chen et al. 2019; Liu et al. 2019; Zhu et al. 2019; Xing et al. 2020). Among them, sci-CAR, SNARE-seq, and Paired-seq show high cell throughputs (tens of thousands), rendering them suitable for organism-scale measurements, such as cell atlas (Cao et al. 2018; Chen et al. 2019; Zhu et al. 2019). However, their high-throughput nature results in rare sequencing reads for each profiled cell and extensive loss of information (Xing et al. 2020). In comparison, scNMT-seq and scCAT-seq are applicable for studies with the intention of mining in-depth molecular distinctions between subpopulations (Clark et al. 2018; Liu et al. 2019). However, chromatin accessibility libraries prepared by scNMT-seq suffer from extremely low mapping percentage and DNA mutations, which most likely resulted from the bisulfite treatment (Xing et al. 2020). Additionally, both methods involve separation of rare gDNA and RNA before any amplifications, which may result in significant loss of the lowly abundant genes and diploid genomic regions (Xing et al. 2020). Apart from this, this step also makes them less compatible with the automated platforms, thereby requiring remarkable manpower per experiment (Xing et al. 2020).
Here, we describe an automated technique with greater sensitivity, assay for single-cell transcriptome and accessibility regions (ASTAR-seq), which concurrently measures whole-cell transcriptome and epigenome accessibility within a single cell. To benchmark the technique, we applied it to deconstruct the heterogeneity of metastable mESCs under various cell states, human cell types of distinct lineage origins, and primary cells undergoing dynamic differentiation. We unraveled the cell state–specific transcriptomic and epigenetic programs, discovered unique pairs of cis-regulatory elements and putative target genes, and uncovered interactome pathways and upstream TFs associated with the heterogeneity observed.
Results
Simultaneous measurement of transcriptome and chromatin accessibility within a single cell
In the ASTAR-seq protocol, individual cells are first isolated at distinct cell capture sites linked with a series of reaction compartments on a Fluidigm C1 microfluidic chip (Fig. 1A). Open chromatin regions of each cell are then tagmented with Tn5 transposase, during which accessible DNA regions (ATAC-DNA) are integrated with sequencing adaptors. Next, mRNA is reverse transcribed to double-stranded cDNA, which is then labeled with biotin during PCR amplification process. Biotinylation of cDNA enables its separation from ATAC-DNA using streptavidin beads. Lastly, the separated ATAC-DNA and cDNA fractions are further processed for library preparation and sequenced in parallel. The earlier prototype, in which reverse transcription was performed before transposition, was not successful (Supplemental Fig. S1A). This could be attributed to Tn5 transposase digesting single-stranded cDNA, which resulted in failure to separate cDNA from ATAC-DNA (Supplemental Fig. S1B–D). ASTAR-seq protocol was first optimized and tested on 1000 BJ cells on benchtop. The optimal condition yielded abundant cDNA and achieved clear separation of ATAC-DNA and cDNA (Supplemental Fig. S1E–G).
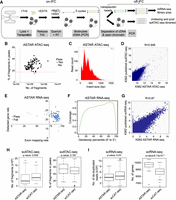
Assay for single-cell transcriptome and accessibility regions (ASTAR-seq). (A) Overview of ASTAR-seq protocol. (B) Dotplot revealing the percentage of fragments in peaks (y-axis) against the number of fragments (x-axis) of each K562 ASTAR ATAC-seq library. Red dotted lines represent the threshold values set for each criterion. (C) Histogram showing the frequency (y-axis) of fragments with the indicated insert size (x-axis). (D) Dotplot showing Pearson's correlation between K562 ASTAR ATAC-seq and the published K562 scATAC-seq libraries. (E) Dotplot revealing detected gene rate (y-axis) of each K562 ASTAR RNA-seq library plotted against its exon mapping rate (x-axis). Blue dots represent the libraries that pass QC, whereas gray dots represent the libraries of low quality. (F) Line plot representing the coverage ratio (y-axis) of K562 ASTAR RNA-seq reads over the gene bodies of housekeeping genes (x-axis). (G) Dotplot showing Pearson's correlation between K562 ASTAR RNA-seq and the published K562 scRNA-seq libraries. (H) Boxplots showing the number of fragments (left) and percentage of fragments in peaks (right) of scATAC-seq libraries prepared by the scCAT-seq and ASTAR-seq protocol. Two-tailed Student's t-test is used to calculate P-values. (I) Boxplots showing the number of de-duplicated reads (left) and genes (right) detected in scRNA-seq libraries prepared by the scCAT-seq and ASTAR-seq protocol. Two-tailed Student's t-test is used to calculate P-values.
As a proof of concept, we first applied ASTAR-seq to an ENCODE cell line, K562. Of 96 ASTAR ATAC-seq libraries sequenced, 92 libraries (95.8%) passed the chromVAR QC thresholds (Fig. 1B; Supplemental Table 1). A median library size of 142,886 was detected, and 27.9% of fragments were in peaks, indicating high signal-to-noise of the ASTAR ATAC-seq libraries (Fig. 1B). Insert-size distribution of the ASTAR ATAC-seq libraries showed clear nucleosomal periodicity, a characteristic pattern of an ATAC-seq library (Fig. 1C; Buenrostro et al. 2015). In addition, ASTAR ATAC-seq libraries showed a Pearson's correlation of 0.946 with the published scATAC-seq libraries (Buenrostro et al. 2015), indicative of its high similarity to the unimodal libraries (Fig. 1D). On the other hand, 83 out of 96 ASTAR RNA-seq libraries (86.5%) passed the QC thresholds set for gene detection rate (≥15%) and exon mapping rate (≥75%) (Fig. 1E; Supplemental Table 1). A median of 4182 genes was detected (Supplemental Fig. S2A). In addition, scRNA-seq reads spread across the entire gene bodies, without biasing toward either ends of the mRNA (Fig. 1F). Moreover, ASTAR RNA-seq showed a high Pearson's correlation (R = 0.87) with the published unimodal K562 scRNA-seq (Fig. 1G; Pollen et al. 2014).
To make a fair comparison with a similar bimodal technique scCAT-seq, we sequenced the K562 ASTAR-seq libraries at a comparable sequencing depth (40 single-cell libraries per lane of HiSeq 4000) (Supplemental Table 1). scCAT-seq libraries showed a significantly lower alignment rate to the human genome in comparison with ASTAR-seq (scATAC-seq: 67.6% vs. 85.8%; scRNA-seq: 54.9% vs. 73.8%) (Supplemental Fig. S2B). Additionally, a significantly higher percentage of ASTAR-seq libraries passed the QC thresholds for both the scATAC-seq and scRNA-seq than the scCAT-seq libraries (ASTAR-seq: 75.4%; scCAT-seq: 43.2%) (Supplemental Fig. S2C). For scATAC-seq libraries that passed QC, scCAT-seq and ASTAR-seq displayed comparable performance in terms of library complexity and signal-to-noise ratio (Fig. 1H). On the other hand, comparable numbers of de-duplicated reads were detected for scRNA-seq libraries (scCAT-seq: 4,507,504; ASTAR-seq: 4,047,857), in line with their comparable sequencing depths (Fig. 1I), whereas ASTAR-seq detected 1014 more genes than scCAT-seq (ASTAR-seq: 9739; scCAT-seq: 8725) (Fig. 1I).
To comprehensively review other bimodal single-cell techniques, we systematically compared ASTAR-seq with scCAT-seq, sci-CAR, SNARE-seq, and Paired-seq in terms of profiled cells, QC rate, estimated cost per paired good-quality libraries, and QC matrices (Supplemental Fig. S2D–F). Among them, ASTAR-seq and scCAT-seq were of lower throughput, and ASTAR-seq showed the highest QC rate (Supplemental Fig. S2D). Correspondingly, owing to their high sequencing depth, ASTAR-seq and scCAT-seq displayed a higher cost per cell than the high-throughput methods (Supplemental Fig. S2E). Despite the comparable overall cost per experiment, the estimated cost per paired good-quality ASTAR-seq libraries is 2.1 times lower than that of scCAT-seq (Supplemental Fig. S2E). On the other hand, ASTAR-seq and scCAT-seq showed a significantly higher number of detected genes (approximately 10-fold) and accessible sites (approximately 100-fold) than the high-throughput bimodal techniques (Supplemental Fig. S2F), whereas the compared techniques did not show specific trends in terms of signal-to-noise ratio (Supplemental Fig. S2F). Taken together, these data indicate the reliability of ASTAR-seq technique and show its superior performance in various aspects.
Deconstruction of heterogeneity in mESCs under distinct cellular states
We next applied ASTAR-seq to 192 E14 mESCs cultured in serum + LIF and 2i + LIF medium, which were named as mESCs and 2i cells throughout the study. All the sequenced scATAC-seq libraries passed the QC thresholds (Fig. 2A; Supplemental Table 2). scATAC-seq reads displayed an insert-size distribution with nucleosomal pattern and high enrichment at transcription start sites (TSSs) (Supplemental Fig. S3A,B). Moreover, these libraries showed a significantly higher signal-to-noise ratio than the unimodal mESC scATAC-seq libraries (Supplemental Fig. S3C; Buenrostro et al. 2015). Additionally, mESCs and 2i cells can be accurately distinguished by confusion matrix analysis based on their highly accessible regions (HARs) (Supplemental Fig. S3D). We then clustered mESCs and 2i ASTAR ATAC-seq libraries based on the overall accessibility profiles and accessibility of mouse JASPAR motifs (Fig. 2B; Supplemental Fig. S3E,F). mESCs and 2i cells were mostly clustered separately, but a certain degree of overlapping was observed (Fig. 2B; Supplemental Fig. S3F). Of note, chromatins containing motif sequences of KLF4, RARG, ZFX, KLF12, and MLXIP showed significant variability in terms of accessibility (P-value <0.05) (Supplemental Fig. S3E; Supplemental Table 2). For instance, KLF4 motif was highly accessible in 2i cells, whereas ZFX showed the opposite trend (Supplemental Fig. S3G). In the previous studies, not only was Klf4 reported to be up-regulated in mESCs under naive state compared with the primed state but also its overexpression facilitated cellular reprogramming of primed EpiSCs to naive ESCs (Guo et al. 2009; Jeon et al. 2016). Conversely, despite favoring self-renewal, mESCs with ZFX overexpression failed to efficiently generate teratoma and chimera, which is in line with naive ESCs with low ZFX activity presenting high chimera formation rate (Galan-Caridad et al. 2007).
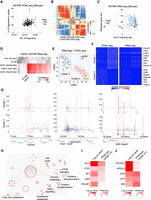
Transcriptomic and epigenetic heterogeneity within primed and naive mESCs. (A) Dotplot revealing the percentage of fragments in peaks (y-axis) and number of fragments (x-axis) of each mouse ASTAR ATAC-seq library. Red dotted lines represent the thresholds set for each criterion. (B) Heatmap showing the correlation among mESCs and 2i cells based on their ASTAR ATAC-seq libraries. (C) Dotplot revealing the detected gene rate (y-axis) of each mouse ASTAR RNA-seq library plotted against its exon mapping rate (x-axis). Blue dots represent the libraries that pass QC, whereas gray dots indicate the low-quality libraries. (D) Heatmap revealing the correlation of each mESC ASTAR RNA-seq library (x-axis) to various lineages of MCA panel (y-axis). Color indicates the correlation level, ranging from gray (low) to red (high). Two-cell-like (2C-like) mESCs are boxed with a dotted line. (E) NMF clustering of mESCs and 2i cells based on the correlative signals of their ASTAR ATAC-seq and ASTAR RNA-seq libraries. (F) Heatmaps revealing pairs of accessible regulatory regions (left) and the corresponding target genes (right) that are differentially enriched between the NMF clusters. Each column represents a library, whereas each row represents a chromatin region (left) or a gene (right). Color indicates the accessibility (left) and expression (right) levels, ranging from blue (low) to red (high). Representative genes are indicated on the right. (G) Line plots showing the differential coaccessibility links between the highlighted regions and its surrounding regions, identified using Cicero. Top plots are constructed from ASTAR ATAC-seq libraries of cluster 1 cells, whereas bottom plots are constructed from that of cluster 2 cells. Peak heights (y-axis) denote the coaccessibility scores. (H) Interactome analysis revealing the top pathways enriched by cluster 2–specific genes. (I,J) Heatmaps showing the enrichment (left columns) of TF motifs on cluster 1–specific (I) and cluster 2–specific (J) accessible regions and their relative expressions (right columns).
Likewise, the majority of mouse ASTAR RNA-seq libraries (80.7%) passed the QC thresholds, in which a median of 2645 and 2429 genes were detected for mESCs and 2i libraries, respectively (Fig. 2C; Supplemental Fig. S3H; Supplemental Table 2). In addition, ASTAR RNA-seq libraries also showed full gene body coverage for the detected transcripts (Supplemental Fig. S3I). Moreover, mESC ASTAR-seq libraries highly correlated (R = 0.86) with the published unimodal scRNA-seq libraries (Supplemental Fig. S3J; Svensson et al. 2017). Similarly, the confusion matrix illustrated that transcriptomic profiles detected by ASTAR-seq can accurately distinguish mESC and 2i cells (Supplemental Fig. S3K). To study the heterogeneity within mESCs, we correlated mESC ASTAR RNA-seq libraries to a mouse cell atlas (MCA) panel (Han et al. 2018). Analysis revealed three types of cells, including mESCs, ICM-like mESCs, and two-cell like (2C-like) mESCs, in agreement with a previous report (Fig. 2D; Macfarlan et al. 2012), whereas MCA analysis of 2i ASTAR RNA-seq libraries showed absence of a 2C-like population (Supplemental Fig. S3L). PCA also revealed a separate cluster of the minority 2C-like cells (Supplemental Fig. S3M). To confirm the presence of 2C-like cells in our mESCs culture, we used the 2C::tdTomato reporter constructed in the previous study (Macfarlan et al. 2012). Indeed, ∼1%–2% of mESCs consistently showed activation of the 2C reporter, which was significantly increased upon depletion of Ehmt2, a H3K9me3 methyltransferase, as previously described (Supplemental Fig. S3N; Macfarlan et al. 2012).
Integrative analysis of ASTAR-seq uncovered cluster-specific cis-regulatory elements and their target genes
To show the advantage of joint profiling, we then clustered mESCs and 2i ASTAR-seq libraries on the basis of bimodal data sets using coupled nonnegative matrix factorization (NMF) (Duren et al. 2018). Of note, two distinct clusters were always observed when clustering was based on either the differentially expressed genes or the differentially accessible regions that were identified by coupled NMF analysis (Supplemental Fig. S4A). Cluster 1 was mostly composed of mESCs, whereas cluster 2 was majorly composed of 2i cells. In comparison, joint clustering based on the NMF cluster–specific genes and accessible regions together showed superior performance in distinguishing the subpopulations, as seen from the clear and accurate separation of mESCs and 2i cells (Fig. 2E). In addition, correlation between accessibility and gene expression enabled us to identify regulatory networks specific to each NMF cluster (Fig. 2F; Supplemental Table 2). For instance, Dnmt3l, lefty1, Utf1, and Zscan10 were highly accessible and expressed in cells of cluster 1, whereas Scd1, Gdf3, Dppa3, Dppa5a, Sp5, and Tfcp2l1 were highly expressed in cluster 2 cells with greater accessibility (Fig. 2F; Supplemental Fig. S4B). These genes were previously reported to be important for naive and primed pluripotency, respectively. For example, higher expressions of Dnmt3l, lefty1, and Utf1 were observed for mESCs under naive state than primed state, whereas the opposite was observed for Dppa5a and Tfcp2l1 (Chen and Lai 2015; Choi et al. 2017; Ghimire et al. 2018). ZSCAN10 regulates epiblast-like cells through interaction with POU5F1, whereas SP5 facilitates the conversion of epiblast to naive ESCs (Buecker et al. 2014; Ye et al. 2016).
In addition to this, Cicero coaccessibility analysis (Pliner et al. 2018) showed that the Dnmt3l gene and its surrounding loci displayed open chromatin architecture with high interaction frequency in cluster 1; on the contrary, more 3D genomic interactions were observed for Scd1, Gdf3, and Dppa3 in cluster 2 (Fig. 2G). The expression of Dnmt3l was reported to be reduced in naive ESCs, resulting in a global DNA demethylation (Leitch et al. 2013). On the contrary, SCD1 is a lipid metabolic enzyme involved in lipogenesis, and proteins associated with fatty acid metabolism were reported to be highly expressed in naive mESCs (Taleahmad et al. 2015, 2018). Similarly, Dppa3 and Gdf3 were known to be important for naive pluripotency (Chen et al. 2015; Sang et al. 2019). Altogether, this highlights the differential interactive networks in which the cluster-specific putative regulatory elements result in differential expression of the respective target genes.
Identification of cluster-specific interactome pathways and TF-centered regulatory networks associated with naive and primed pluripotency
NMF cluster–specific genes showed extensive interaction among themselves by involving in similar biological processes (Fig. 2H; Supplemental Fig. S4C; Supplemental Table 2). Specifically, cluster 2 genes were majorly associated with metabolic pathways, including oxidative phosphorylation, glutathione metabolism, and fatty acids metabolism, which were reported to be key characteristics of naive pluripotency (Fig. 2H). For example, in terms of catabolism, naive ESCs show wide energy substrate usage through glycolysis, oxidative phosphorylation, and fatty acid oxidation, whereas primed ESCs mainly use glucose as energy source (Mathieu and Ruohola-Baker 2017). Glutathione is an antioxidant preventing cellular damage from the toxic molecules produced under oxidative stress during oxidative phosphorylation, which were found to be highly active in 2i cells compared with mESCs (Taleahmad et al. 2015, 2018). Similarly, anabolism processes, including amino acids and nucleotides synthesis, were reported to be highly active in the naive ESCs, which were also enriched in cluster 2–specific genes, such as tyrosine phosphorylation pathway and purine metabolism (Taleahmad et al. 2015, 2018).
Apart from the metabolic process, mTOR and Hippo signaling pathways were also enriched by the 2i cluster–specific genes (Fig. 2H). Attenuation of Hippo signaling was observed along the developmental axis from early blastocyst to epiblast, whereas depletion of YAP/TAZ, whose nuclear localization and regulatory function are repressed by Hippo signaling, caused differentiation specifically in mESCs but not in 2i cells (Lian et al. 2010; Azzolin et al. 2014; Weinberger et al. 2016; Hashimoto and Sasaki 2019). Likewise, proteins involved in the mTOR signaling pathway were highly expressed in naive ESCs, whereas disruption of Mtor led to early postimplantation lethality and prohibited ESCs development (Gangloff et al. 2004; Taleahmad et al. 2015). Mechanistically, mTOR positively regulated mitochondrial function and oxidative metabolism, which were highly active in naive ESCs (Yu and Cui 2016). On the other hand, cluster 1 genes showed specific association with glucose metabolism and TGFB signaling (Supplemental Fig. S4C). TGFB/activin signaling was shown to be required for epiblast induction, whereas its inhibition resulted in conversion of epiblast to naive state (Han et al. 2011; Taleahmad et al. 2018).
There are also pathways commonly enriched for 2i and mESC clusters, such as ribosome complex, RNA splicing, and cell cycle (Fig. 2H; Supplemental Fig. S4C; Supplemental Table 2). However, the component genes of those complexes are distinct. For instance, genes related to the G1/S phase, such as Cdca3 and Mad2l1, were enriched in the 2i cluster (Supplemental Table 2), whereas genes associated with the G2M phase, including Cep76, Csnk1e, and Tubb3, were enriched in the cluster-specific genes of mESCs (Supplemental Table 2). They are in line with the prolonged G1/S phase and abbreviated G2M phases of naive mESCs compared with primed mESCs (Ter Huurne et al. 2017; Taleahmad et al. 2018).
To further examine the TFs responsible for the differential regulatory networks, we performed motif enrichment analysis for the cluster-specific accessible sites. KLF3, CTCF, SP1 and MAZ motifs were enriched on cluster 1–specific accessible regions, which were also highly expressed in cluster 1 cells (Fig. 2I; Supplemental Table 2). An earlier study reported lower genomic looping frequencies in 2i cells compared with mESCs (Krijger et al. 2016), supporting the higher CTCF regulon activity detected in cluster 1. On the contrary, motifs of TFCP2L1, NFE2L2, KLF6, TCF7, and SP5 were enriched on cluster 2–specific accessible regions, majority of which displayed higher expression in the cells of cluster 2 (Fig. 2J; Supplemental Table 2).
ASTAR-seq recapitulates 2C-like population in mESCs
To further examine the capability of ASTAR-seq recapitulating known biology, we prepared ASTAR-seq libraries for mESCs in which DUX was overexpressed for 24 h in two batches (Supplemental Fig. S5). DUX overexpression was known to induce 2C-like fate in mESCs (Hendrickson et al. 2017). One hundred eighty-six out of 192 ASTAR ATAC-seq libraries passed QC, and a median of 2353 genes were detected in ASTAR RNA-seq libraries (Supplemental Fig. S5A,B). Of note, replicates of ASTAR-seq libraries showed distinctively high correlation between themselves in terms of both transcriptomic profile and chromatin accessibility (R = 0.987; 0.99) (Supplemental Fig. S5C). ASTAR RNA-seq libraries showed two clusters that were free of technical bias (Supplemental Fig. S5D). Cluster 2 showed a higher expression of genes associated with 2C-like fate, including Zscan4a, Zscan4c, Zscan4d, Zscan4f, Usp17la, Usp17lb, Usp17lc, and Eif4a3l1 (Supplemental Fig. S5E; Macfarlan et al. 2012; Hendrickson et al. 2017). Likewise, ASTAR ATAC-seq libraries distinguished different populations with no technical bias (Supplemental Fig. S5F). One population showed higher accessibility for DUX binding sites and MERVL loci (Supplemental Fig. S5G). This goes in agreement with the earlier studies reporting the reactivation of MERVL in 2C-like mESCs and the crucial role of DUX in maintaining 2C-like fate (Macfarlan et al. 2012; Hendrickson et al. 2017). Indeed, mESCs with a 2C-like transcriptomic signature showed a higher accessibility for MERVL loci and DUX binding sites (Supplemental Fig. S5H). Taken together, these data indicate the reproducibility and reliability of ASTAR-seq in recapitulating known biological phenomenon.
Cell type–specific programs revealed by ASTAR-seq
To expand the applicability, we also prepared ASTAR-seq libraries for various human cell lines, including BJ cells in adherent culture, and JK1 and Jurkat cells in suspension culture (Fig. 3). To characterize the molecular distinction among the hematopoietic cells, 96 K562 ASTAR-seq libraries sequenced at a similar depth were also included for the following analysis. Out of 384 libraries profiled, 375 ASTAR ATAC-seq libraries passed the QC thresholds for chromVAR (Supplemental Fig. S6A; Supplemental Table 3). In median, a library size of 55,193 was captured, and 35% of fragments was in peaks (Supplemental Fig. S6A,B). Insert-size distribution of ASTAR ATAC-seq libraries also showed characteristic nucleosomal pattern of ATAC-seq libraries (Supplemental Fig. S6C; Buenrostro et al. 2015). In addition, ASTAR ATAC-seq libraries also showed a high similarity to the published scATAC-seq libraries (R = 0.897) (Supplemental Fig. S6D; Buenrostro et al. 2015). Confusion matrix analysis showed that the HARs detected by ASTAR-seq libraries can distinguish these cell lines with high accuracy (96.5%) (Supplemental Fig. S6E). These indicate that the prepared ASTAR ATAC-seq libraries are of good quality.
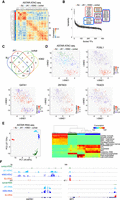
Application of ASTAR-seq on human cell lines. (A) Clustering of BJ, JK1, K562, and Jurkat ASTAR ATAC-seq libraries based on the human JASPAR motif deviation scores calculated over the HARs. Color indicates the correlation level among the libraries, ranging from blue (no) to red (high). Side color bar (y-axis) indicates the identity of each cell. (B) Variability plot indicating the variable TF motifs across the ASTAR ATAC-seq libraries of four human cell lines. The y-axis represents the variability score assigned to each JASPAR motif, whereas the x-axis represents the motif rank. Top variable motifs are classified based on their enrichment scores across the cell lines and colored accordingly. (C) Multi-Venn diagram showing the shared and unique TFs across the cell lines. (D, top left) t-SNE clustering of BJ, JK1, K562, and Jurkat ASTAR ATAC-seq libraries based on the deviation scores of human JASPAR motifs. Colors represent the cell lines. (Top right and bottom) Superimposition of motif enrichment scores for FOSL1, GATA1, ZBTB33, and TEAD3 on the t-SNE plot. Colors represent the motif enrichment levels, ranging from blue (no) to red (high). (E, left) PCA clustering of ASTAR RNA-seq libraries based on their correlation to the RCA panel. (Right) Heatmap showing the lineages that each cell correlates to. (F) UCSC screenshots indicating the chromatin accessibility (top) and expression (bottom) levels of GATA1 (left) and SP1 (right) across the cell lines.
Next, we clustered ASTAR ATAC-seq libraries based on enrichment of human JASPAR motifs and observed four clusters, among which BJ cells displayed distinct accessibility profiles and clustered away from the cells of blood lineage (Fig. 3A). Variability analysis identified TF motifs determining the cell type identities and their distinctions (Fig. 3B; Supplemental Table 3). Consistent with its distinct cluster, 16 TF motifs were specifically accessible in BJ cells, such as FOS-JUN and NFE2, which were reported to display tissue-specific activities, especially in fibroblasts (Fig. 3B–D; Supplemental Fig. S6F; Wilkinson et al. 1989). On the other hand, BJ cells also extensively shared motifs with cells of myeloid lineage: K562 (79 motifs), including ETS1 and ZBTB33, and JK1 (28 motifs), including families of NFY, MEF2, and SP factors, compared with cells of lymphoid lineage—Jurkat (six motifs), including the TEAD family (Fig. 3B–D; Supplemental Fig. S6F). In addition, extensive overlap (46 motifs) was seen between cells of myeloid lineage (JK1 and K562), including TFs of GATA-TAL family (Fig. 3B–D). These enriched factors were reported to play important roles in the respective lineages. For instance, ZBTB33 is specifically important for K562 by binding to methylated DNA, whereas its depletion causes differentiation defects (Cofre et al. 2012; Lin et al. 2019). GATA1 is a master TF of erythropoiesis with specific expression in myeloid lineage but not in the lymphoblastic leukemia cells (Lee et al. 2017; Gutiérrez et al. 2020). On the contrary, inactivation of Hippo kinase modules, such as MST1/2, causes lymphopenia in human and results in translocation of YAP/TAZ to nucleus, which activates regulatory activity of TEAD TFs (Cheng et al. 2018; Yamauchi and Moroishi 2019). The gathered literature reports suggest inactive Hippo pathway in lymphoblast Jurkat cells and the resultant activation of TEAD activity.
Meanwhile, of 384 ASTAR RNA-seq libraries, 296 libraries (77.1%) passed QC and showed full coverage for the expressed transcripts (Supplemental Fig. S6G–I; Supplemental Table 3). A total of 291 ASTAR-seq libraries (75.8%) passed the QC filtration for both the scATAC-seq and scRNA-seq libraries (Supplemental Table 3). In addition, ASTAR-seq libraries showed high similarity (R = 0.82) to the unimodal scRNA-seq libraries (Supplemental Fig. S6J; Pollen et al. 2014). Besides, transcriptomic profiles detected by ASTAR RNA-seq can distinguish among distinct cell lines with high accuracy (99.63%) (Supplemental Fig. S6K). Furthermore, reference component analysis (RCA) analysis (Li et al. 2017) showed distinct correlation of BJ cells to foreskin fibroblasts and muscle lineage, JK1 cells to erythroblasts, K562 cells to leukemia K562, and Jurkat cells to leukemia lymphoblast (Fig. 3E). Consistent with their differential regulatory activities, GATA1 was uniquely accessible and expressed in myeloid JK1 and K562 cells, whereas SP1 was accessible and expressed in BJ, JK1, and K562 cells (Fig. 3F). Taken together, ASTAR-seq enables the identification of TFs responsible for the distinctions among distinct cell types.
ASTAR-seq reveals dynamics in transcriptome and epigenome along the pseudotemporal axis of erythroblast differentiation
To measure its capability in capturing the dynamic changes, we applied ASTAR-seq to the primary cells undergoing erythroblast differentiation. We harvested a total of 480 cells at day 6 (D6), day 8 (D8), day 10 (D10), and day12 (D12) of erythroblast differentiation, which was induced from mononuclear cells isolated from umbilical cord blood, for ASTAR-seq library preparation (Fig. 4A). Of the 480 ASTAR-seq libraries, 273 cells (56.9%) presented good-quality libraries for both scATAC-seq and scRNA-seq (Supplemental Table 4). Comparatively, the QC rate is lower than that of the cell lines profiled earlier, which is likely associated with the inherent difficulty in culturing the primary cells. We next investigated the trajectories of erythroblast differentiation using pseudotemporal analysis (Trapnell et al. 2014; Qiu et al. 2017). The resultant trajectories consisted of two branching events and five pseudotemporal states (Fig. 4B,C). Pseudotime highly correlated with the actual differentiation time points. For instance, cells of earlier time points, such as D6 and D8, were mostly at states 1–3, whereas D10 cells were mostly at states 4 and 5 (Fig. 4B,C). On the other hand, the majority of D12 cells belonged to state 5 and located at the endpoint of pseudotime (Fig. 4B,C). Consistently, HBA2, a hemoglobin gene, showed elevated expression in cells of state 5 compared with the others (Fig. 4B).
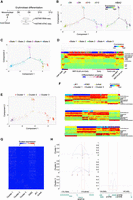
Interactive analysis of transcriptome and chromatin accessibility during erythroblast differentiation. (A) Schematic of erythroblast differentiation time points harvested for ASTAR-seq library preparation. (B, left) Trajectory of erythroblast differentiation identified from ASTAR RNA-seq libraries using DDRTree dimension reduction. Colors represent time points. (Right) Superimposition of HBA2 expression on the trajectory. Colors represent expression levels. (C) Trajectory plot revealing the pseudotemporal states. Colors represent pseudotemporal states. (D) RCA heatmap showing the clustering of cells undergoing erythroblast differentiation, based on their correlation to the cells of different lineage origins in RCA panel. Color indicates correlation value, ranging from blue (low) to red (high). Each row indicates one lineage, whereas each column represents an ASTAR RNA-seq library. Pseudotemporal state of each cell is indicated on top. Cellular differentiation status is determined based on their correlation to the cells of RCA panel and indicated below. (E) Superimposition of NMF clusters on the trajectory plot. (F) RCA clustering of JK1, K562, and Jurkat cells with the cells of NMF cluster 1 (top), cluster 2 (middle), and cluster 3 (below), respectively. Color indicates correlation value, ranging from blue (low) to red (high). Each row indicates one lineage, whereas each column represents an ASTAR RNA-seq library. Cell identities are indicated on top. (G) Heatmap showing the expression of NMF cluster–specific genes across NMF clusters and the hematopoietic cell lines. Color indicates expression level, ranging from blue (no) to red (high). (H) Line plots indicating the Cicero coaccessibility links between the regions highlighted in red and the distal sites in the surrounding regions. Height indicates the coaccessibility score of the connected peaks. The links are constructed from ASTAR ATAC-seq libraries of cluster 1 (top), cluster 2 (middle), and cluster 3 (bottom), respectively.
We then performed RCA analysis to examine the differentiation status of various pseudotemporal states. Generally, the majority of cells showed strong correlation to the myeloid or erythroid lineages (Fig. 4D). As the differentiation time point increased, transitions in the order of common myeloid progenitors (CMPs), myeloid erythroid progenitors (MEPs), erythroblast progenitors (Eryth pro), early erythroblasts, and late erythroblasts were observed (Fig. 4D). Specifically, the majority of states 1–3 cells showed a strong correlation to Eryth pro cells and certain degrees of correlation to MEPs and early erythroid (Fig. 4D). Cells of state 5 can be broadly classified into three groups, including groups with correlation to early erythroid but not to MEPs, with strong early erythroid identities, and with significant late erythroid fate (Fig. 4D). On the other hand, cells of state 4 bifurcated from the differentiation trajectory and acquired alternative fate resembling granulocyte monocyte progenitor (GMP) (Fig. 4D). In addition to this, GO analysis also showed that genes abundantly expressed in cells of state 5 were associated with oxygen transport, hydrogen peroxide catabolic process, and erythrocyte differentiation process, whereas state 4 cells highly expressed genes related to innate immune response and antigen processing and presentation (Supplemental Fig. S7A). On the contrary, compared with state 1, genes that were repressed in state 5 were associated with stem cell population maintenance, positive regulation of H3-K4 methylation, and chromatin organization, whereas genes related to erythrocyte maturation, oxygen transport, and NF-kB signaling were repressed in state 4 cells (Supplemental Fig. S7B). Altogether, state 5 represents cells attaining erythroid fate, whereas state 4 cells deviate from erythroblast differentiation path, instead acquiring GMP identities.
To identify the genes and regulatory regions responsible for the progression of erythroblast differentiation, we used coupled NMF analysis, which uncovered three clusters (Fig. 4E). Superimposition of NMF clusters on the erythroblast differentiation trajectory showed that the majority of cluster 1 cells belonged to states 1–2, whereas cells of cluster 3 were the major constituent of state 5 (Fig. 4E). Cluster 2 cells scattered across the various states (Fig. 4E). Consistent with the cell fate of pseudotemporal states, cluster 1 cells showed characteristics of proerythroblasts and early erythroid cells and clustered closer to K562 cells, whereas cluster 2 cells showed stronger early erythroid identities and started to develop late erythroid characteristics (Fig. 4F). In addition, cluster 2 cells showed a higher similarity to JK1 than to K562 cells, reflecting their advanced differentiation compared with cluster 1 cells (Fig. 4F; Koeffler and Golde 1980), whereas cluster 3 cells displayed the strongest late erythroid identities and were clustered intimately with JK1 cells (Fig. 4F). Supporting this notion, differentially expressed genes of NMF cluster 1 were highly represented in K562 cells, whereas cluster 3 genes were specifically expressed in JK1 cells (Fig. 4G; Supplemental Table 4). This was further substantiated by CTen analysis (Supplemental Fig. S7C,D; Shoemaker et al. 2012). On the other hand, the accessibility of chromatins containing motif sequences of GATA1:TAL1 and MEF2D was highly variable across the NMF clusters (Supplemental Fig. S7E,F; Supplemental Table 4). In addition, cluster 1 accessible regions were highly enriched with HOXB4 and FOXA3 motifs, cluster 2 with ONECUT1 motif, and cluster 3 with POU2F2 and RUNX2 motifs, implicating the importance of these TFs in regulating the genes crucial for progression of erythroblast differentiation (Supplemental Fig. S7G; Supplemental Table 4). Moreover, the cluster-specific genes showed differential coaccessibility with its surrounding regulatory regions. For example, NOP16 was highly expressed and accessible in cluster 1 with multiple genomic interactions with the nearby genes, whereas the cluster 3–specific gene GYPB showed a high interaction frequency with GYPE only in cluster 3 (Fig. 4H).
Discussion
In this study, we presented an automated bimodal single-cell technology, ASTAR-seq, which allows for parallel profiling of whole-cell transcriptome and chromatin accessibility within the same single cell, with a greater sensitivity. Among the multimodal single-cell techniques for transcriptome and chromatin accessibility, sci-CAR, SNARE-seq, and Paired-seq are more suitable for organism-scale measurements, owing to their high-throughput profiling and low cost (Cao et al. 2018; Chen et al. 2019; Zhu et al. 2019). However, high-throughput profiling meanwhile results in a major drawback, massive loss of information, owing to the low sequencing depth. Apart from this, scRNA-seq libraries prepared by these high-throughput techniques specifically capture transcriptome within nucleus and are biased toward the 3′ end of mRNA. In sum, because of these drawbacks, high-throughput multimodal techniques are not suitable for studying lowly abundant transcripts, isoforms, and transcriptional maturation.
However, these could be achieved by using high-depth multimodal single-cell assays, such as scCAT-seq (Liu et al. 2019) and ASTAR-seq. scCAT-seq and ASTAR-seq display much greater detection sensitivity than the high-throughput multimodal techniques. Comparatively, ASTAR-seq presents the following advantages compared with scCAT-seq. ASTAR-seq shows better performance in terms of genome alignment, gene detection sensitivity, and QC rate than scCAT-seq. In addition, ASTAR-seq measures transcriptomic profile within a whole cell, whereas scCAT-seq captures cytoplasmic transcriptome. Furthermore, owing to the integration with automated devices, ASTAR-seq displays higher consistency and requires less manual handling time and manpower. Therefore, the pros and cons of these multimodal assays should be carefully considered to better suit your application (Xing et al. 2020). Altogether, ASTAR-seq is a powerful integrated approach to understand the connectivity between transcription and epigenetic regulation.
Methods
Cell culture
mES-E14TG2a and 2i cells were cultured with the routinely used medium. DUX overexpressed mESCs was obtained following the previously described procedures (Hendrickson et al. 2017). mESCs were routinely propagated, passaged using trypsin, and replated onto 0.1% gelatin-coated plates every 3–4 d. BJ, K562, JK-1, and Jurkat were cultured with standard medium. Detailed medium recipes and induction methods can be found in the Supplemental Methods.
ASTAR-seq
Single-cell suspension was loaded onto Fluidigm C1 Open App microfluidic chips, and single-cell capture efficiency was assessed using a Nikon automated microscope. ASTAR-seq on-IFC steps were automatically performed on C1 machine using the custom built “ASTAR- ASTAR (1861x/1862x/1863x)” scripts. ASTAR-seq on-IFC reactions include lysis and transposition, EDTA (inactivate Tn5), MgCl2 (quench excess EDTA) and RT, and cDNA-PCR (biotinylation and amplification). Off-IFC steps are composed of streptavidin bead separation of cDNA and open chromatins (ATAC-DNA), PCR amplification of cDNA, and ATAC-seq and mRNA-seq library preparation. Detailed ASTAR-seq protocol can be found in the Supplemental Methods. ASTAR-seq scripts can be found in Supplemental Codes, ASTAR script.
ASTAR-seq library sequencing
One hundred ninety-two ASTAR RNA-seq libraries were sequenced in a lane of HiSeq 4000 sequencer using a 101-bp pair-end sequencing parameter. One hundred ninety-two ASTAR ATAC-seq libraries were sequenced in a lane of HiSeq 4000 sequencer using 50-bp pair-end sequencing parameter.
Mapping of ASTAR-seq libraries
ASTAR-seq libraries were mapped to mm9 (for mouse libraries) and hg19 (for human libraries) using STAR aligner (Dobin et al. 2013; see Supplemental Methods). For scRNA-seq libraries, we allowed up to two mismatches and removed reads that map to more than one locus. The option “–outSAMstrandFiled intronMotif” was used to make BAM outputs of STAR compatible with subsequent analyses. For scATAC-seq libraries, options ‐‐alignIntronMax 1 and ‐‐alignEndsType EndToEnd were used. Detailed scripts can be found in Supplemental Code. ASTAR-seq library filtration criteria can be found in the Supplemental Methods.
Quantification and normalization of scRNA-seq libraries
GTF files were generated using the “genePredToGtf” tool designed by UCSC. BAM files and generated GTF files were used as inputs for Cuffquant (Trapnell et al. 2010). Options -u and -m were included in the Cuffquant script. The abundances files were then used as inputs for Cuffnorm (Trapnell et al. 2010). The classic-fpkm normalization style was used. The detailed scripts can be found in Supplemental Code.
Clustering of scRNA-seq libraries
FPKM table was uploaded to Seurat (Butler et al. 2018) as input for t-SNE clustering. For UMAP generation, the raw counts generated by featureCounts were uploaded to Seurat v3. Data were then scaled, and variable genes were detected using default parameters. PCA was performed using the variable genes, and then UMAP or t-SNE were generated. The FeaturePlots function of Seurat was used to superimpose gene expression over UMAP plot. VlnPlot function was used to determine the number of genes detected per cell for all samples. The detailed script can be found in Supplemental Code.
scRNA-seq analysis
MCA (Han et al. 2018) and RCA (Li et al. 2017) analyses were performed as previously described. Details can be found in the Supplemental Methods, and the detailed script for RCA can be found in Supplemental Code.
Pseudotime analysis
FPKM table generated by Cuffnorm was used as an input for Monocle (Trapnell et al. 2014). FPKM values were converted to mRNA counts using the “relative2abs” function (Qiu et al. 2017). The expression family was set to negbinomial while creating the CellDataSet. Then, size factors and dispersions were estimated with default parameters. ReduceDimension function was used with method “DDRTree”. This was followed by cell ordering (orderCells) and trajectory plotting. The detailed script can be found in Supplemental Code.
Determination of HARs
Human and mouse ASTAR ATAC-seq libraries were merged independently using SAMtools merge (Li et al. 2009). Duplicates were removed using the MarkDuplicates module of Picard (https://broadinstitute.github.io/picard/). Peak calling was performed using MACS2 (Zhang et al. 2008), and the ‐‐nomodel ‐‐nolambda ‐‐keep-dup all ‐‐call-summits options were used. The narrowPeaks output of MACS2 was considered as the HARs. The detailed script can be found in Supplemental Code.
chromVAR analysis
scATAC-seq libraries were subjected to chromVAR analysis as previously described (Schep et al. 2017). Details can be found in the Supplemental Methods, and the detailed script can be found in Supplemental Code.
Motif analysis
The findMotifsGenome.pl script of HOMER (Heinz et al. 2010) was executed in order to identify the known motifs enriched in the differentially accessible regions.
Integrative analysis
Coupled NMF (Duren et al. 2018) was used to cluster the cells based on the integration of both scATAC-seq and scRNA-seq libraries, as previously described. Details and downstream analysis can be found in the Supplemental Methods. The detailed script can be found in Supplemental Code.
Prediction of cis-regulatory interactions
ASTAR ATAC-seq libraries belonging to each NMF cluster were merged using SAMtools merge to determine the HARs of each cluster as described above. Then coverage of each library belonging to a particular cluster over the HARs of its corresponding cluster was measured using DepthOfCoverage as described above. The raw coverage table was used as an input for Cicero (Pliner et al. 2018). Cicero CDS were created using make_cicero_cds, and “run_ cicero” was then performed (with default settings). The plot_connections function was used to visualize the predicted regulatory elements of NMF-identified genes. Coordinates used were 10× zoomed-out from the precise coordinates of the genes of interest. The detailed script can be found in Supplemental Code.
Interactome analysis
Genes specific to each NMF cluster were uploaded to STRING (Szklarczyk et al. 2019). The text-mining option was disabled. The network formed was downloaded in tabular format, and then the tables were uploaded to Cytoscape (Shannon et al. 2003) for visual formatting.
Confusion matrix generation
The confusion matrixes were generated using MLSeq (Zararsiz et al. 2017). Details can be found in the Supplemental Methods, and the detailed script can be found in Supplemental Code.
External data
External data sets used in this study were downloaded from the following database under the specified accession numbers: BJ scRNA-seq (NCBI Sequence Read Archive [SRA]: SRP041736), K562 scRNA-seq (NCBI SRA: SRP041736), mESCs scRNA-seq (ArrayExpress database: E-MTAB-5484, E-MTAB-5485), BJ scATAC-seq (NCBI Gene Expression Omnibus [GEO]: GSE65360), K562 scATAC-seq (GEO: GSE65360), mESCs scATAC-seq (GEO: GSE65360), scCAT-seq (CNGB Nucleotide Sequence Archive [CNSA]: CNP0000213).
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE113418.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Haitong Fang, Naresh Waran Gnanasegaran, and Sudhagar Samydurai for the technical assistance. H.L. is supported by National Instiutes of Health (AG056318, AG61796, and CA208517), the Glenn Foundation for Medical Research, Mayo Clinic Center for Biomedical Discovery, Center for Individualized Medicine, Mayo Clinic Cancer Center, and the David F. and Margaret T. Grohne Cancer Immunology and Immunotherapy Program. Y.H.L. is supported by the National Research Foundation Investigatorship award (NRFI2018-02), Agency for Science, Technology and Research JCO Development Program Grant (1534n00153 and 1334k00083), and Singapore National Research Foundation under its Cooperative Basic Research grant administered by the Singapore Ministry of Health's National Medical Research Council (NMRC/CBRG/0092/2015). We thank the Biomedical Research Council, Agency for Science, Technology and Research, Singapore for research funding.
Author contributions: Q.R.X. developed the ASTAR-seq protocol, performed experiments, and wrote the paper. C.A.E.F. performed most of the bioinformatics analyses and wrote the paper; Y.Y.Z. performed part of the bioinformatics analyses; Y.Y., T.W., and P.G. assisted with the manuscript writing; J.J.C., J.X., P.D., C.G.K., H.L., and L.F.Z. analyzed data; and Y.H.L. designed the study, analyzed data, and wrote the paper.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.257840.119.
-
Freely available online through the Genome Research Open Access option.
- Received October 2, 2019.
- Accepted June 25, 2020.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








