
PRAM: a novel pooling approach for discovering intergenic transcripts from large-scale RNA sequencing experiments
- 1Department of Biostatistics and Medical Informatics, University of Wisconsin, Madison, Wisconsin 53706, USA;
- 2Department of Cell and Regenerative Biology, Wisconsin Blood Cancer Research Institute, Carbone Cancer Center, University of Wisconsin School of Medicine and Public Health, University of Wisconsin, Madison, Wisconsin 53705, USA;
- 3Department of Computer Sciences, University of Wisconsin, Madison, Wisconsin 53706, USA;
- 4Department of Statistics, University of Wisconsin, Madison, Wisconsin 53706, USA
Abstract
Publicly available RNA-seq data is routinely used for retrospective analysis to elucidate new biology. Novel transcript discovery enabled by joint analysis of large collections of RNA-seq data sets has emerged as one such analysis. Current methods for transcript discovery rely on a ‘2-Step’ approach where the first step encompasses building transcripts from individual data sets, followed by the second step that merges predicted transcripts across data sets. To increase the power of transcript discovery from large collections of RNA-seq data sets, we developed a novel ‘1-Step’ approach named Pooling RNA-seq and Assembling Models (PRAM) that builds transcript models from pooled RNA-seq data sets. We demonstrate in a computational benchmark that 1-Step outperforms 2-Step approaches in predicting overall transcript structures and individual splice junctions, while performing competitively in detecting exonic nucleotides. Applying PRAM to 30 human ENCODE RNA-seq data sets identified unannotated transcripts with epigenetic and RAMPAGE signatures similar to those of recently annotated transcripts. In a case study, we discovered and experimentally validated new transcripts through the application of PRAM to mouse hematopoietic RNA-seq data sets. We uncovered new transcripts that share a differential expression pattern with a neighboring gene Pik3cg implicated in human hematopoietic phenotypes, and we provided evidence for the conservation of this relationship in human. PRAM is implemented as an R/Bioconductor package.
Transcript discovery and characterization are essential to unravel genomic functional elements. Genomic locations and splicing patterns of transcripts provide fundamental information for dissecting RNA functions. Multiple databases have been annotating transcripts for decades (Harrow et al. 2012; O'Leary et al. 2016; Yates et al. 2016). Yet, their collections are incomplete, mainly due to complex and variable expression patterns under different cellular conditions and limited coverage of transcript libraries (Mudge and Harrow 2016).
In the last decade, RNA-seq has revolutionized experimental transcript discovery, which had previously been performed through technologies such as cDNA and expressed sequence tag sequencing. RNA-seq provides a snapshot of the whole transcriptome with sequence data that often cover the entire length of transcripts. Annotation databases such as RefSeq, Ensembl, and GENCODE have all incorporated RNA-seq data for transcript discovery (Harrow et al. 2012; O'Leary et al. 2016; Yates et al. 2016), leading to major increases in the numbers of transcripts they harbor. For example, the number of transcripts in GENCODE version 7 increased by 45% after utilizing ENCODE RNA-seq data sets (Djebali et al. 2012). Although efforts to repurpose public RNA-seq data sets for biological discoveries are accelerating (Bernstein et al. 2017; Collado-Torres et al. 2017; Lachmann et al. 2018; Pertea et al. 2018), major opportunities exist to innovate and deploy tools that leverage vast RNA-seq data from multiple consortia (The International Cancer Genome Consortium 2010; Djebali et al. 2012; The GTEx Consortium 2013) to discover new transcripts and therefore new biological mechanisms.
A number of computational tools have been developed for reconstructing transcripts from a single RNA-seq data set (The RGASP Consortium et al. 2013; Shao and Kingsford 2017). Cufflinks (Trapnell et al. 2010), one of the first of these methods, predicts transcript models by using a minimum chain decomposition formalism and was employed by the ENCODE Consortium to expand the collection of transcripts (Djebali et al. 2012). StringTie, a more recent method, improved prediction accuracy and led to faster run times via a network flow-based approach (Pertea et al. 2015). Several meta-assembly computational methods have also been developed to utilize multiple RNA-seq data sets (Trapnell et al. 2012; Pertea et al. 2015; Niknafs et al. 2017). Their applications led to the discovery of a large number of new transcripts (Cabili et al. 2011; Hezroni et al. 2015; Iyer et al. 2015). A common feature of these approaches is a ‘2-Step’ process that first builds transcript models from individual RNA-seq data sets by one algorithm and then merges different sets of transcript models into a single unified set by another algorithm. An intuitive alternative to this type of 2-Step method, which relies on two distinct algorithms, is a 1-Step method that builds transcript models directly on pooled RNA-seq data sets. While Trapnell et al. (2012) argued that a 2-Step approach avoids high computational costs and complicated splicing patterns resulting from pooling RNA-seq data sets, benchmark comparisons have not been reported. Moreover, the application of a 1-Step approach to transcript discovery in intergenic regions has not been explored.
Here, we present a new computational framework named Pooling RNA-seq and Assembling Models (PRAM) that employs a 1-Step approach for intergenic transcript discovery. PRAM was well supported by a benchmarking experiment that assessed the relative performances of the 1-Step and 2-Step methods. We employed PRAM to build a master set of unannotated human transcript models in intergenic regions and computationally validated them with RAMPAGE and histone modification ChIP-seq data. In a case study that focused on the hematopoietic system, we applied PRAM to predict and characterize unannotated transcripts in mouse and human intergenic regions. We validated PRAM transcripts by qRT-PCR and identified new transcripts that were supported by external genomic data and conserved features between human and mouse.
Results
The 1-Step strategy outperforms the 2-Step strategy in transcript discovery
For benchmarking 1-Step and 2-Step methods, we prepared ‘noise-free’ RNA-seq data sets for a subset of GENCODE transcripts (version 24). This data set contained only RNA-seq fragments that were consistent with and sufficient for the reconstruction of the target transcripts. In this setting, a perfect prediction method would have the entire set of target transcripts reconstructed correctly. To build this benchmark, we downloaded 30 ENCODE poly(A) RNA-seq data sets that were all strand-specific, paired-end, and from untreated human cell lines (Supplemental Table 1). We selected a target subset of GENCODE transcripts based on these 30 RNA-seq samples. We defined target transcripts as those that (1) were multi-exon with genomic span of at least 200 nucleotides, (2) belonged to a single-transcript gene on Chromosomes 1 to 22 or X, (3) did not overlap with any other gene on either strand, and (4) had every exonic base and splice junction covered by at least one RNA-seq fragment from any of the 30 samples (no requirement for overhang length). While requirement (2) constrains the target set to single-transcript genes, this is consistent with the fact that most newly discovered GENCODE transcripts were from different genes (Supplemental Fig. 1) and removes complications due to alternative splicing. Due to frequently incomplete RNA-seq coverage of 5′ and 3′ ends of transcripts, we ignored coverage of the first and last 200 nt of exons. These criteria resulted in a set of 1256 target transcripts. We next constructed the inputs for our benchmark test by selecting only those alignments from the 30 RNA-seq data sets that mapped to our targets. This allowed us to build a noise-free benchmark from real RNA-seq data. We refrained from including noisy RNA-seq reads as such task largely depends on the completeness of the transcript annotation and a definition of “transcriptional noise,” a consensus for which is lacking in the literature. All of the 30 input RNA-seq data sets and target transcript annotations are available at GitHub (https://github.com/pliu55/PRAM_paper).
In the benchmark test, we assessed two 1-Step and three 2-Step methods. The two 1-Step methods involved pooling RNA-seq alignments from the 30 data sets, followed by the application of Cufflinks (‘pooling + Cufflinks’) or StringTie (‘pooling + StringTie’) to the pooled alignments. The three 2-Step methods involved building transcript models from individual data sets by Cufflinks, followed by an assembly merging step with Cuffmerge (‘Cufflinks + Cuffmerge’) or TACO (‘Cufflinks + TACO’); or building models by StringTie followed by StringTie-merge (‘StringTie + merging’). We excluded the StringTie and TACO combination because TACO was found to perform the best with Cufflinks (Niknafs et al. 2017). All five methods were evaluated by their precision and recall in predicting three features of a transcript: exon nucleotides; individual splice junctions; and transcript structure (i.e., whether all splice junctions within a transcript were reconstructed in a model). For exon nucleotides, all five methods had nearly perfect precision, while the two 1-Step methods and StringTie + merging had the highest recall (Fig. 1A; Supplemental Fig. 2). For detection of individual splice junctions and overall transcript structures, both of the two 1-Step methods had markedly higher recall than the three 2-Step methods and had higher precision than two out of the three 2-Step methods, especially for lowly expressed transcripts (Fig. 1A; Supplemental Fig. 2). The imperfect precisions on splice junctions for the three Cufflinks-based methods were caused by false positive predictions from Cufflinks (Supplemental Note 1; Supplemental Table 2; Supplemental Figs. 3, 4). Cufflinks and StringTie predictions on individual RNA-seq data sets without further merging resulted in far lower precision and recall than the five meta-assembly methods (Supplemental Fig. 2). Overall, pooling + Cufflinks outperformed Cufflinks + Cuffmerge and Cufflinks + TACO for all three metrics, and pooling + StringTie surpassed StringTie + merging, demonstrating the strength of 1-Step methods. Stratifying target transcripts by their expression levels showed that 1-Step methods had marked advantages for lowly expressed transcripts.
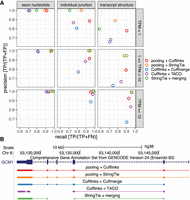
1-Step outperforms 2-Step reconstruction methods. (A) Precision and recall of five meta-assembly methods in a benchmark test on target transcripts stratified by their maximum TPMs in the 30 ENCODE RNA-seq data sets: (1) TPM < 1 (413 transcripts); (2) 1 ≤ TPM < 10 (515 transcripts); and (3) TPM ≥ 10 (328 transcripts). (B) Comparison of target transcript GCM1 and predicted models by five meta-assembly methods.
The benchmark experiment revealed that 1-Step methods have a notable advantage over 2-Step methods at learning the overall splicing structure of transcripts. In-depth comparison of the number of transcripts that had their structures correctly predicted confirmed this feature (Supplemental Table 3). To further elucidate the advantage of 1-Step methods, we examined all 18 transcripts that were predicted by both 1-Step methods and missed by all three 2-Step methods (Fig. 1B; Supplemental Fig. 5). In this set of transcripts, GCM1, a chorion-specific transcription factor, contains the largest number of splice junctions (five in total). Both of the two 1-Step methods modeled the transcript structure of GCM1 correctly, whereas all of the three 2-Step methods missed its first splice junction (Fig. 1B). Detailed examination of the input RNA-seq fragments from all data sets revealed the existence of a single RNA-seq fragment from the data set ENCFF782TAX that provided information for GCM1’s first junction. This fragment was disconnected from the rest of the ENCFF782TAX fragments (Supplemental Fig. 6). Consequently, neither Cufflinks nor StringTie predicted this junction in their transcript models, leading all of the 2-Step methods to miss this junction (Supplemental Fig. 6). Pooling all of the data sets brings this particular fragment together with other fragments spanning this junction and enables both of the two 1-Step methods to detect it. In summary, this benchmark test established clear advantages of 1-Step methods over 2-Step methods.
Next, we performed a second benchmark experiment based on simulated RNA-seq data and incorporated noise in the form of RNA-seq reads coming from a background noise component (Supplemental Note 2). In this benchmark, 1-Step methods missed fewer targets than 2-Step methods (Supplemental Table 4; Supplemental Fig. 7). Transcripts predicted from noisy RNA-seq fragments or alignments tended to have lower expression levels than correctly detected transcripts and supported an expression-based filtering step to remove noisy transcript models (Supplemental Figs. 8, 9). For target transcripts that were predicted by all five methods, precision and recall were similar across all methods, except for Cufflinks + Cuffmerge which had lower precision (Supplemental Fig. 10).
Pooling RNA-seq fragments by 1-Step methods from diverse samples could result in prediction of chimeric transcripts (i.e., combination of partially overlapping transcripts that are expressed under different conditions). To investigate this issue, we considered ‘newly discovered’ transcripts that were annotated by GENCODE version 24, but not by GENCODE version 20 (earliest available version for hg38). Only 5% (51 of 963) of the transcripts were partially overlapping on the same strand and could potentially lead to chimeric transcripts (Supplemental Note 3; Supplemental Table 5). This indicated a very small fraction of chimeric loci for transcript discovery. To formally evaluate this, we performed a third benchmark experiment using 40 ENCODE RNA-seq data sets encompassing a diverse set of nineteen human tissues from five different donors (Supplemental Note 3; Supplemental Table 6; Supplemental Fig. 11). This experiment yielded, at most, 16 chimeric transcripts among the set of more than 1000 newly discovered transcripts for 1-Step methods (Supplemental Fig. 12). This was largely due to incomplete RNA-seq coverage of chimeric loci and suggested that chimeric transcripts are not typical predictions from 1-Step methods. Furthermore, 6–12 chimeric transcripts were also predicted by the 2-Step method StringTie + merging, depending on the number of input data sets (Supplemental Fig. 12).
PRAM: an R package for applying the 1-Step approach to transcript discovery
Motivated by the benchmark results, we organized the 1-Step approach into a computational pipeline named PRAM (Supplemental Code) to discover transcripts in intergenic regions from multiple RNA-seq data sets. PRAM's workflow contains four steps (Fig. 2A). First, PRAM defines its search space as the intergenic genomic regions defined by an existing transcript annotation and a user-supplied minimum distance to genes. Next, it extracts all of the alignments that reside in these intergenic regions from multiple RNA-seq data sets. Then, it builds transcript models using the 1-Step method pooling + Cufflinks, which was selected because it had the highest recall for individual junctions and transcript structures in our benchmark test. In the final step, PRAM filters transcript models by their numbers of exons and lengths with user-specified parameters. These transcript models represent the master set and serve as the entry point for investigator-specific queries. The PRAM package is available at Bioconductor (https://bioconductor.org/packages/pram) and supports parallel computing. To increase the package's functionality, PRAM also includes implementations of the other 1-Step method, pooling + StringTie, as well as three 2-Step methods, Cufflinks + Cuffmerge, Cufflinks + TACO, and StringTie + merging. In addition, if a user only provides a single RNA-seq data set as the input, PRAM accommodates building intergenic transcript models using either Cufflinks or StringTie. Evaluation of the computational requirements on the 30 ENCODE RNA-seq data sets underlined that PRAM markedly reduced input size and had competitive computing time and memory cost for intergenic transcript discovery (Supplemental Note 4; Supplemental Tables 7–9). Analysis of the trade-off between accuracy and execution time suggested that pooling + Cufflinks with a large number of input data sets resulted in the fewest missed targets, while pooling + StringTie with about 30 input data sets required less computing time and yielded high precision (Supplemental Figs. 13–15).
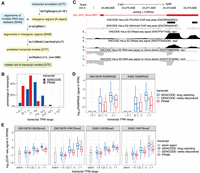
PRAM as a new computational framework predicts a valid master set of transcript models in human intergenic regions. (A) PRAM's workflow of input (cyan), intermediate (yellow), and output (green) files, with format labeled in brackets. PRAM's R functions and example parameters for each step are displayed next to arrows. (B) Distribution of GENCODE and PRAM transcripts in terms of expression levels across seven ENCODE cell lines. (C) PRAM transcript with the highest TPM had multiple complementary genomic features supporting its existence. The model ‘plcf_chr5_minus.2607.1’ had an average TPM of 245 in HeLa-S3 cells. It had high DNase-seq signals around its 5′ exon, suggesting high chromatin accessibility, and had multiple H3K4me3 ChIP-seq peaks, suggesting active transcription. Moreover, it had two RNA Pol II ChIP-seq peaks in close proximity to its transcription start site. All of these external genomic data supported the existence of this highly expressed PRAM transcript. (D,E) RAMPAGE (D) and histone modification ChIP-seq (E) signals of GENCODE and PRAM transcripts stratified by their expression levels together with ‘silent genomic regions’ defined based on H3K27me3 peaks as negative controls in all of GM12878 or K562's data sets. RAMPAGE and ChIP-seq values were derived from replicate 1 in their corresponding data sets (Supplemental Tables 13, 14). Transcripts with promoter or genomic span mappability <0.8 were excluded from D or E, respectively, due to uncertainty in their RAMPAGE or epigenetic signals. RAMPAGE and ChIP-seq signals were calculated as reads per million (RPM) and reads per kilobase per million (RPKM), respectively.
PRAM discovers new human transcripts supported by external genomic data
Given PRAM's pooling + Cufflinks predictions on the 30 RNA-seq data sets, we filtered transcript models for those with at least two exons and a minimum genomic span (total length of all exons and introns) of 200 bp. This screening resulted in a master set of 14,226 transcript models grouped into 10,372 gene models. All of the transcripts are available in a session named ‘PRAM_master_set’ at the UCSC Genome Browser (http://genome.ucsc.edu/cgi-bin/hgPublicSessions). The genomic coordinates of all models and their expression levels in the 30 RNA-seq data sets are available at GitHub (https://github.com/pliu55/PRAM_paper).
We compared the expression levels of these new transcripts with those of GENCODE-annotated transcripts in the 30 RNA-seq data sets from seven cell lines. To simplify the comparison, a transcript's expression level in each cell line was first summarized as the average of its TPM values across all the RNA-seq data sets from that cell line. If a transcript was not expressed (TPM = 0) in any RNA-seq data set of a cell line, we considered this transcript as unexpressed in this cell line and assigned it a TPM of 0 instead of taking the average. Then, a transcript's overall expression level was defined as the maximum expression level across all the cell lines. Transcripts that were not expressed in any of the seven cell lines were excluded from the comparison (Supplemental Table 10 displays two examples). For a fair comparison, we also excluded GENCODE transcripts that had only one exon or had a genomic span shorter than 200 bp. These filters resulted in 109,275 GENCODE transcripts (from a total of 198,201 GENCODE transcripts on Chromosomes 1 to 22 and X) and 5389 PRAM transcripts (Supplemental Table 11). Out of the 5389 PRAM transcripts, 2938 (55%) had TPM ∈ [0.1, 1), whereas out of the 109,275 GENCODE transcripts, only 32,950 (30%) fell into the same range, suggesting relatively lower expression levels for PRAM transcripts (Fig. 2B). The same trend was observed if we calculated a transcript's overall expression level as the maximum TPM across all 30 RNA-seq data sets regardless of cell line identity (Supplemental Fig. 16). Splitting GENCODE transcripts into ‘newly discovered’ and ‘long-standing’ showed that PRAM transcripts had similar expression levels to those of ‘newly discovered’ transcripts (Supplemental Fig. 17). Two PRAM transcripts had an average TPM > 100 and were supported by complementary genomic assays (Fig. 2C; Supplemental Fig. 18).
Further analysis on PRAM transcripts showed that they had fewer but longer exons and shorter introns than GENCODE transcripts (Supplemental Fig. 19), had higher repeat composition than newly discovered and long-standing transcripts (Supplemental Fig. 20), and were unlikely to be enhancer RNAs (Supplemental Note 5; Supplemental Fig. 21; Kim et al. 2010; The FANTOM Consortium et al. 2014) or upstream open read frames (Supplemental Note 5; McGillivray et al. 2018),
PRAM transcripts were built on biological RNA-seq data sets that were prone to contamination with technical noise, which we define as sequencing fragments not originating from true transcripts. We investigated the potential impact of such technical noise on PRAM transcript predictions by examining promoter activities and epigenetic signals of H3K36me3 and H3K79me2, the two histone marks that best correlated with gene expression (Dong et al. 2012). The premise of this analysis relied on the widely observed association of the expression levels of actual transcripts with their promoter activities and epigenetic signals (Dong et al. 2012) as opposed to little or no association for transcript models arising from technical noise. We stratified PRAM transcripts into three groups with respect to their expression levels as TPM < 0.1, TPM ∈ [0.1, 1), and TPM ≥ 1 across all RNA-seq data sets from GM12878 or K562 (Supplemental Table 12). In addition, we included GENCODE transcripts (version 24) as a positive control and further split them into long-standing and newly discovered classes (Supplemental Table 12). This enabled us to examine if PRAM transcripts share similar features with newly discovered GENCODE transcripts. We quantified transcript promoter activities by computing the RAMPAGE signals (Supplemental Table 13) in the 500-bp regions flanking transcription start sites. In both GM12878 and K562, PRAM transcripts showed the same trend as GENCODE transcripts in that higher expression levels associated with higher promoter activities (Fig. 2D; Supplemental Fig. 22). Moreover, interquartile ranges of PRAM transcript RAMPAGE signals were more similar to those of newly discovered transcripts (Fig. 2D; Supplemental Fig. 22), indicating that PRAM transcripts exhibited promoter activities that were consistent with those of newly discovered transcripts. We also evaluated H3K36me3 and H3K79me2 ChIP-seq signals (Supplemental Table 14) over genomic spans of transcripts. Similar to promoter activities, PRAM transcripts exhibited the same trend over TPM ranges as GENCODE transcripts and had an interquartile range similar to that of newly discovered GENCODE transcripts (Fig. 2E; Supplemental Fig. 23). Moreover, they had significantly higher signals than those of ‘silent regions’ defined by ChIP-seq peaks of a transcriptional repressive mark H3K27me3 (Fig. 2E; Supplemental Fig. 23). The positive correlation of expression levels with promoter activities and epigenetic signatures suggested that PRAM transcripts are unlikely to have been built from RNA-seq technical noise. The resemblance of PRAM transcripts to newly discovered GENCODE transcripts further support the biological relevance of our PRAM models.
In addition to supporting the biological relevance of PRAM transcripts by their RAMPAGE and histone modification signals, we also asked whether a comparison of PRAM transcripts with the latest GENCODE annotation (which was not utilized in PRAM) and an investigation of their conservation and protein-coding potential could provide any further support for their potential functionality. Since PRAM transcripts were multi-exonic with a minimum genomic span of 200 bp and resided in intergenic regions that were at least 10 kb away from any GENCODE version 24 genes or pseudogenes, we asked whether the latest GENCODE annotation (version 29, as of Jan. 2019) had multi-exonic transcripts satisfying these requirements. Indeed, the latest GENCODE annotation had 272 such transcripts. Of these, 48% (131 out of 272) overlapped with PRAM transcripts and automatically validated these 131 PRAM transcripts according to the metric of appearing in the latest GENCODE annotations. Comparison with the GENCODE regularly updated data (as of Nov. 26, 2019) revealed that 23 GENCODE transcripts fell within intergenic regions and 11 of them (48%) overlapped with PRAM transcripts (Supplemental Fig. 24).
In terms of conservation, 64.4% (9164 of 14,226) (Supplemental Table 15) of the PRAM transcripts mapped to the same strand on the same chromosome in mouse (mm10). This percentage was within the range of newly discovered GENCODE transcripts (53.7%) and long-standing GENCODE transcripts (72.5%) (Supplemental Table 15), suggesting that PRAM transcripts were as conserved as GENCODE transcripts. Of 14,226 PRAM transcripts, 1170 (8.2%) overlapped with GENCODE mouse transcripts (vM19), a percentage that is similar to that of newly discovered GECODE transcripts (16.7%) but far lower than that of long-standing GENCODE transcripts (64.5%) (Supplemental Table 15), indicating that PRAM transcripts had features similar to those of newly discovered GENCODE transcripts. Analysis by phastCons scores based on 100 vertebrate species also suggested a similar degree of conservation between PRAM transcripts and newly discovered GENCODE transcripts, and their levels of conservation were higher than expected by chance (Supplemental Fig. 25).
To assess the coding potential of PRAM transcripts, we used BLAST (Camacho et al. 2009) and PhyloCSF scores (Supplemental Note 6; Supplemental Tables 16–19; Supplemental Figs. 26–31; Mudge et al. 2019). The distributions of the number of BLAST-matched proteins for PRAM and newly discovered transcripts are similar (Supplemental Fig. 26). As only 6% of newly discovered GENCODE transcripts are classified as protein-coding, these BLAST results suggest that the vast majority of PRAM transcripts are also noncoding. Nevertheless, the BLAST and PhyloCSF results for a number of PRAM transcripts are suggestive of protein-coding potential, and we highlight three such transcripts. One repeat-free PRAM transcript, plcf_chr2_minus.9034.2, with >100 BLAST hits, has an ORF with a positive PhyloCSF score (Supplemental Fig. 30). Two other PRAM transcripts have >70% of their exons overlapped with PhyloCSF-predicted coding regions on the same strand regardless of frame (Supplemental Table 19; Supplemental Fig. 31).
Comparison with Pacific Biosciences (PacBio) long reads revealed that a substantial fraction of PRAM transcripts overlapped with these long reads and had matching splice junctions (Supplemental Fig. 32). Collectively, these comparisons with multiple complementary data sources support the biological relevance of the PRAM transcripts.
As a final assessment of PRAM transcripts, we used RAMPAGE and histone modification ChIP-seq data to validate 1-Step-predicted transcripts that were missed by 2-Step methods (Supplemental Note 7; Supplemental Table 20). We found that these transcripts had well-supported promoter activities (Supplemental Fig. 33) and epigenetic signals (Supplemental Fig. 34), suggesting that 1-Step methods outperformed 2-Step methods.
New transcripts were discovered by PRAM from mouse hematopoietic RNA-seq data sets and validated experimentally
As a case study, we applied PRAM on 32 hematopoietic mouse ENCODE RNA-seq data sets (Supplemental Table 21) to predict unannotated transcripts in intergenic regions (Fig. 3A). Specifically, we used the 1-Step method of pooling + Cufflinks, which had the highest recall for splice junction and splice pattern identification as well as comparable recall for exon nucleotide detection in our benchmark test. PRAM built 6969 gene models containing 8652 spliced transcripts. Focusing on genes that could be most easily validated, we selected the 2657 gene models, corresponding to 3189 transcripts, with mappability ≥0.8 and not overlapping with GENCODE or RefSeq genes on either strand (Fig. 3A). We further screened transcript models by selecting those that were differentially expressed in at least two of the following hematopoiesis-related RNA-seq data sets (Supplemental Table 22): (1) wild type versus Gata2 +9.5 enhancer-mutant aorta-gonad-mesonephros (AGM, which includes hematopoietic stem cells) (Gao et al. 2013); (2) wild type versus Gata2 −77 enhancer-mutant fetal liver containing hematopoietic stem and progenitor cells (Johnson et al. 2015); and (3) untreated G1E-ER-GATA1 versus ß-estradiol-induced (48 h to induce erythroid maturation) G1E-ER-GATA1 proerythroblast-like cells (Tanimura et al. 2016). GATA2 is a master transcriptional regulator of hematopoiesis (Tsai et al. 1994; Katsumura et al. 2017). The +9.5 Gata2 enhancer triggers hematopoietic stem cell (HSC) generation in the AGM (Gao et al. 2013; Soukup et al. 2019), and both the +9.5 and −77 enhancers confer differentiation potential to myelo-erythroid progenitor cells (Johnson et al. 2015, 2020; Mehta et al. 2017). In addition to the three data sets, we analyzed wild type versus Exosc10 mutant pluripotent embryonic stem cells (Pefanis et al. 2015). This selection step removed most of the gene models and resulted in 10 gene models (corresponding to 18 transcript models). Further filtering by conservation between mouse and human, as well as mappability of the exons to ensure qRT-PCR primer design (see Methods; Fig. 3A; Supplemental Table 23), narrowed down the gene models to six (corresponding to 13 transcript models) for experimental validation (Table 1; Supplemental Fig. 35). We evaluated this resulting list for potential regulatory activity by occupancy of GATA2 and TAL1, which often colocalizes with GATA2 on chromatin (Wozniak et al. 2008; Fujiwara et al. 2009; Wilson et al. 2010). All of the gene models had at least one GATA2 peak nearby based on a large collection of ChIP-seq data sets (Table 1; Supplemental Table 24). Moreover, four out of six models had GATA2 peaks overlapping with predicted enhancers identified during mouse blood formation (Table 1; Lara-Astiaso et al. 2014). Two models, CUFFm.chr12.33668 and CUFFp.chr12.15498, had GATA2 and TAL1 peaks overlapping with each other in three ChIP-seq data sets: two in G1E cells and one in HPC7 cells, an immortalized cell line that mimics multipotent hematopoietic precursors (Table 1; Fig. 3B; Wilson et al. 2010). In two data sets, G1E and HPC7, the overlapping GATA2-TAL1 peaks harbored a “+9.5-like” composite element CANNTG-[N6-14]-AGATAA (N represents A, C, T, or G; the spacer in between ranged from 6 to 14 nucleotides) (Table 1; Fig. 3B). This element resembles that found in the +9.5 Gata2 intronic enhancer, which is required for hematopoietic stem cell genesis (Gao et al. 2013) and is also found on diverse hematopoietic-regulatory genes (Wadman 1997; Hewitt et al. 2015, 2017). All ChIP-seq, motif, and enhancer features supported biological relevance of these six gene models.
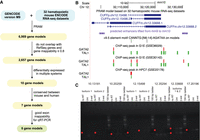
Genomic features and experimental validations of PRAM mouse transcripts. (A) Workflow of applying PRAM to discover transcripts from mouse hematopoiesis-related RNA-seq data sets: input (cyan), intermediate results (yellow), and output (green). (B) PRAM transcripts CUFFp.chr12.15498 and CUFFm.chr12.33668 had multiple supporting genomic features from external data sets. (C) Semi-qRT-PCR measurements of the six PRAM models in untreated (Unt) and 48-h ß-estradiol (ß-est)-treated G1E-ER-GATA1 cells. Red dots demarcate anticipated transcript sizes. Isoforms with splice junctions distant from each other were measured separately. Gene model name prefixes were removed for brevity.
Genomic features of PRAM-predicted mouse gene models
To experimentally validate these six models, we performed semiquantitative reverse transcription PCR (semi-qRT-PCR) in untreated and 48-h ß-estradiol-treated G1E-ER-GATA1 cells (Supplemental Tables 22, 25; Supplemental Fig. 36). We chose this system because it had the largest number of computationally inferred expressed models (TPM ≥ 1 in all RNA-seq replicates of a condition): two in untreated and four in treated cells (Supplemental Table 26). In contrast, at most, one model was inferred as expressed in each of the two conditions of the other three systems (AGM, fetal livers, and ES in Supplemental Table 26). In untreated cells, semi-qRT-PCR showed that CUFFm.chr12.33668, CUFFm.chr17.20196, CUFFp.chr10.20259, and CUFFp.chr12.15498 (faint band for isoform 2) were expressed, while the other two models were not detected or yielded nonspecific amplification (Fig. 3C). This result echoes the computationally estimated status for five of the six models: CUFFm.chr12.33668 and CUFFp.chr10.20259 had TPM > 1, CUFFp.chr12.15498 had TPM slightly lower than 1, and two out of the other three models had TPM < 1 in all three replicates (Supplemental Table 26). Only the expression status of CUFFm.chr17.20196 was discordant between semi-qRT-PCR and computational inference. In cells treated by ß-estradiol for 48 h, all of the four models, CUFFm.chr12.33668, CUFFm.chr17.20196, CUFFp.chr10.20259, and CUFFp.chr12.15498, which had TPM > 1 in all three replicates, were detected by semi-qRT-PCR, whereas the other two models that had TPM < 1 in all three replicates were either not detected or had nonspecific amplification (Fig. 3C; Supplemental Table 26). These results not only confirmed the existence of PRAM transcripts but also illustrated PRAM's strength in characterizing their splicing structures. We further remark that only two of the four validated gene models were successfully predicted by 2-Step methods (Supplemental Note 8; Supplemental Tables 27, 28), highlighting the higher sensitivity of the 1-Step approach over the 2-Step approach.
Expression of PRAM models correlates with neighboring gene Pik3cg
Since we had detected CUFFm.chr12.33668, CUFFm.chr17.20196, CUFFp.chr10.20259, and CUFFp.chr12.15498 in untreated and 48-h ß-estradiol-treated G1E-ER-GATA1 cells, we decided to validate their expression levels under the corresponding condition. All four of them had higher average expression levels in treated cells than in untreated ones (Fig. 4A; Supplemental Fig. 37). In particular, CUFFm.chr12.33668, CUFFp.chr12.15498, and CUFFm.chr17.20196 were significantly differentially expressed (Fig. 4A; Supplemental Fig. 37). CUFFm.chr12.33668 and CUFFp.chr12.15498's upstream and downstream neighbors, Pik3cg and Prkar2b, were also significantly differentially expressed (Fig. 4A). Moreover, both genes and both gene models had fold changes greater than two in the RNA-seq data sets between the same two conditions and in the RNA-seq data sets of wild type versus +9.5 enhancer-mutant AGM (Fig. 4B). The expression patterns of our gene models and their neighboring genes instigated us to further investigate their expressions during erythroid maturation of fetal liver cells. Both gene models were differentially expressed (Supplemental Fig. 38), which again confirmed their existence and PRAM's strength in predicting unannotated transcripts. Pik3cg and Prkar2b were not differentially expressed in fetal liver cells (Supplemental Fig. 38), indicating that their relationship with our gene models is system-dependent. We examined protein-coding potential for the two transcripts of CUFFm.chr12.33668 and a transcript of CUFFp.chr12.15498 that was differentially expressed in untreated and treated G1E cells (Fig. 4A). All of them matched at least one mammalian protein containing ≥60 amino acids with ≥75% of its sequence aligned under a BLASTX e-value cutoff of 10−15 (Supplemental Table 29). CUFFm.chr12.33668.2 and CUFFp.chr12.15498.2 aligned to multiple protein segments longer than 100 amino acids, whereas CUFFm.chr12.33668.1, the longest transcript of the three, only aligned to a protein segment of 69 amino acids. Comparison with a recent mouse GENCODE annotation (vM18) showed that CUFFm.chr12.33668.1 and CUFFm.chr12.33668.2 overlapped with newly annotated transcripts in similar 5′ and 3′ ends (Supplemental Fig. 39). The new GENCODE annotation suggested again the existence of PRAM mouse transcripts.
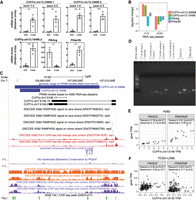
Expression of PRAM transcripts correlate with the neighboring gene Pik3cg in mouse and human. (A) Expression levels of PRAM transcripts and their neighboring genes in untreated (Unt) and 48-h ß-estradiol (ß-est)-treated G1E-ER-GATA1 cells. CUFFp.chr12.15498's isoform 1 was not detected by semi-qRT-PCR and thus was not measured here. Two-tailed Student's t-test; (**) P-value < 0.01, (***) P-value < <0.001. (B) Fold changes of PRAM mouse transcripts and their neighboring genes in the RNA-seq data sets of untreated and 48-h ß-estradiol-reated G1E-ER-GATA1 cells (G1E) and wild type versus deletion of Gata2 +9.5 enhancer aorta-gonad-mesonephros (AGM). (C) Counterparts of PRAM mouse transcripts in human with their supporting genomic features. (D) Semi-qRT-PCR measurement of PRAM human transcripts and their neighboring genes. Gene model name prefixes were removed for brevity. (E,F) Correlation of gene expression levels between CUFFm.chr7.6148 with PIK3CG and PRKAR2B in K562 cells (E) and TCGA-LAML patients (F).
Given CUFFm.chr12.33668 and CUFFp.chr12.15498's interesting features, we asked whether they had a human counterpart and, if so, whether they neighbored the same genes and coexpressed as in mouse. We collected all ENCODE RNA-seq data sets for human K562 erythroleukemia cells (Supplemental Table 30) and applied PRAM to predict transcript models. Two PRAM models, CUFFm.chr7.6148 and CUFFp.chr7.6106 overlapped with the lifted genomic span of CUFFm.chr12.33668 (Fig. 4C; Supplemental Fig. 40). Neither model overlapped with any transcript in the latest GENCODE (version 29) or CHESS annotation (version 2.1) (Pertea et al. 2018). Both of them neighbored with PIK3CG and PRKAR2B (Supplemental Fig. 41), displaying conserved synteny between mouse and human at this locus. Moreover, PIK3CG, PRKAR2B, and both PRAM models resided within chromosome segment 7q22, of which deletions had been identified in myeloid leukemias (Fischer et al. 1997). CUFFm.chr7.6148 had an estimated TPM > 1 and expected fragment counts >500 in a large fraction of K562 RNA-seq data sets (Supplemental Fig. 42A,B), supporting its expression in K562. In contrast, CUFFp.chr7.6106 was not expressed in K562 (Supplemental Note 9; Supplemental Figs. 42, 43). K562 RAMPAGE and RNA Pol II ChIP-seq data sets also supported this observation (Fig. 4C). Both RAMPAGE replicates had a peak near the 5′ end of CUFFm.chr7.6148, suggesting a potential transcription start site, whereas no RAMPAGE peak was observed for CUFFp.chr7.6106 (Fig. 4C). Similarly, both RNA Pol II ChIP-seq replicates had a strong peak around the 5′ end of CUFFm.chr7.6148, and no peak was observed around CUFFp.chr7.6106 (Fig. 4C). Following this analysis, we carried out experiments to assess whether CUFFm.chr7.6148 was a bona fide expressed transcript in K562. Our semi-qRT-PCR detected the two junctions of isoform 1 of CUFFm.chr7.6148, while not detecting the unique splice junction for the second isoform (Fig. 4D), indicating that isoform 1 was expressed in K562. This isoform matched to multiple mammalian proteins under the same criteria we used for the three mouse transcripts (Supplemental Table 31). Taken together, our computational and experimental results as well as evidence from ENCODE data all revealed the existence of CUFFm.chr7.6148 in K562.
We further considered the potential biological relevance of CUFFm.chr7.6148 by using additional genomic analysis. A predicted enhancer that resided in the intron of the first isoform and downstream of the second isoform of mouse transcript CUFFm.chr12.33668 (Fig. 3B) successfully lifted over to the intron of human CUFFm.chr7.6148 (Fig. 4C). This enhancer region was highly conserved across vertebrates, had high chromatin accessibility as suggested by DNase-seq, and had ChIP-seq signal for enhancer mark H3K27ac (Fig. 4C), indicating relevance of this predicted enhancer in K562. Moreover, this predicted enhancer was also occupied by TAL1 in the corresponding ChIP-seq experiment, as were its counterparts in mouse (Figs. 3B, 4C; Supplemental Table 32), further suggesting its potential involvement in the hematopoietic system. We assessed coexpression of CUFFm.chr7.6148 with its two neighboring genes, PIK3CG and PRKAR2B, both in K562 and relevant The Cancer Genome Atlas (TCGA) samples. In K562, expression of CUFFm.chr7.6148 significantly correlated with both PIK3CG and PRKAR2B (Fig. 4E). In TCGA Acute Myeloid Leukemia patients (TCGA-LAML) (https:// portal.gdc.cancer.gov/projects/TCGA-LAML), CUFFm.chr7.6148 was expressed at low levels (Supplemental Fig. 42C,D); however, its expression significantly correlated with that of PIK3CG (Fig. 4F). In mouse, Pik3cg encodes a catalytic subunit of PI3K, and mice lacking this subunit have reduced thymocyte survival and defective T lymphocyte activation (Sasaki et al. 2000). The expression pattern of PIK3CG and CUFFm.chr7.6148 together with a potentially active enhancer harbored in CUFFm.chr7.6148's intron indicates possible involvement of PRAM models in hematopoiesis.
Discussion
Transcript discovery and characterization opens up new dimensions in cell regulation across a broad spectrum of research fields. There is strong support for the existence of many unannotated transcripts in the well-characterized human and mouse genomes (Mudge and Harrow 2016). To innovate new strategies to identify such transcripts, we developed a computational framework named PRAM that pools multiple RNA-seq data sets to build transcript models as a master set independent of cell type or condition. We demonstrated that PRAM's 1-Step transcript reconstruction approach outperforms the conventional 2-Step approach in data-driven computational experiments. In our application of PRAM to mouse and human genomes, we discovered unannotated transcripts in hematopoietic cell systems, which were supported by multiple lines of genomic data evidence and validated by semi-qRT-PCR and differential expression experiments. Moreover, one transcript shared an expression pattern with its neighboring genes in both mouse and human. Collectively, our experiments indicate that PRAM provides an efficient and reliable method to extend existing technologies to discover transcripts.
To discover new transcripts in intergenic regions, PRAM pools multiple RNA-seq data sets first and then builds transcript models. This new approach increases both the depth and coverage of input sequencing data for predicting transcript models and therefore enables PRAM to have higher recall than other methods (Fig. 1A). PRAM is computationally feasible since it leverages the observation that the number of RNA-seq fragments aligning to intergenic regions is much smaller than that aligning to known genes (Supplemental Tables 7, 8). Moreover, stratifying model building by chromosome and strand for parallel computing makes the computational cost of 1-Step methods only slightly higher or comparable to 2-Step methods (Supplemental Table 9).
We have not yet explored whether PRAM is useful for predicting additional transcripts at known genic regions. We anticipate that the pooling feature of PRAM would increase the detection power for novel transcripts that partially overlap with existing annotations. Furthermore, PRAM's parallel-processing capability and its ability to run on specified genomic regions make it computationally feasible to apply PRAM to predict novel transcripts at genic regions. However, the issue of chimeric transcript prediction is likely to be more significant at known genic regions, due to the highly overlapping nature of alternative isoforms. Nevertheless, the higher detection rate enabled by pooling within intergenic regions suggests that it would be worthwhile to investigate whether a 1-Step approach may improve detection of alternatively spliced isoforms.
Methods
Benchmark test on 1-Step and 2-Step methods
For each alignment file, we only considered fragments that had both mates properly paired and uniquely mapped to the same chromosome (1 to 22 and X) to avoid any ambiguity during counting. When different fragments aligned to the same genomic region, we kept, at most, 10 of them for each of the uni- and multimapping fragments to speed up transcript assembly. Based on the 1256 target transcripts, we filtered RNA-seq data sets by keeping spliced fragments that matched to any transcript junction and nonspliced fragments that solely aligned to exons.
In all five transcript reconstruction methods, we used 0.1 as the minimum isoform fraction cutoff. We removed transcript models that did not have strand assignments (not labeled as ‘+’ or ‘−’) or that were labeled with a strand inconsistent with the input RNA-seq data (e.g., a model built from RNA-seq alignments on ‘+’ strand, but labeled as ‘−’ strand). For Cufflinks (version 2.2.1) and StringTie (version 1.3.3), we allowed 100% multireads per transcript and required at least one fragment to report a transfrag. Cufflinks provided options that enabled us to use bias correction by human genome sequences and a ‘rescue method’ for multimapping fragments. For StringTie-merge and TACO (version 0.7.0), we allowed transcript models to be reported regardless of their expression levels (Cuffmerge does not have an option for this purpose). Because the input reads were strand-specific, we used TACO's option to disable assembly of unstranded transfrags.
Prediction and validation of master set human intergenic transcript models
After a master set of transcript models was built, we merged them with GENCODE (version 24) transcript annotations and quantified their expression using ENCODE's STAR-RSEM protocol (https://github.com/ENCODE-DCC/long-rna-seq-pipeline). RAMPAGE signals (in RPM) for a transcript's promoter were calculated by ENCODE's RAMPAGE processing pipeline (https://github.com/ENCODE-DCC/long-rna-seq-pipeline/blob/v2.3.0/dnanexus/rampa ge/rampage-signals/resources/usr/bin/rampage_signal.sh). ChIP-seq alignments labeled as unmapped, not passing filters (e.g., platform/vendor quality controls), or PCR/optical duplicates were removed. We kept, at most, five strand-specific identical alignments to avoid PCR artifacts and calculated ChIP-seq signals (in RPKM) over all of a transcript's exons and introns.
Discovery and characterization of new mouse and K562 transcripts
We defined ‘intergenic regions’ as genomic intervals that were 10 kb away from any known genes on either strand of Chromosomes 1 to 19, X, and Y according to GENCODE annotation version M9. We aligned each RNA-seq FASTQ file by STAR (Dobin et al. 2013) with ENCODE's protocol. In order to detect more novel splice junctions for predicting transcript models, we ran STAR in its ‘2-pass’ mode. To look for models that were more likely to be true transcripts, we removed single-exon models and models with genomic span <200 nt. We also removed models that were labeled with the incorrect strand (e.g., built from ‘+’ strand RNA-seq data but were labeled as ‘−’ strand). To make sure transcript models could be validated experimentally, we removed models with overall (exons and introns) mappability score <0.8, all exons’ mappability score <0.8, or any exon's mappability score <0.001. Since we were interested in transcripts that were conserved between mouse and human, we only kept models that had all of their exons and introns mapped to the same strand on the same chromosome after being lifted over from mouse genome (mm10) to human genome (hg38).
Abundances were estimated by STAR and RSEM using ENCODE's protocol. Differential expression analysis was carried out with EBSeq (Leng et al. 2013). Differentially expressed transcript models were selected at a false discovery rate (FDR) of 0.05 with additional requirements of fold change ≥2 and normalized fragment counts ≥10 in all RNA-seq replicates from at least one condition. For ChIP-seq data with multiple replicates and control, PRAM called peaks using SPP (Kharchenko et al. 2008) and IDR (Li et al. 2011) by ENCODE's protocol with an IDR threshold of 0.05. For ChIP-seq data with no replicate and with control, peaks were called by SPP only. For ChIP-seq data without control, all ChIP-seq replicates were pooled into one, and peaks were called by MOSAiCS (Kuan et al. 2011) with a fragment length of 200 nt and a bin size of 200 nt. A one-sample MOSAiCS model was fitted with estimated background, and peaks were called by a two-signal-component model with default MOSAiCS options. DNA motifs and enhancers were searched within 10 kb of transcript models. PRAM used UCSC Genome Browser's liftOver to examine the conservation of transcript models and nearby predicted enhancers between genomes (e.g., mouse and human). PRAM also used UCSC Genome Browser's bigWigSummary and available mappability files to calculate mappability scores of the transcript models at the exon and transcript level. Since the predicted enhancers were published in mouse genome version mm9, we lifted them over to mm10. We predicted and characterized K562 transcript models by PRAM following the same procedure used for mouse. Intergenic regions were defined based on GENCODE version 25. Enhancers were lifted from mm9 to mm10 first and then lifted to hg38 due to lack of direct conversion between mm9 to hg38.
Experimental validation
G1E-ER-GATA1 cells were maintained as described previously (Tanimura et al. 2018). ER-GATA1 activity was induced by addition of 1 μM β-estradiol (Steraloids) to the medium for 48 h. K562 cells were maintained in RPMI medium (Gibco) with 10% fetal calf serum (Gemini). Fetal liver (FL) precursors were collected from E14.5 embryos and lineage-depleted as described previously (McIver et al. 2018). Cells were cultured in a humidified incubator at 37°C (5% carbon dioxide) for 48 h. FACS of erythroid maturation was conducted on a FACSAriaII (BD Biosciences) using CD71 and Ter119 markers (antibody catalog number: PE-CD71 BioLegend #113808 and APC-Ter119 BioLegend #116212). R1: CD71lowTer119−; R2: CD71hiTer119− C; R3: CD71hiTer119+; R4: CD71low/−Ter119+.
Total RNA was purified with TRIzol (Thermo Fisher Scientific). DNase (Thermo Fisher Scientific) treatment was performed on 0.1–1 μg RNA at 25°C for 15 min, followed by addition of 2.5 mM EDTA at 65°C for 10 min. cDNA was prepared by annealing with 250 ng of a 1:5 mixture of random hexamer and oligo (dT) primers incubated with m-MLV Reverse Transcriptase (Thermo Fisher Scientific) with 10 mM DTT, RNasin (Promega), and 0.5 mM dNTPs at 42°C for 1 h, and heat-inactivated at 95°C for 5 min. For confirmation of transcripts, PCR reactions were performed with GoTaq polymerase (Promega) according to the manufacturer's instructions prior to running on a 2% agarose gel. For qPCR, cDNA was analyzed in reactions (20 μL) containing 2 μL of cDNA, primers (Supplemental Table 25; Supplemental Fig. 36), and 10 μL of Power SYBR Green (Applied Biosystems) by real-time RT-PCR with a Viia7 real-time RT-PCR cycler (Applied Biosystems). Standard curves of serial 1:5 dilutions of cDNAs were prepared from control cDNA with the highest predicted gene expression. Values were normalized to the standard curve and 18S control. Results are displayed as mean ± SEM. Statistical comparisons were performed using two-tailed Student's t-tests (two conditions) or Tukey's multiple comparison test (multiple conditions) in GraphPad Prism.
Software availability
The PRAM package is available in Supplemental Code as well as at Bioconductor (https://bioconductor.org/packages/pram).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Institutes of Health (NIH) grant HG007019 to S.K., C.N.D., and E.H.B.; NIH AI117924 to S.K. and C.N.D.; NIH HG003747 to S.K.; National Heart, Lung, and Blood Institute T32 HL07899 to A.A.S.; National Institute of Diabetes and Digestive and Kidney Diseases DK050107 and Carbone Cancer Center P30CA014520 to E.H.B. We thank the three anonymous reviewers for their insightful comments and suggestions.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.252445.119.
-
Freely available online through the Genome Research Open Access option.
- Received May 12, 2019.
- Accepted August 27, 2020.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








