
Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses
- Bérénice A. Benayoun1,5,6,7,
- Elizabeth A. Pollina1,8,
- Param Priya Singh1,
- Salah Mahmoudi1,
- Itamar Harel1,9,
- Kerriann M. Casey2,
- Ben W. Dulken1,
- Anshul Kundaje1,3 and
- Anne Brunet1,4
- 1Department of Genetics, Stanford University School of Medicine, Stanford, California 94305, USA;
- 2Department of Comparative Medicine, Stanford University School of Medicine, Stanford, California 94305, USA;
- 3Department of Computer Science, Stanford University, Stanford, California 94305, USA;
- 4Paul F. Glenn Laboratories for the Biology of Aging, Stanford University, Stanford, California 94305, USA
Abstract
Aging is accompanied by the functional decline of tissues. However, a systematic study of epigenomic and transcriptomic changes across tissues during aging is missing. Here, we generated chromatin maps and transcriptomes from four tissues and one cell type from young, middle-aged, and old mice—yielding 143 high-quality data sets. We focused on chromatin marks linked to gene expression regulation and cell identity: histone H3 trimethylation at lysine 4 (H3K4me3), a mark enriched at promoters, and histone H3 acetylation at lysine 27 (H3K27ac), a mark enriched at active enhancers. Epigenomic and transcriptomic landscapes could easily distinguish between ages, and machine-learning analysis showed that specific epigenomic states could predict transcriptional changes during aging. Analysis of data sets from all tissues identified recurrent age-related chromatin and transcriptional changes in key processes, including the up-regulation of immune system response pathways such as the interferon response. The up-regulation of the interferon response pathway with age was accompanied by increased transcription and chromatin remodeling at specific endogenous retroviral sequences. Pathways misregulated during mouse aging across tissues, notably innate immune pathways, were also misregulated with aging in other vertebrate species—African turquoise killifish, rat, and humans—indicating common signatures of age across species. To date, our data set represents the largest multitissue epigenomic and transcriptomic data set for vertebrate aging. This resource identifies chromatin and transcriptional states that are characteristic of young tissues, which could be leveraged to restore aspects of youthful functionality to old tissues.
The functional decline of organs and tissues is a hallmark of aging, and it is accompanied by changes in gene expression and chromatin modifications across cell types and tissues (Benayoun et al. 2015; Booth and Brunet 2016; Pal and Tyler 2016; Sen et al. 2016). Aging is the primary risk factor for a variety of chronic diseases, including neurodegeneration, cardiovascular disease, and cancer. Several conserved pathways are misregulated during aging, defining hallmarks or pillars of aging (López-Otín et al. 2013; Kennedy et al. 2014). One such hallmark is the accumulation of epigenetic alterations, defined here as changes to gene regulation by chromatin modifications. Perturbation in chromatin-modifying enzymes can extend lifespan in invertebrate models (Benayoun et al. 2015; Pal and Tyler 2016; Sen et al. 2016), suggesting that loss of chromatin homeostasis drives aspects of aging. As chromatin marks are relatively stable and can even persist through cell division (Kouskouti and Talianidis 2005), sustained alterations to the chromatin landscape may mediate the propagation of age-associated functional decline.
Age-dependent changes in chromatin marks (e.g., DNA methylation, histone modifications) have been observed in multiple species and tissues (Benayoun et al. 2015; Booth and Brunet 2016; Pal and Tyler 2016; Sen et al. 2016). However, most of this knowledge has relied on DNA methylation or global assessments of histone modification changes (e.g., mass spectrometry, western blot) rather than locus-specific evaluation (e.g., ChIP-seq) (Horvath 2013; Benayoun et al. 2015; Wagner 2017; Cheung et al. 2018). Several genome-wide studies have interrogated locus-specific changes in histone modifications and chromatin states, as well as changes in gene expression in several cell and tissue types with mammalian aging (e.g., tissue stem cells, liver cells, pancreatic beta cells, neurons, and T cells) (Rodwell et al. 2004; Cheung et al. 2010; Liu et al. 2013; Shulha et al. 2013; Bochkis et al. 2014; Sun et al. 2014; Avrahami et al. 2015; White et al. 2015; Zheng et al. 2015; Moskowitz et al. 2017; Stegeman and Weake 2017; Ucar et al. 2017; Nativio et al. 2018). While these studies have provided important insights into genome-wide chromatin and transcriptome remodeling with age, they have remained restricted to specific cell types and/or a single histone mark. Thus, whether general rules and patterns govern age-related chromatin and transcriptional changes with age—and how they are linked—remains largely unknown.
Results
A genome-wide epigenomic and transcriptomic landscape of four tissues and one primary cell type during mouse aging
To understand how chromatin and transcriptional profiles change during aging, we collected several tissues and cells from C57BL/6N male mice at three different time points: youth (3 mo), middle age (12 mo), and old age (29 mo). We focused on a subset of tissues—heart, liver, cerebellum, and olfactory bulb—that are known to display age-related functional decline and are clearly anatomically defined. We also derived primary cultures of neural stem cells (NSCs) from these young, middle-aged, and old mice. For each tissue or cell culture from all three ages, we generated transcriptomic maps (i.e., RNA-seq) and epigenomic maps (i.e., ChIP-seq of total Histone 3 [H3] for normalization, trimethylation of Histone 3 at lysine 4 [H3K4me3], and acetylation of Histone 3 at lysine 27 [H3K27ac]), yielding 143 data sets (Fig. 1A,B; Supplemental Fig. S1A,B; Supplemental Table S1A). We chose H3K4me3 and H3K27ac because both marks are associated with ‘active chromatin’ and because spread of these marks is linked to cell identity and specific transcriptional states. Indeed, H3K4me3 and H3K27ac are preferentially enriched at active promoters and active enhancers, respectively (Heintzman et al. 2007). Broad H3K4me3 domains that spread beyond the promoter region are known to mark genes that are important for cell identity and function (Bernstein et al. 2006; Benayoun et al. 2014; Chen et al. 2015) and exhibit increased transcriptional levels (Chen et al. 2015) and consistency (Benayoun et al. 2014). Large clusters of H3K27ac-enriched enhancers that spread beyond a simple enhancer region are known as ‘super-enhancers’ (Hnisz et al. 2013) or ‘stretch-enhancers’ (Parker et al. 2013), and they mark enhancers of genes that are cell- or tissue-specific and highly transcribed in that specific cell or tissue. Importantly, ChIP-seq data sets for the H3K4me3 and H3K27ac histone mark were normalized to paired total Histone H3 ChIP-seq data to account for potential changes in the local nucleosome landscape. Quality metrics and correlation assessment indicated that most samples were of good quality and well-correlated across tissues and ages (Supplemental Table S1B–E; see Supplemental Material).
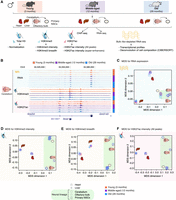
A genome-wide epigenomic and transcriptomic landscape in four tissues and one cell type during mouse aging. (A) Experimental setup (see Supplemental Table S1). (B) Example genome browser region showing tracks of data sets in cerebellum tissue at different ages. (Chr) Chromosome. (C–F) Multidimensional scaling analysis results across data sets based on RNA expression (C), H3K4me3 peak intensity (D), H3K4me3 peak breadth (E), or H3K27ac peak intensity at all peaks (F). For RNA-seq data, the input was a matrix of log2-transformed DESeq2 1.6.3 normalized counts. For chromatin marks, the most intense or broadest peak associated with a gene was used when more than one peak was present, and the log2-transformed DESeq2 1.6.3 normalized intensity or breadth was used as input.
To visualize the similarity of our genomic samples, we used multidimensional scaling (MDS) (Chen and Meltzer 2005). MDS analysis on RNA, H3K4me3 intensity (i.e., read density of H3K4me3 ChIP-seq per base pair normalized to H3 ChIP-seq density), H3K4me3 breadth (i.e., genomic spread of the H3K4me3 peaks), H3K27ac intensity (i.e., read density of H3K27ac ChIP-seq per base pair normalized to H3 ChIP-seq density), or H3K27ac super-enhancers (i.e., intensity of H3K27ac ChIP-seq at enhancer clusters) (Hnisz et al. 2013) revealed that, as expected, the main source of sample separation is the nature of the tissue, regardless of age (Fig. 1C–F; Supplemental Fig. S1D,E). Principal component analysis (PCA), another method to visualize similarity of genomic samples (Ringnér 2008), yielded similar results (Supplemental Fig. S1F,G). Thus, we used MDS analysis for subsequent analyses. Our results are consistent with the observation that RNA profiles, H3K4me3, and H3K27ac are associated with cell identity (Hnisz et al. 2013; Benayoun et al. 2014; Wagner et al. 2016) and indicate that overall, tissue and cell identities remain quite stable during aging.
To understand how age impacts the global epigenomic and transcriptomic landscapes in each tissue or cell type, we performed MDS or PCA on individual tissues/cell types (Fig. 2A–J; Supplemental Fig. S2A–T). In all tissues and for all features (RNA, H3K4me3 intensity and breadth, H3K27ac intensity and breadth), there was a progressive separation based on age, with the young samples clustering closer to the middle-aged samples and further away from the old samples (Fig. 2A–J; Supplemental Fig. S2A–T). For primary NSC cultures, there was also a separation with age for H3K4me3 intensity, H3K4me3 breadth, and H3K27ac super-enhancers (Supplemental Fig. S2F,I,O). However, the transcriptome and H3K27ac intensity of NSCs did not separate well with respect to age (Supplemental Fig. S2C,L), possibly because of technical noise. Our observations are consistent with previous reports of age-associated epigenetic changes at enhancers in liver tissue and pancreatic beta cells (Avrahami et al. 2015; Cole et al. 2017). Thus, genome-wide RNA and features of H3K4me3 and H3K27ac deposition can distinguish between ages across tissues and cells.
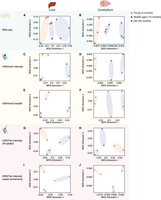
Separation of samples across tissues and cell types as a function of age. Multidimensional scaling analysis results across samples derived from specific tissues, liver and cerebellum, based on RNA expression (A,B), H3K4me3 peak intensity (C,D), H3K4me3 peak breadth (E,F), H3K27ac peak intensity (all peaks: G,H; super-enhancers only: I,J). For RNA-seq data, the input was log2-transformed DESeq2 1.6.3 normalized counts. For chromatin marks, the most intense or broadest peak associated with a gene was used when more than one peak was present, and the log2-transformed normalized intensity or breadth was used as input.
Machine learning reveals that age-related epigenomic changes can predict transcriptional changes
To understand how age-related changes in the epigenome predict age-related transcriptional changes, we took advantage of machine learning (Fig. 3A; Supplemental Fig. S3A). Using four algorithms (i.e., neural networks [NNETs], support vector machines [SVMs], gradient boosting [GBM], and random forests [RFs]), we trained models to discriminate between transcriptional changes with age—up-regulated, down-regulated, or unchanged gene expression (Fig. 3A; Supplemental Fig. S3A). As potential predictors for the models, we used, for each gene, features from chromatin data sets generated in this study (e.g., H3K4me3 signal at the promoter, or breadth of H3K4me3), features from mouse ENCODE ChIP-seq data sets in heart, liver, cerebellum, and olfactory bulb in young mice (e.g., POLR2A, H3K27me3, etc.) (Supplemental Table S2A; Shen et al. 2012; Yue et al. 2014), and features from the underlying DNA sequence (e.g., %CpG in promoter, etc.). The models were trained with sets of features that were either (1) “dynamic”—reflecting the age-related changes in the chromatin environment of genes (e.g., age-related changes in H3K4me3 breadth), (2) “static”—describing the youthful state of the chromatin or DNA sequence at the gene (e.g., H3K27ac in young tissue), or (3) both (Fig. 3A; Supplemental Fig. S3A). Because absolute gene expression levels can influence the ability to call differential gene expression (Oshlack and Wakefield 2009), we also included, as a feature, the average expression level of the genes in the young samples.
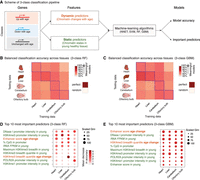
Machine-learning analysis reveals that changes in enhancer score and H3K4me3 domain breadth with age can predict transcriptional aging. (A) Scheme of the three-class machine-learning pipeline. (NNET) Neural network, (SVM) support vector machine, (RF) random forest, (GBM) gradient boosting machine. (B,C) Balanced classification accuracy over the three classes across tissues for random forest models (B) or gradient boosting machine models (C). The accuracy of the model trained in a specific tissue on the same tissue (e.g., the liver-trained model on liver data) is measured using held-out validation data. For cross-tissue validation, the entire data of the tested tissue were used. ‘Random’ accuracy illustrates the accuracy of a meaningless model (∼50%). All tests were more accurate than random. The robustness of the prediction is supported by the fact that samples for RNA and chromatin profiling were collected from independent mice at two independent times (Supplemental Table S1A). Balanced accuracy across the three classes is reported. (D,E) Feature importance from random forest models (D; Gini score and mean decrease in accuracy) or gradient boosting machine models (E; Gini score). High values indicate important predictors. See two-class models in Supplemental Figure S3. Note that two-class models, though containing less biological information, outperformed three-class models, which is consistent with the increased complexity of a classification problem with the number of classes to discriminate.
All machine-learning algorithms assigned genes to the correct transcriptional change with age 67%–81% of the time on average, significantly above that of a random classification (50%) (Fig. 3B,C; Supplemental Fig. S3B,C; Supplemental Table S3A,B). Models derived using tree-based algorithms (GBM and RF) performed slightly better than other models (69%–81% vs 67%–75%) (Supplemental Table S3B). The accuracy was similar whether validation was performed within or across tissues (Fig. 3B,C; Supplemental Table S3B; Supplemental Fig. S3B,C). These observations suggest that genes that are misregulated with age share common epigenomic signatures, even if these genes and loci are different across tissues (see below) (Fig. 4). Models trained with only static or dynamic features also had predictive power, but models trained with both types of features performed better (Supplemental Table S3B; Supplemental Fig. S3F–I). Key predictors of age-related transcriptional changes in all tissues were dynamic features, including changes in the amount of H3K27ac at enhancers or changes in the breadth of the H3K4me3 domains during aging (Fig. 3D,E; Supplemental Fig. S3D,E). Other predictors of age-related transcriptional changes were static features, describing the young chromatin context (e.g., H3K4me3 promoter intensity, H3K4me3 domain breadth) (Fig. 3D,E). While this may result from incomplete accounting for differences in gene expression (The ENCODE Project Consortium 2012), the active chromatin context of a gene in youth might predict changes in expression of that gene with age, perhaps because active loci are more impacted by stresses throughout life. Thus, static and dynamic chromatin states can both predict age-dependent changes in transcription.

Misregulated pathways during aging reveal the activation of an inflammatory innate immune signature. (A,B) Venn diagram for the overlap of significantly up-regulated (A) or down-regulated (B) genes with aging in each tissue called by DESeq2 1.6.3 at FDR < 5%. (C–F) Functional pathway enrichments (C,E–G) and transcription factor (TF) target enrichments (D) using the minimum hypergeometric test for differential RNA expression (C,D), H3K4me3 intensity (E), H3K4me3 breadth (F), and H3K27ac intensity (all enhancers) (G). Enriched pathways were plotted if four out of the six tests (RNA) or three out of the five tests (chromatin marks) were significant (FDR < 5%). (H) Heat maps of expression for all repetitive elements with significant differential expression with aging (TEtranscripts quantification and DESeq2 1.16.1 statistical test at FDR < 5%). Analysis of repetitive elements using HOMER, and overlap with TEtranscripts, is reported in Supplemental Table S6A–E.
Immune response pathways are robustly up-regulated during mouse aging
We asked if some genes are consistently misregulated with age across tissues (Fig. 4A,B; Supplemental Fig. S4A–E; Supplemental Fig. S5A–F). We identified 16 genes whose expression was up-regulated with aging in all tissues (and 0 down-regulated) when each tissue was assessed separately (FDR < 5%) (Fig. 4A,B). These genes encode complement and coagulation factors (i.e., C1QA, C1QC, C4), interferon-response related proteins (i.e., IFI27l1, IFIT3, IFITM3), and a protein involved in leukocyte trans-endothelial migration (i.e., ITGB2) (Guan et al. 2015). Though the overlap is small, these results suggest that a common response to aging across tissues is related to immunity. Genes up-regulated with age in our data set overlap with genes up-regulated with age in mouse liver (Bochkis et al. 2014; White et al. 2015) and astrocytes (Boisvert et al. 2018), and common genes are also involved in the interferon response and the complement and coagulation cascade (Supplemental Fig. S6A–I). Analyzing age-related transcriptional changes combining all tissues and ages, but including tissue of origin as a covariate, identified 771 genes up-regulated in a tissue-independent manner (and 174 genes down-regulated with age; FDR < 5%) (Supplemental Fig. S4F; Supplemental Table S4). These genes also include complement and interferon response genes. Thus, a subset of immune response genes is commonly up-regulated across tissues during aging.
In contrast, we did not observe any recurrently misregulated loci across tissues with age at the epigenomic level (H3K4me3 or H3K27ac marks, FDR < 5%) (Supplemental Fig. S5A–F). The observation that transcriptomic changes, but not epigenomic changes, are recurrent with age between tissues could be due to differences in sensitivity between read-outs or to the fact that the same gene can be regulated by different regulatory elements in diverse tissues.
Next, we investigated whether specific pathways are recurrently misregulated with age across tissues (Fig. 4C–G; Supplemental Fig. S4G–I). Several pathways were consistently misregulated with aging, not only at the transcriptome level, but also at the chromatin level (Fig. 4C,E–G; Supplemental Fig. S4G–I). Down-regulated pathways included mitochondrial function (e.g., ‘oxidative phosphorylation’), consistent with previous work in mouse and human tissues (Zahn et al. 2006, 2007). Up-regulated pathways included protein homeostasis (e.g., ‘lysosome’, ‘ribosome’) or immune signaling pathways (e.g., ‘inflammatory response’, ‘interferon alpha response’) (Fig. 4C,E-G; Supplemental Fig. S4G–I), in line with previous observations in a number of aging tissues or cells (Stegeman and Weake 2017), such as choroid plexus (Baruch et al. 2014), kidney (Rodwell et al. 2004; O'Brown et al. 2015), liver (Bochkis et al. 2014; White et al. 2015), and astrocytes (Boisvert et al. 2018). Complement and coagulation-related pathways were also significantly up-regulated with age across tissues and cell types (Fig. 4C; Supplemental Fig. S4G), consistent with findings in aging astrocytes (Boisvert et al. 2018). However, the strongest signal came from the interferon response pathways, which were significantly induced across aged tissues at both the transcriptional and chromatin levels (Fig. 4C,E–G; Supplemental Fig. S4G–I). The transcriptional activation of the interferon response was confirmed by Ingenuity Pathway Analysis (e.g., IFNG, IFNB1, IFNAR) (Supplemental Table S5A). This enrichment for immune signaling pathways (and other pathways) was also observed when re-analyzing previously published RNA-seq data sets in aging liver (Bochkis et al. 2014; White et al. 2015) and astrocytes (Supplemental Fig. S6J,K; Boisvert et al. 2018). While an age-related inflammatory response has been reported at the transcriptional level (Stegeman and Weake 2017; Boisvert et al. 2018), this is the first time it is observed at both transcriptional and chromatin levels.
Interferon response activation can stem from (1) exogenous viral infection, (2) reactivation of endogenous transposable elements (TEs) (De Cecco et al. 2013; Wood and Helfand 2013), and (3) aberrant cytosolic DNA detection by the cyclic GMP-AMP synthase (cGAS) pathway (Sun et al. 2013; West et al. 2015). As old and young mice were kept in specific-pathogen-free facilities and were documented to not have viral infection, we first queried TE expression in the different tissues during aging using our RNA-seq data sets. Increased TE activity has been reported with aging in several species (Maxwell et al. 2011; De Cecco et al. 2013; Wood and Helfand 2013; Van Meter et al. 2014). Using the TEtranscripts (Jin et al. 2015) and HOMER (Heinz et al. 2010) repeats pipelines, we identified repeats whose transcription levels were significantly changed—mostly up-regulated—with aging (Fig. 4H; Supplemental Table S6A–E). The most significantly up-regulated repetitive elements belonged to endogenous retrovirus (ERV) families (Fig. 4H). Consistently, H3K4me3 and H3K27ac intensity at several of these repeat families was remodeled during aging (Supplemental Fig. S5G,H; Supplemental Table S6F–I).
The interferon signaling pathway up-regulation is also compatible with the significant up-regulation of the “cytosolic DNA-sensing pathway,” corresponding to cGAS activation (Supplemental Fig. S4G). The cGAS pathway is up-regulated in senescent cells in response to aberrant cytoplasmic chromatin (Dou et al. 2017) and deficient mitochondrial DNA—a known consequence of aging (West et al. 2015). Thus, activation of the cGAS pathway by endogenous DNA may also play a role in the age-related increase in the interferon response.
Consistent with functional pathway enrichment results, target genes of pro-inflammatory transcription factors IRF8 and TCF3 were significantly up-regulated with aging (Fig. 4D). Similarly, targets of pro-inflammatory transcription factors IRF3, IRF5, and IRF7 were up-regulated across tissues according to Ingenuity Pathway Analysis (Supplemental Table S5A). Targets of FOXO factors were also up-regulated with aging (Fig. 4D). As FOXO factors are known to be pro-longevity genes (Martins et al. 2016) and to modulate innate immunity (Seiler et al. 2013), this up-regulation may result from a compensatory mechanism and could contribute to the up-regulation of the innate immune response with aging. MYC targets were also up-regulated (Fig. 4D), consistent with MYC's reported pro-aging effects (Hofmann et al. 2015). Finally, targets of the RNA binding protein TARDBP (also known as TDP-43) were significantly down-regulated with age across tissues (Fig. 4D). Mutations in human TARDBP (also known as TDP-43) are involved in the pathogenesis of amyotrophic lateral sclerosis and frontotemporal dementia (Scotter et al. 2015), and TDP-43 has been suggested to play a role in retrovirus suppression by host cells (Ou et al. 1995) and in microglia activation (Zhao et al. 2015). Thus, misregulation of targets of several transcription factors and RNA binding proteins may be critical for the induction of innate immune response pathways with age.
Finally, we asked if the transcriptional increase in immune pathways in tissues with aging could result from the transcriptome of infiltrated immune cells or from other changes in cellular composition (Rodwell et al. 2004; Lumeng et al. 2011; Pinto et al. 2014; O'Brown et al. 2015; Teschendorff and Zheng 2017). Using CIBERSORT to perform de-convolution of aging tissue RNA-seq data sets (Newman et al. 2015), no significant change could be detected in the proportions of predicted inflammatory cell signatures (Supplemental Fig. S7A–E; Supplemental Table S7A–D). However, some known immune markers were up-regulated with age (Supplemental Fig. S7F,G). Thus, a portion of the observed inflammatory response might be due to a low amount of infiltrated immune cells in old tissues. Our de-convolution analysis did not detect significant changes in the proportion of other cell types in tissue samples (e.g., fibroblasts, astrocytes/neurons, hepatocytes, cardiomyocytes, etc.) (Supplemental Table S7D), though we cannot exclude that actual changes exist below the sensitivity threshold of the algorithm (Teschendorff and Zheng 2017). Single-cell RNA-seq will be needed to fully address the impact of cell composition on transcriptomic changes in aging tissues. Thus, several factors may contribute to the up-regulation of inflammatory responses in old tissues.
Conservation of age-regulated transcriptional trajectories across vertebrate species
To investigate whether the age-related changes observed across mouse tissues are conserved in other vertebrate species, we used publicly available aging transcriptome data sets in rat (Yu et al. 2014), human (The GTEx Consortium 2015), and the naturally short-lived African turquoise killifish (Baumgart et al. 2014, 2016). We identified rat, human, and killifish orthologs for each mouse gene that was significantly misregulated with age. Notably, the interferon alpha and gamma response pathways were significantly misregulated in rat, human, and killifish samples (Fig. 5A; Supplemental Fig. S8A). In addition, similar aging trajectories (i.e., up-regulation or down-regulation with age) were observed for the same genes in similar tissues across vertebrate species (Fig. 5B; Supplemental Fig. S8B). These trajectories were less conserved in the GTEx human data, perhaps because other factors (e.g., environment, diseases) overshadow aging differences in human tissues. Indeed, when accounting for body mass index and metabolic status in an independent human liver microarray data set (GSE61260) (Horvath et al. 2014), gene expression trajectories with aging were more similar between mouse and human (Supplemental Fig. S8C,D). Collectively, these data indicate that core signatures of innate immune responses are consistently up-regulated with aging across vertebrate species.
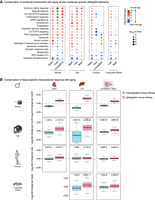
Age-related transcriptional signatures are overall conserved across vertebrate species. (A) Functional enrichments using the minimum hypergeometric test for differential RNA expression with aging in mouse, rat, human, and killifish samples. The mouse data are a subset of Figure 4C and are plotted as a reference. (B) DESeq2 1.6.3 normalized log2 fold changes per unit of time for genes orthologous to differentially expressed mouse genes in rat, human, and killifish samples. The mouse data are plotted for comparison. P-values were calculated using a one-sample, one-sided Wilcoxon test to test the differences between observed fold changes and 0 (i.e., no change with age). Only male data are plotted. Data with the contribution of females (when available) are in Supplemental Figure S8B.
Discussion
A resource for the study of aging
To understand the effect of aging on genomic regulation and chromatin identity with aging, we have generated transcriptomic and epigenomic maps in young, middle-aged, and old mice from a variety of tissues and cells known to functionally decline with age (i.e., heart, liver, cerebellum, olfactory bulb, primary NSCs). To our knowledge, this data set is the largest epigenomic and transcriptomic data set for mammalian aging to date and will serve as a resource for the aging community. It is one of the rare cases with a middle-aged point, in addition to young and old time points, which helps understand epigenomic and transcriptomic aging as trajectories rather than end-point results. Thanks to the inclusion of this middle-age time point, we find that progressive changes accumulate throughout mouse lifespan not only at the transcriptional level but also at the level of several chromatin features. The progressive accumulation of remodeling of histone modifications with aging is reminiscent of the DNA methylation clock paradigm (Horvath 2013; Cole et al. 2017; Quach et al. 2017; Stubbs et al. 2017; Wang et al. 2017). The existence of these progressive changes is compatible with the existence of chromatin modification clocks. Additional time points and individuals will be required to build such clocks and compare their performance to that of the well-established DNA methylation clock. This data set could also be integrated to future studies with additional marks. The potential interaction between different molecular clocks could provide key insights into the regulation of cellular and organismal aging.
The transcriptomic arm of our data set is consistent with the wealth of published transcriptional aging data sets (Stegeman and Weake 2017), at least at the pathways level, with an increase in inflammation and a decrease in mitochondria function. Several studies have started to interrogate genome-wide chromatin remodeling with vertebrate aging (Cheung et al. 2010; Liu et al. 2013; Shulha et al. 2013; Bochkis et al. 2014; Sun et al. 2014; Avrahami et al. 2015; Zheng et al. 2015; Cole et al. 2017; Moskowitz et al. 2017; Ucar et al. 2017), with concomitant changes in transcription. What is unique to our study is the combination of cross-tissue assessment (i.e., heart, liver, cerebellum, olfactory bulb, primary NSCs), multiple chromatin feature profiling (i.e., total H3, H3K4me3, H3K27ac), and the inclusion of three ages, thereby allowing us to conduct an integrated study of conserved and coordinated genomic misregulation with mammalian aging. This resource will help identify candidate regulators that affect age-dependent dysfunction across multiple tissues in vertebrates.
Machine learning as a powerful tool to study aging epigenomics
By using machine learning, we show that age-related epigenomic remodeling is predictive of age-related transcriptional changes. This is consistent with the ‘histone code hypothesis,’ whereby the chromatin context may direct transcriptional outputs (Jenuwein and Allis 2001), and it supports the idea that the rules that govern the relationship between the chromatin landscape and transcriptional outputs are mostly preserved throughout life. However, these models cannot be used to infer directionality or causative nature of the flow of information between chromatin and transcriptional changes, and age-related transcriptional changes might precede or even guide observed chromatin changes. What machine-learning models do indicate is that changes at the chromatin and transcription levels with aging are to some extent coordinated and that the breakdown of gene regulation with age is a complex process. In this study, we focused on active chromatin marks (H3K4me3 and H3K27ac) because of their association with cell identity in several organisms and with lifespan in invertebrates (Benayoun et al. 2015). Machine-learning models built with these active marks are fairly accurate, in line with the observation that active chromatin marks are the most predictive of expression levels by the ENCODE Consortium (The ENCODE Project Consortium 2012). Nevertheless, changes in constitutive and/or facultative heterochromatin marks (e.g., DNA methylation, H3K9me3, H3K27me3) are major events during aging (Tsurumi and Li 2012; Benayoun et al. 2015). Thus, information about age-related remodeling of heterochromatin marks may further improve the predictive power of machine-learning models. Key predictors of the relationship between chromatin and transcriptional aging could provide relevant candidates for future functional studies of aging epigenomics. These models and their features may also have a broader applicability in other contexts, including development and disease.
Innate immune pathways are broadly induced during aging across tissues and species
Our analyses reveal that immune pathways are broadly up-regulated with aging across tissues and species. This increase in immune activity in the absence of exogenous pathogens is consistent with the concept of “inflamm-aging” (Xia et al. 2016). We find that interferon response pathways, both alpha and gamma, are recurrently and robustly activated with vertebrate aging across tissues. Although interferon signaling is traditionally associated with the response to viral infection, the interferon pathway can also be induced in response to mitochondrial DNA stress and cytosolic DNA detection (Sun et al. 2013; West et al. 2015) and reactivation of endogenous transposable elements (TEs) (De Cecco et al. 2013; Wood and Helfand 2013). Our analyses find evidence for induction of cytosolic DNA-sensing pathway genes as well as a significant up-regulation of several families of TEs. Many TEs can retain viral characteristics, including the ability to replicate, form viral particles, and trigger host immune responses (Kassiotis and Stoye 2016). Thus, the global increase in innate immunity signals across tissues with aging may be mediated through the detection of endogenous aberrant DNA or reactivated endogenous retroviral particles. The increased interferon response could also be due to infiltrated immune cells. Further studies, especially at the single-cell level, will be needed to disentangle the relative contribution of infiltrated immune cells and endogenous detection of aberrant DNA. Our study indicates that inflammation is a conserved hallmark of aging, and it identifies candidate factors that could be involved in this phenomenon. This information should help define strategies to counter aging and age-related diseases.
Methods
Chromatin preparation, quantification, and immunoprecipitation
We performed ChIP experiments on different tissues and one cell type from independent animals (Supplemental Table S1A). ChIP experiments were performed on tissues and cells using a standard protocol (Webb et al. 2013; Benayoun et al. 2014). For liver, heart, and cerebellum, chromatin content was measured and equalized for all ages to enable comparison across samples of a tissue. We used 20 µg of chromatin for the H3 ChIPs, 50 µg for the H3K4me3 ChIPs, and, respectively, 70 µg (heart) or 100 µg (liver, cerebellum) for the H3K27ac ChIPs. For the olfactory bulb, chromatin from ∼30 mg of tissue was used for immunoprecipitation with anti-H3 antibody, and 60 mg was used for immunoprecipitation with anti-H3K4me3 and -H3K27ac antibodies. For NSCs, we used chromatin from ∼250,000 cells for the H3 ChIPs, ∼750,000 cells for the H3K4me3 ChIPs, and ∼1,000,000 for the H3K27ac ChIPs. Immunoprecipitations were performed with the following antibodies: 5 µg H3K4me3 antibody (Active Motif #39159, lot #1609004), 5 µg Histone H3 (Abcam #1791, lot #GR178101-1), and 7 µg H3K27ac (Active Motif #39133, lot #1613007) (see Supplemental Material for more details).
Next-generation sequencing ChIP library generation
For olfactory bulb libraries and the first set of NSCs libraries, libraries were generated with the Illumina TruSeq kit (IP-202-1012) according to the manufacturer's instructions. Briefly, repaired and adapter-ligated DNA was size-selected in the range of 250–400 bp and PCR-amplified for 16 (H3), 17 (H3K4me3), and 18–19 (H3K27ac) cycles. After the TruSeq kit became backordered, we generated libraries for the liver, heart, cerebellum, and the second set of H3 and H3K4me3 NSCs libraries using the NEBNext DNA library prep kit (E6040L). Repaired and adapter-ligated DNA was size-selected in the range of 250–400 bp using agarose gel electrophoresis and PCR-amplified for 14 (H3), 16–17 (H3K4me3), or 17–18 (H3K27ac) cycles. Library quality was assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies). Single-end 101-bp reads were generated on Illumina HiSeq 2000 machines at the Stanford Genome Center.
ChIP-seq data processing
ChIP-seq data sets were processed using a standard data processing pipeline, including quality trimming, mapping to the mm9 assembly, and duplicate removal. Significant peaks were called using MACS2 2.0.8 (Zhang et al. 2008) with the --broad option to enable detection of wider enrichment regions (see Supplemental Material for more details).
Statement on the use of the mm9 assembly
We used the GRCm37 (mm9) assembly to map all sequencing reads from mouse origin in this study (RNA-seq and ChIP-seq) because many programs did not yet support the mm10 build when we started our study. Because our study compares samples across different ages and does not perform absolute analyses, realigning the reads to GRCm38 (mm10) should not significantly affect our conclusions.
H3K4me3 breadth remodeling analysis
To compare changes in the breadth of H3K4me3 domains, we improved upon our pipeline to computationally adjust samples such that the signal-to-noise ratio across all peaks is equalized between samples (Benayoun et al. 2014). We created a reference peakset for all comparative analyses using pooled QC reads from all ages and replicates (hereafter referred to as ‘metapeaks’). To match the signal-to-noise ratios across all aging samples, we down-sampled reads separately in each H3K4me3 ChIP-seq biological sample to match the coverage histogram across all samples over the metapeaks intervals, similar to Benayoun et al. (2014). This procedure matches the “height” of the peaks from the peak caller's point of view. The appropriate down-sampling rate that allows the coverage histogram of higher sensitivity H3K4me3 ChIP-seq samples to be equal or lower than that of the lowest sensitivity H3K4me3 ChIP-seq sample was determined by minimizing the P-value of the Kolmogorov-Smirnov test (comparison to the sample with lowest H3K4me3 ChIP-seq sensitivity). To limit the effect of variations in input sample depth, we also matched the effective depth of H3 input samples to that of the lowest available sample. Final H3K4me3 domain breadth calls per samples were performed by using MACS2 2.08 with the same parameters as above. IntersectBed (BEDTools 2.16.1) (Quinlan and Hall 2010) was used to estimate the length coverage of the sample peaks over the reference metapeaks. This pipeline increases the likelihood that called gains/losses of breadth result from a change in breadth of the enriched region and not simply from an underlying difference in H3K4me3 intensity. Differential breadth was estimated using R (R Core Team 2018) and the DESeq2 R package (DESeq2 1.6.3) (Love et al. 2014).
Super-enhancer calling
Super-enhancers were called as outlined in Hnisz et al. (2013). Briefly, MACS2 H3K27ac peaks were stitched together if within 12.5 kb of one another (Hnisz et al. 2013), using mergeBed from BEDTools 2.16.0. Reads mapping within these peaks were counted using intersectBed from BEDTools 2.16.0, and the ROSE algorithm (Hnisz et al. 2013) was used to determine the H3K27ac intensity inflexion point determining typical versus super-enhancers.
H3K4me3 and H3K27ac intensity remodeling analysis
Similar to the above, we created reference peak sets (i.e., “metapeaks”) for all comparative analyses using pooled QC reads from all ages and replicates. Intensity signals for histone H3 modifications normalized to the local H3 occupancy were obtained using the “DiffBind” R package (DiffBind 1.12.3) (Ross-Innes et al. 2012). Normalized intensities were then used to estimate differential intensities as a function of age using the DESeq2 R package (DESeq2 1.6.3) (Love et al. 2014).
Cell and tissue isolation for RNA purification
For RNA isolation, we used a new cohort of aging male C57BL/6N mice (same ages as the ChIP-seq cohort), and RNA-seq data sets were generated at a later time than the ChIP-seq data sets (Supplemental Table S1A). For RNA extraction on tissues: Olfactory bulbs were microdissected from 3-mo-, 12-mo-, and 29-mo-old C57BL/6N male mice and weighed, and tissues from two independent mice of the same age were pooled per biological replicate. Cerebellum samples were dissected, weighed, and samples from two mice of the same age were pooled per biological replicate. For the liver, the leftmost part of upper left lobe of the liver was dissected and weighed from an individual mouse and was used for a single biological replicate. For the heart, following removal of blood, the bottommost part of heart ventricles from an individual mouse was dissected, weighed, and used as a single biological replicate. All tissue samples were flash-frozen in liquid nitrogen until further processing. Tissues were resuspended in 600 µL of RLT buffer (RNeasy Plus Mini kit, Qiagen) supplemented with 2-mercaptoethanol, then homogenized on Lysing Matrix D 2-mL tubes (MP Biomedicals) on a FastPrep-24 machine (MP Biomedicals) with a speed setting of 6. Heart tissue was homogenized for 4 × 30 sec, and all other tissues were homogenized for 40 sec. Subsequent RNA extraction was performed using the RNeasy Plus Mini kit (Qiagen) following the manufacturer's instructions. Primary NSC neurospheres (passages 2–3) were dissociated 16–18 h prior to collection and seeded in 12-well plates. Cells were spun down and collected in RLT buffer supplemented with 2-mercaptoethanol and processed as above.
RNA-seq library preparation
For RNA-seq library preparation, 1μg of total RNA was combined with 2 µL of a 1:100 dilution of ERCC RNA Spike-In Mix (Thermo Fisher Scientific). The resulting mix was subjected to rRNA depletion using the NEBNext rRNA Depletion kit (NEB) according to the manufacturer's protocol. Strand-specific libraries were constructed using the SMARTer Stranded RNA-Seq kit (Clontech) according to the manufacturer's protocol. Paired-end 75-bp reads were generated on the Illumina NextSeq 500 platform.
RNA-seq analysis pipeline
Paired-end 75-bp reads were trimmed using Trim Galore! 0.3.1 (github.com/FelixKrueger/TrimGalore) to retain high-quality bases with phred score >15 and a remaining length >35 bp. Read pairs were mapped to the UCSC mm9 genome build using STAR 2.4.0j (Dobin et al. 2013). Read counts were assigned to genes using subread 1.4.5-p1 (Liao et al. 2014) and were imported into R to estimate differential gene expression as a function of age using the DESeq2 R package (DESeq2 1.6.3) (Love et al. 2014). Because no overt variation of ERCC spike-in levels were observed from sample to sample within a tissue/cell type, and because their use can increase technical noise, ERCC reads were not used after initial quality-checking.
To map repetitive element expression, we used the TEtranscripts 1.5.1 software (Jin et al. 2015), with mm9 RepeatMasker data (Smit 1996–2005) downloaded from the UCSC Table Browser. Read counts were imported into R to estimate differential gene expression as a function of age using the DESeq2 R package (DESeq2 1.6.3) (Love et al. 2014). We also used the “analyzeRepeats.pl” functionality of the HOMER software (Heinz et al. 2010). In that case, read counts were imported into R (R Core Team 2018) to estimate differential gene expression as a function of age using the DESeq2 R package (DESeq2 1.16.1) (Love et al. 2014). A more recent version of DESeq2 was used for this because these analyses were run at a later time than the rest of the study. Because there are no major changes between these versions, the overall results should not be significantly affected.
Machine-learning analysis
Machine-learning models were built for each tissue, but not in NSCs since no gene was found to be significantly misregulated by RNA-seq in these cells. We built classification models in each tissue independently using four different classification algorithms as implemented through R package ‘caret’ (caret 6.0-80). Classification algorithms for neural nets (NNET; “pcaNNet”) are directly implemented in the caret 6.0-80 package. Auxiliary R packages were used with caret to implement random forests (‘randomForest’ 4.6-14), gradient boosting (‘gbm’ 2.1.3) and radial support vector machines (kernlab 0.9-27). These package versions for machine learning are used throughout our machine-learning analyses. Using 10-fold cross validation, caret optimized model parameters on the training data. Accuracies, sensitivities, and specificities for all classifiers in their cognate tissue were estimated using a test set of randomly held-out 1/3 of the data (not used for training) obtained using the “createDataPartition” function (Supplemental Table S3). Feature importance estimation was only done for RFs and GBMs, as other algorithms do not allow for it. The Gini score for feature importance was computed by caret 6.0-80 for each feature in the GBM and RF models, and the maximum in each model was scaled to ‘100’ for ease of visualization. For each gene in each tissue, we extracted two types of ‘features’: (1) dynamic features, which reflect changes to the chromatin landscape with age; and (2) static features, which reflect the state of the chromatin and transcriptional landscape in young animals. Models were built with (1) all features, (2) static features only, and (3) dynamic features only. Details of feature extraction are reported in the Supplemental Material.
Data access
ChIP-seq and RNA-seq data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA281127. All code for this study is available in Supplemental Code and at the Benayoun Laboratory GitHub repository (https://github.com/BenayounLaboratory/Mouse_Aging_Epigenomics_2018). The coordinates of significantly changed chromatin peaks can also be found in this repository.
Acknowledgments
We thank Aaron Newman and Ash Alizadeh for advice on the use of CIBERSORT for RNA-seq de-convolution and Art Owen for advice on statistical analyses. We thank Ashley Webb for assistance in tissue dissection for ChIP-seq and RNA-seq and Katja Hebestreit for advice on ChIP-seq and RNA-seq analyses. We thank Lauren Booth, Kévin Contrepois, Aaron Daugherty, C. David Lee, Dena Leeman, John Tower, Marc Vermulst, and Robin Yeo for feedback on analyses and manuscript. We thank Matthew Buckley, Brittany Demmitt, Andrew McKay, and Robin Yeo for independent code-checking. Illumina HiSeq 2000 sequencing was performed at the Stanford Genome Sequencing Service Center, and Illumina NextSeq 500 sequencing was performed at the Stanford Functional Genomics Facility, supported in part by National Institutes of Health (NIH) P30 CA124435 through the use of the Genetics Bioinformatics Service Center (Stanford Cancer Institute Shared Resource). Support for this work was provided by NIH DP1 AG044848 (A.B.), NIH P01 AG036695 (A.B. and A.K.), a generous gift from Tim and Michele Barakett (A.B.), NIH R00 AG049934 (B.A.B.), the Hanson-Thorell family fellowship (B.A.B.), and NIH F31 AG043232 (E.A.P.).
Author contributions: B.A.B., E.A.P., and A.B. designed the study. B.A.B. and E.A.P. generated the ChIP-seq and RNA-seq data sets for this study. B.A.B. processed the data and performed the analyses. P.P.S. mapped and quantified the killifish RNA-seq data sets and generated homology tables between killifish and mouse genes. I.H. and B.W.D. helped with data set generation, and I.H. processed tissues for histological analysis. P.P.S., S.M., and B.W.D. helped with independent code-checking. S.M. performed Ingenuity Pathway Analysis. K.M.C. performed the histopathology analysis. A.K. advised on data processing pipelines and on machine learning. B.A.B. and A.B. wrote the paper. All authors edited and commented on the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.240093.118.
-
Freely available online through the Genome Research Open Access option.
- Received May 31, 2018.
- Accepted January 25, 2019.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








