
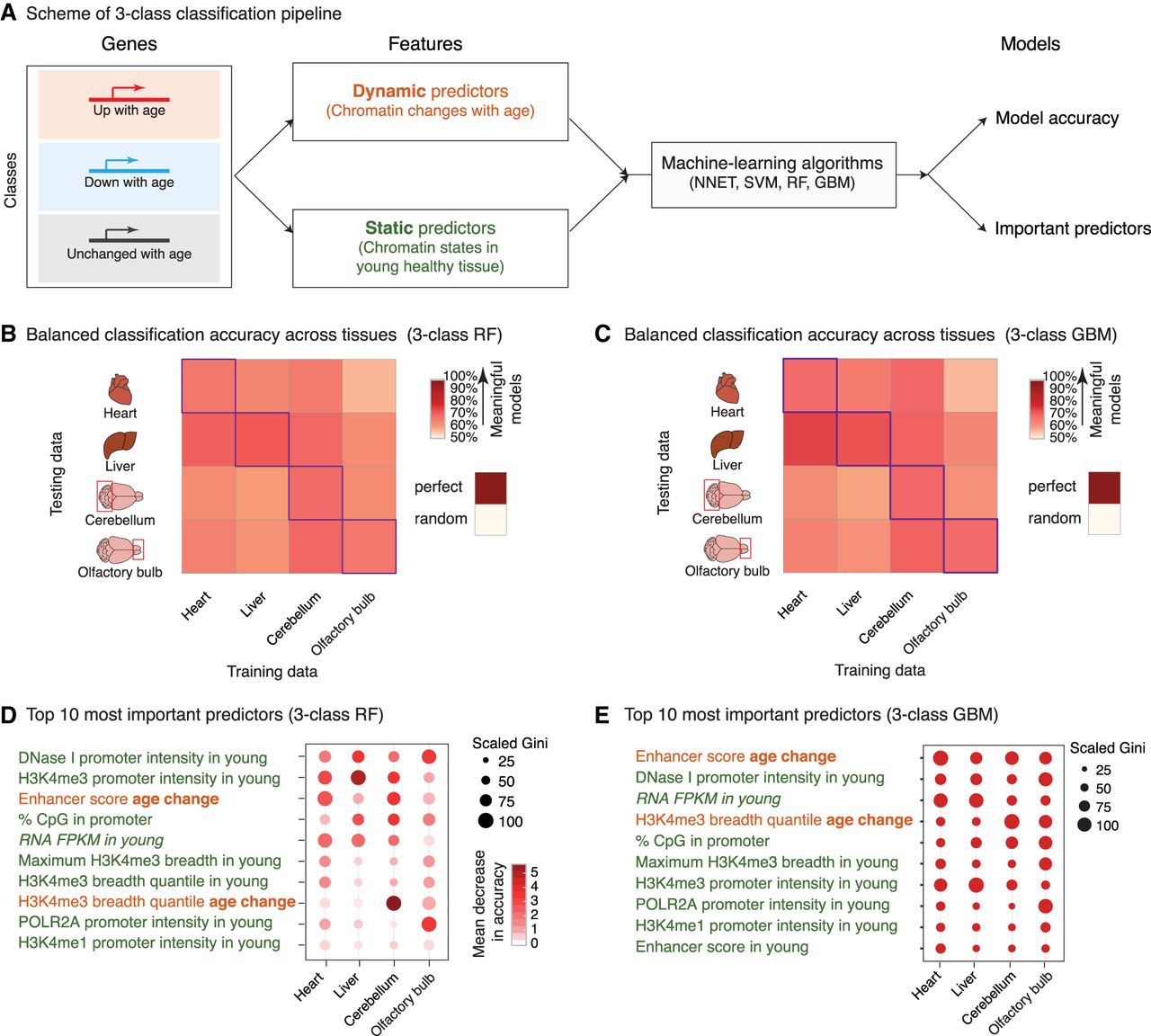
Machine-learning analysis reveals that changes in enhancer score and H3K4me3 domain breadth with age can predict transcriptional aging. (A) Scheme of the three-class machine-learning pipeline. (NNET) Neural network, (SVM) support vector machine, (RF) random forest, (GBM) gradient boosting machine. (B,C) Balanced classification accuracy over the three classes across tissues for random forest models (B) or gradient boosting machine models (C). The accuracy of the model trained in a specific tissue on the same tissue (e.g., the liver-trained model on liver data) is measured using held-out validation data. For cross-tissue validation, the entire data of the tested tissue were used. ‘Random’ accuracy illustrates the accuracy of a meaningless model (∼50%). All tests were more accurate than random. The robustness of the prediction is supported by the fact that samples for RNA and chromatin profiling were collected from independent mice at two independent times (Supplemental Table S1A). Balanced accuracy across the three classes is reported. (D,E) Feature importance from random forest models (D; Gini score and mean decrease in accuracy) or gradient boosting machine models (E; Gini score). High values indicate important predictors. See two-class models in Supplemental Figure S3. Note that two-class models, though containing less biological information, outperformed three-class models, which is consistent with the increased complexity of a classification problem with the number of classes to discriminate.








