
Systematic interrogation of human promoters
- Shira Weingarten-Gabbay1,2,3,4,
- Ronit Nir1,2,3,5,
- Shai Lubliner1,2,
- Eilon Sharon1,2,6,7,
- Yael Kalma1,2,8,
- Adina Weinberger1,2 and
- Eran Segal1,2
- 1Department of Computer Science and Applied Mathematics, Weizmann Institute of Science, Rehovot 76100, Israel;
- 2Department of Molecular Cell Biology, Weizmann Institute of Science, Rehovot 76100, Israel
-
↵3 These authors contributed equally to this work.
Abstract
Despite much research, our understanding of the architecture and cis-regulatory elements of human promoters is still lacking. Here, we devised a high-throughput assay to quantify the activity of approximately 15,000 fully designed sequences that we integrated and expressed from a fixed location within the human genome. We used this method to investigate thousands of native promoters and preinitiation complex (PIC) binding regions followed by in-depth characterization of the sequence motifs underlying promoter activity, including core promoter elements and TF binding sites. We find that core promoters drive transcription mostly unidirectionally and that sequences originating from promoters exhibit stronger activity than those originating from enhancers. By testing multiple synthetic configurations of core promoter elements, we dissect the motifs that positively and negatively regulate transcription as well as the effect of their combinations and distances, including a 10-bp periodicity in the optimal distance between the TATA and the initiator. By comprehensively screening 133 TF binding sites, we find that in contrast to core promoters, TF binding sites maintain similar activity levels in both orientations, supporting a model by which divergent transcription is driven by two distinct unidirectional core promoters sharing bidirectional TF binding sites. Finally, we find a striking agreement between the effect of binding site multiplicity of individual TFs in our assay and their tendency to appear in homotypic clusters throughout the genome. Overall, our study systematically assays the elements that drive expression in core and proximal promoter regions and sheds light on organization principles of regulatory regions in the human genome.
In contrast to the significant progress made in identifying the DNA elements involved in transcriptional regulation, our understanding of the rules that govern this process, namely, how the arrangement and combination of elements affect expression, remains mostly unknown (Shlyueva et al. 2014; Weingarten-Gabbay and Segal 2014a). Advances in DNA synthesis and sequencing technologies have led researchers to tackle these questions using high-throughput approaches, yet most studies have focused on enhancers. Thus, the core promoter region, which contains the transcription start site (TSS), and the proximal promoter region, which harbors specific transcription factor (TF) binding sites, have not been thoroughly characterized, and we have not yet achieved an in-depth understanding of their function, architecture, and cis-regulatory sequences.
Transcription initiation occurs in both promoters and enhancers, and generally generates divergent transcripts that differ in their stability (Core et al. 2008, 2014; Seila et al. 2008; Neil et al. 2009). Traditionally, the core promoter region was viewed as a universal stretch of DNA that directs the preinitiation complex (PIC) to initiate transcription. However, core promoters are structurally and functionally diverse regulatory sequences composed of a variety of DNA elements, including CpG islands, TATA-box, initiator (Inr), upstream and downstream TFIIB recognition elements (BREu and BREd), motif ten element (MTE), and downstream core promoter element (DPE) (Lagrange et al. 1998; Lim et al. 2004; Deng and Roberts 2005; Sandelin et al. 2007; Juven-Gershon and Kadonaga 2010; Kadonaga 2012). Hence, in the analysis of gene expression, it is necessary to understand and to incorporate the specific components of the core promoter (Juven-Gershon and Kadonaga 2010; Kadonaga 2012). With the growing appreciation of the importance of core promoters in determining gene expression, two recent studies measured the autonomous promoter activity of random native sequences genome-wide in human and Drosophila (Arnold et al. 2017; Cvetesic and Lenhard 2017; van Arensbergen et al. 2017). These approaches provided a large collection of endogenous promoter sequences and uncovered that autonomous promoter activity is widely distributed across the genome. However, due to their native nature, any two promoters differ in many sequence elements, and thus, the precise contribution of a single motif such as a specific core promoter element cannot be inferred solely by observing native genomic sequences. To achieve this goal, a large number of designed sequences in which specific elements are systematically varied in a highly controlled setting should be assayed.
Other pivotal components of functional promoters are TF binding sites. Despite a remarkable characterization of the sequence specificities of these transcriptional building blocks in vitro and in vivo (Berger et al. 2008; Badis et al. 2009; Jolma et al. 2013), much less is known about the effect on expression of each of the hundreds of sites identified. Moreover, the spatial organization of TF binding sites in the genome is an important feature of transcriptional regulation. Multiple binding sites for a single TF, known as homotypic clusters of TF binding sites (HCTs), are statistically enriched in proximal promoters and distal enhancers (Gotea et al. 2010). This architecture may have several mechanistic advantages such as cooperativity binding (Hertel et al. 1997), lateral diffusion between adjacent binding sites (Kim et al. 1987; Khoury et al. 1990), and functional redundancy (Somma et al. 1991; Papatsenko et al. 2002). However, it is not clear if increased number of homotypic sites for any TF always results in higher expression levels or, rather, if the effect on expression is TF-specific. Another important question is the range within which adding another site still has a substantial effect on expression and the maximal expression levels that can be achieved for different TFs. Here too, a large set of designed promoters in which binding sites for specific TFs are carefully added in various positions, orientations, and distances is needed to quantitatively characterize the relationship between TF binding sites and expression.
To address fundamental questions in gene expression, we and others have developed massively parallel reporter assays (MPRAs) probing the expression of various regulatory regions (Kwasnieski et al. 2012; Melnikov et al. 2012; Patwardhan et al. 2012; Sharon et al. 2012; Shalem et al. 2015). However, since these measurements are performed using episomal plasmids, they are limited in their ability to mimic the genomic context. Progress in this direction was recently made by integrating the reporter constructs into the human genome using lentiviruses (Weingarten-Gabbay et al. 2016; Inoue et al. 2017; Maricque et al. 2017). However, lentivirus-mediated integration occurs in random locations along the genome and is thus susceptible to the effects of local chromatin environment and interaction with neighboring enhancers. The latter is of high importance to the measurements of promoters due to core-promoter-enhancer specificity resulting in variability in core promoter activity when placed near different sets of enhancers (Zabidi et al. 2015).
Here, we present a new high-throughput method for accurately measuring approximately 15,000 fully designed sequences from a fixed and predefined locus in the human genome. By using this system, we set to decipher the sequence determinants of core promoters and proximal promoter regions, from broad aspects of mapping their location and orientation in the genome to in-depth characterization of the cis-regulatory elements driving their expression, including core promoter elements and TF binding sites.
Results
Accurate measurements of about 15,000 designed promoters from a fixed locus in the human genome
To broaden our understanding of human promoters, we designed a library of 15,753 oligonucleotides representing native and synthetic sequences. We included 508 PIC binding regions and 1875 core promoters of coding genes from the human genome. In addition, we designed synthetic sequences aimed at systematic investigation of the cis-regulatory sequences driving transcription, including core promoter elements, 133 TF binding sites, and nucleosome disfavoring sequences (see Methods) (Fig. 1A). To accurately measure promoter activity in the genomic context, we developed a high-throughput method for assaying the activity of thousands of sequences from a fixed locus in the human genome using site-specific integration into the “safe harbor” AAVS1 site (see Methods) (Fig. 1B; Urnov et al. 2005; DeKelver et al. 2010). Briefly, we obtained a mixed pool of oligonucleotides, 200 bp in length, to match our designed sequences and cloned it upstream of an eGFP reporter. We integrated the library into the AAVS1 site in K562 erythroleukemia cells by inducing a double-strand break using specific zinc finger nucleases (ZFNs) followed by genomic integration of the reporter cassette by homologous recombination. We used fluorescence-activated cell sorting (FACS) to select cells with a single integrated cassette according to mCherry expression driven from a constant EF1alpha promoter. We sorted the resulting pool into 16 bins according to eGFP expression normalized by mCherry. In the last step, we used deep sequencing to determine the distribution of reads across the different bins for each oligo. For each designed promoter, we computed its activity levels from the mean of the reads distribution and cell-to-cell variability (noise) from the standard deviation (see Methods).
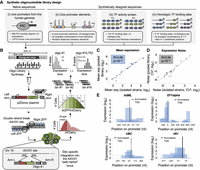
Construction and measurements of 15,753 designed oligonucleotides for promoter activity using site-specific integration technology. (A) Illustration of the design of the main sets composing the synthetic library. (B) We synthesized 15,753 designed ssDNA oligos 200 nt in length on Agilent programmable arrays and harvested them as a single pool. Oligos were amplified by PCR using constant primers and cloned into pZDonor plasmid upstream of eGFP. The plasmid pool was conucleofected with mRNAs encoding zinc finger nucleases (ZFNs) targeting the AAVS1 site into a modified K562 cell line containing only two (of three) copies of the AAVS1 site (see Methods). mCherry expression driven from a constitutive EF1alpha promoter was used to select cells with a single integration by FACS. Cells were then sorted into 16 bins according to eGFP/mCherry ratio. Oligos were amplified from each bin and submitted for deep sequencing. Finally, the distribution among expression bins was determined for each oligo, and mean expression and noise were computed. (CV) Coefficient of variation. (C,D) Accuracy of expression measurements. Twenty-one clones, each expressing a single oligo, were isolated from the library pool and identified by Sanger sequencing. eGFP/mCherry ratio was measured for each clone individually by flow cytometry. Shown are comparisons between these isolated measurements and those calculated from the pooled expression measurements for mean expression (C; R = 0.98, Pearson's correlation, P < 10−15) and noise (D; R = 0.94, Pearson's correlation, P < 10−10). (E) Detection of autonomous core promoter activity. Sequences of four full-length promoters were partitioned in-silico into 153-nt fragments with a large overlap of 103 nt between oligos. The positions of the annotated transcription start sites (TSSs) from the literature are denoted, and the positions on the x-axis are relative to the TSSs. Dashed lines represent the activity threshold determined by the empty vector measurements (Methods).
To assess the accuracy of our measurements from site-specific integration in comparison to a traditional retrovirus-based technique, we integrated a single promoter construct multiple times using each system. As expected, in our ZFN system, where all constructs are integrated into the same genomic location, the variability between cells was lower than in the retroviral system, where integration occurs at random locations, spanning a range of about one and two orders of magnitude in expression, respectively (P < 10−20) (Supplemental Fig. S1A). Moreover, the expression of independently isolated clones was highly similar in the ZFN system, whereas it varied more in the retroviral system (Supplemental Fig. S1B). To evaluate the accuracy of our assay in comparison with each oligo's individual measurement, we isolated 21 clones from the library pool and measured the expression of each isolated clone using flow cytometry. We found excellent agreement between these measurements and those extracted from the massively parallel assay for both mean expression (R = 0.98, P < 10−15) (Fig. 1C) and noise (R = 0.94, P < 10−10) (Fig. 1D). To gauge the reproducibility of our measurements, we designed replicates for different promoters with 10 unique barcodes. For each promoter, we examined the distribution of deep sequencing reads among the 16 expression bins for all 10 barcodes for which synthesis, cloning, sorting, and sequencing were independent, and found very good agreement between different barcodes (Supplemental Fig. S2). To ensure that mCherry expression driven from the constant EF1alpha promoter is not influenced by the cloned oligos, we examined the expression of eGFP and mCherry in the 16 expression bins. While eGFP levels increase, mCherry expression remains constant across the 16 expression bins with similar levels to those of the empty vector (Supplemental Fig. S3A,B). In addition, we examined the relationship between eGFP and mCherry in the isolated clones. Here too, we find no correlation between eGFP and mCherry expression, with constant mCherry levels across the different clones (P > 0.1) (Supplemental Fig. S3C). Finally, to test our ability to detect autonomous core promoter activity, we designed 153-nt-long sequences tiling the entire length of previously characterized promoters (Supplemental Table S1; Van Beveren et al. 1982; Hansen and Sharp 1983; Adachi et al. 1986; Hennighausen and Fleckenstein 1986; Adra et al. 1987; Eisenstein and Munro 1990; Moriyama et al. 1994; Nenoi et al. 1996; Lay et al. 2000; Wang et al. 2008) with a 103-bp overlap between oligos and measured their activity in the pooled assay. Our assay accurately detects the core promoter region in 10 of 11 promoters for which TSSs were previously reported. (Fig. 1E; Supplemental Fig. S4; Supplemental Table S2).
Together, these results demonstrate that our method enables highly accurate measurements of autonomous promoter activity for thousands of fully designed sequences in parallel from a fixed location within the human genome.
Functional measurements of PIC binding sequences in promoters and enhancers
Emerging evidence from recent studies suggests that in contrast to the decades-long wide-held belief, transcription initiation is not restricted to promoters. Nascent RNA measurements uncovered thousands of TSSs in promoters and enhancers with similar architecture (Core et al. 2014). Moreover, a genome-wide binding assay identified thousands of PIC-bound regions across the human genome, including enhancers (Venters and Pugh 2013). However, several questions remain unclear, including whether PIC binding sequences can act as functional core promoters, what is the relationship between binding levels and core promoter activity, and whether divergent transcription is a result of true bidirectionality or two adjacent unidirectional initiation sites.
To investigate the functional activity of PIC binding sequences across the human genome, we designed synthetic oligos to match 508 reported binding regions (Venters and Pugh 2013) and tested their ability to initiate transcription in our reporter assay (Fig. 2A; Supplemental Table S3). Our measurements uncover a positive relationship between PIC binding levels (TFIIB ChIP-exo reads number) and functional core promoter activity such that regions for which PIC binding is higher also drive stronger expression (P < 10−15) (Fig. 2B). To compare the functional transcriptional activity in promoters and enhancers directly, we designed oligos to match the sequences bound by PIC from the two regions. To control for potential differences in expression resulting from PIC binding levels, we selected sequences from the same range of binding scores for the two groups. We find that PIC binding sequences from promoters present a higher fraction of positive sequences (P < 10−5) (Fig. 2C) and activity levels (P < 0.03) (Fig. 2C) that do not stem from differences in binding intensity (P > 0.1) (Fig. 2C).
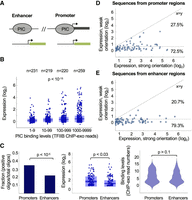
Functional measurements of autonomous core promoter activity of PIC binding sequences from promoters and enhancers. (A) Illustration of the designed sequences matching 508 PIC binding regions in promoters and enhancers that were identified by ChIP-exo measurements in K562 cells (Venters and Pugh 2013). (B) Comparison between core promoter activity of sequences with different PIC binding levels (TFIIB ChIP-exo). Data were binned into four groups according to the number of ChIP-exo reads, and expression measurements were compared between bins (P < 10−15, Kruskal-Wallis test). (C) Comparison between the fraction of positive core promoters for PIC binding sequences from promoters and enhancers (left; P < 10−5, two-proportion z-test) and the activity levels of positive sequences from both groups (middle; P < 0.03, Wilcoxon rank-sum test). To avoid biases in activity stemming from different PIC binding levels, sequences with the same number of ChIP-exo reads were selected in the design process (right; P > 0.1, Wilcoxon rank-sum test). (D,E) Comparison between core promoter activity of PIC binding sequences from promoters (D) and enhancers (E) in two orientations. Each dot represents a distinct PIC binding site that presented positive activity in at least one orientation. Expression measurements of designed sequences are shown for the stronger and weaker orientations of each pair of sequences. The horizontal dashed line represents the activity threshold as determined by empty vector measurements; the diagonal dashed line, a theoretical x = y line expected for promoters with equal expression in the two orientations.
Next, we set to investigate whether PIC binding sequences can drive bidirectional transcription. To this end, for each binding site we designed two oligos representing the core promoter sequence (−103 to +50) on either the plus or minus strand. Our measurements uncover that most of the PIC binding sequences display positive activity in only one of the two orientations tested, with 72.5% and 79.3% unidirectional versus 27.5% and 20.7% bidirectional expression from promoter and enhancer regions, respectively (Fig. 2D,E). The general unidirectional activity is also demonstrated by negative correlation between expression measurements in the plus and minus strands for promoters (R = −0.27, P < 0.02) (Supplemental Fig. S5A) and enhancers (R = −0.33, P < 0.003) (Supplemental Fig. S5B). The sequences of promoters that were active in both orientations are listed in Supplemental Table S4.
While working on this manuscript, the original paper that identified the PIC binding sequences in the human genome has been retracted (Venters and Pugh 2014). The main concerns raised were about the analyses of the core promoter elements downstream from the data acquisition (Siebert and Söding 2014), and in the retraction letter the investigators claim that to the best of their knowledge, the raw and processed ChIP-exo data are valid. However, since some doubts were also raised about the data itself, we set to investigate the validity of the PIC binding sequences tested in our library using independent measurements of transcription initiation sites by GRO-cap technique from the groups of Adam Siepel and John Lis (Core et al. 2014). We find a significant enrichment of the PIC binding regions identified by ChIP-exo in the reported TSSs identified by GRO-cap (32%, hypergeometric P < 10−198) (Supplemental Fig. S6A,B). Although both methods aimed at the detection of TSSs genome-wide, they measure different properties of the initiation region. While GRO-cap measurements rely on the detection of nascent transcripts, ChIP-exo measures the binding of the PIC that may or may not be involved in active transcription. Thus, one would expect that PIC binding regions that were detected by GRO-cap (i.e., lead to higher levels of transcript) will have stronger core promoter activity. Indeed, by comparing our functional measurements of PIC binding sites detected by GRO-cap to those that were not, we find higher promoter activity for the former (P < 10−18) (Supplemental Fig. S6C). Finally, to validate the results that we obtained with the 508 PIC binding sequences, we repeated the analyses comparing the activity and directionality of PIC binding sequences from promoters and enhancers for the 160 sequences that were identified by both ChIP-exo and GRO-cap methods and found similar results to those described above (Supplemental Fig. S6D–F).
Together, our results demonstrate positive relationships between PIC binding and core promoter activity, an intrinsic difference between sequences from promoters and enhancers, and the finding that core promoters mostly drive unidirectional transcription.
Systematic investigation of core promoter elements in synthetic and native sequences
There is no one universal architecture of core promoters. Rather, different core promoters exhibit distinct DNA elements, including CpG islands and local short sequence motifs that interact with the PIC (Sandelin et al. 2007; Kadonaga 2012). Even for the more characterized core promoter elements associated with sharp (focused) promoters such as the TATA-box, Inr, BREu, BREd, MTE, and DPE, the effect on transcription of their arrangement and combination is not fully understood. For example, the BRE motifs have been reported to act both as an activator and a repressor of transcription (Evans et al. 2001; Chen and Manley 2003; Deng and Roberts 2005; Juven-Gershon et al. 2008; Kadonaga 2012). In addition, while the MTE and DPE motifs were mostly investigated in Drosophila, their sequence is conserved from Drosophila to human, suggesting a functional role in higher eukaryotes (Kadonaga 2012). Moreover, although DPE was typically thought to substitute the TATA box in directing the precise TSS selection (Sandelin et al. 2007; Juven-Gershon and Kadonaga 2010) incorporating DPE with TATA, Inr and MTE in the rationally designed “super core promoter” yielded high transcription levels in human cells (Juven-Gershon et al. 2006).
To evaluate the contribution of GC content to promoter activity, we tested the relationship between GC content and our expression measurements of 1875 native core promoters from the human genome. We find that sequences with positive promoter activity have higher GC content than sequences with no promoter activity (P < 10−27) (Supplemental Fig. S7). Examining the distribution of GC content and promoter activity for these two groups uncovers that negative sequences can span the same GC content range as active promoters (Fig. 3A), suggesting that elevated GC content is necessary but not sufficient to enhance promoter activity and that additional sequence motifs, such as core promoter elements, are required.
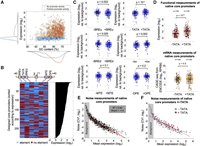
Systematic investigation of core promoter elements in synthetic configurations and native core promoters from the human genome. (A) The relationship between GC content and promoter activity in 1875 native core promoters from the human genome. (Cyan) Sequences with no promoter activity as defined by empty vector measurements; (orange) sequences with positive promoter activity. (B) Three hundred twenty synthetic oligos representing all possible combination of six core promoter elements on five different backgrounds were designed. Each line in the heatmap (left) represents a single designed oligo, and each column represents one of the six elements tested. The configurations were sorted according to the expression measurements (right). (C) Comparison between the expression of all the designed sequences with and without each of the six core promoter elements. Each measurement was normalized by the expression levels of the matching background sequence. Wilcoxon rank-sum tests were performed to determine significant differences in expression, and P-values are denoted. (D) The effect of TATA-box in native human core promoters. (Top) Expression measurements from our functional assay of native core promoters from the human genome with and without a consensus TATA-box. Elevated expression is observed in promoters with TATA element (P < 10−4, two-sample t-test). (Bottom) CAGE-seq measurements in K562 cells for the same promoters from ENCODE (The ENCODE Project Consortium 2012). No significant difference was detected between the two groups (P > 0.5, two-sample t-test). (E) Noise measurements of 990 native core promoters from the human genome as a function of mean expression. A linear fit was performed on oligos with positive core promoter activity as described before (Bar-Even et al. 2006). (F) Comparison of noise measurements of native core promoters with and without a TATA-box.
To systematically test the effect on expression of different core promoter architectures, we designed 320 synthetic core promoters with all possible combinations of the consensus sequences of the TATA-box, Inr, BREu, BREd, MTE, and DPE (Fig. 3B; Supplemental Table S5; Lagrange et al. 1998; Lim et al. 2004; Deng and Roberts 2005; Juven-Gershon et al. 2006; Juven-Gershon and Kadonaga 2010). To examine different contexts, we placed the designed configurations in five backgrounds: ACTB, human cytomegalovirus IE1 promoter (CMV), RTRAF, HIV1, and RPLP0. In each tested background, we mutated existing core promoter elements in the wild-type promoter (Supplemental Table S6). By sorting all 320 designed core promoters according to expression (Supplemental Table S7), we identify patterns of elements that are abundant in high-or low-expressing promoters (Fig. 3B). To quantitatively assay the contribution of each element, we compared the expression levels of all the tested configurations with and without each of the motifs (Fig. 3C). This approach allowed us to average over many combinations of elements and surrounding sequences and thus is not sensitive to a specific context. To account for the differences between the basal levels of the five tested backgrounds, each expression measurement was first normalized by the expression levels of the matching background described above (Supplemental Table S6). Of the six elements tested, the only two sequences that led to a significant increase in expression were TATA-box and Inr, with 45% and 28% increase, respectively (P < 10−5 and P < 10−3) (Fig. 3C). We found that both the BREu and the BREd elements significantly decreased expression by 35% and 20%, respectively (P < 10−3 and P < 10−2) (Fig. 3C). The DPE and MTE elements had no detected effect on expression (P > 0.1 and P > 0.2, respectively) (Fig. 3C), suggesting that they do not play a substantial role in humans or that they require additional context-dependent features.
Although synthetically designed oligos have a tremendous advantage in the investigation of cis-regulatory elements in a controlled setting, their sequences diverge from native promoters in the human genome. To measure the expression of native sequences, we designed 1875 core promoters of coding genes representing constitutive and induces promoters, various endogenous expression levels, interactions with distal DNA elements, and different sequence features (see Methods) (Supplemental Table S8). Next, we set to investigate the effect of the TATA-box in native context using these measurements. By comparing the expression levels of hundreds of native core promoters with and without a consensus TATA-box, we find a significant increase in TATA-containing core promoters (P < 10−4) (Fig. 3D). When we compare CAGE-seq measurements from the ENCODE Project (The ENCODE Project Consortium 2012), which indicate the transcript levels produced from the native genomic locus, we find no significant difference between the two groups (P > 0.5) (Fig. 3D). This finding demonstrates the importance of performing designated functional assays to decipher the autonomous activity of core promoters when isolated from additional factors influencing the transcriptional output such as neighboring enhancers and local chromatin environment.
In addition to regulating mean expression, core promoter elements such as the TATA-box were also shown to have an effect on cell-to-cell variability (expression noise) in yeast (Tirosh and Barkai 2008; Lehner 2010). To investigate the effect of the core promoter sequence on noise, we used the distributions of reads across the expression bins to compute for each oligo the mean and standard deviation. We quantified the noise by the squared coefficient of variation (CV2), which is the variance divided by the square mean (Fig. 1B; Bar-Even et al. 2006). We find that noise is scaled with mean expression with similar dependency as described for yeast (fitted slope of −1.4) (Fig. 3E; Bar-Even et al. 2006). However, in contrast to yeast, in which the noise levels of promoters with similar mean expression can vary more than one order of magnitude (Sharon et al. 2014), here we do not find large differences in noise for the same mean expression, with most of the variability explained by the mean expression (R2 = 0.84) (Fig. 3E). Moreover, we do not find substantial differences between TATA and TATA-less sequences in native core promoters (Fig. 3F) or for any of the six core promoter elements tested in the synthetic sequences (Supplemental Fig. S8). A potential source for the observed difference between yeast and human cells is the generation time. While yeast cells divide every ∼1.5 h, the generation time of most cultured mammalian cells is ∼24 h. Thus, using stable eGFP reporter, as done in our assay, can buffer the effect of rapid fluctuations in mRNA levels (Raj and van Oudenaarden 2008). However, since the median half-life of mammalian proteins is 46 h (Schwanhäusser et al. 2011), the stable eGFP reporter that we use here may better represent the true cell-to-cell variability of most endogenous proteins.
Taken together, our findings demonstrate significant effects of core promoter elements on mean expression, with positive effects for the TATA and the Inr, and with negative effects for the BRE upstream and downstream elements.
TATA and Inr additively increase expression at preferred distances
A key question in the investigation of core promoter elements is the effect of motif combinations on expression. Bioinformatic analyses suggested that core promoter elements act in a synergistic manner to recruit RNA polymerase II (Gershenzon and Ioshikhes 2005). Moreover, previous studies demonstrated that their coordinated effect on transcription depends on the distance between the elements (O'Shea-Greenfield and Smale 1992; Emami et al. 1997).
To investigate the relationship between the TATA and the Inr, both found to positively regulate promoter activity in our assay, we compared the expression of all tested configurations with TATA to those containing TATA and Inr. We found that adding Inr to TATA-containing promoters results in increased expression (P < 10−3) (Fig. 4A). Next, we set to investigate if the two elements act in synergy by comparing the expression levels of oligos containing both elements to the sum of expression of oligos containing TATA or Inr separately (Fig. 4B). We do not find higher expression for oligos with the two elements, suggesting that they act in a partially additive manner and not synergistically to increase transcription. We then analyzed the effect of adding MTE or DPE to Inr. In contrast to the positive effect of TATA, adding MTE or DPE had no significant effect on expression (P < 10−4, P > 0.08, and P > 0.2 for TATA, MTE, and DPE, respectively) (Supplemental Fig. S9). We also tested the effect of adding BREu and BREd to TATA-containing promoters. Here too we find, similar to their negative effect on expression when tested separately, a significant reduction in expression when adding BREu, BREd, and both BRE elements (P < 10−3, P < 0.04, and P < 0.03 for BREu, BREd, and BREu+BREd, respectively) (Supplemental Fig. S10).
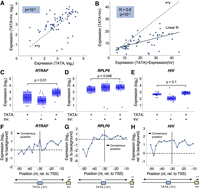
The effect on expression of TATA-box and Initiator (Inr) combinations and relative distances in different backgrounds. (A) Comparison of expression levels of synthetic oligos with TATA to those containing both TATA and Inr. Each dot represents a pair of sequences with either TATA or TATA+Inr elements. An increase in expression is observed when adding Inr (P < 10−3, Wilcoxon signed-rank test). (B) Testing for synergy between TATA and Inr elements. Each dot represents a pair of expression values. On the x-axis, expression was computed as the sum of the expression of separate oligos with either TATA or Inr. The y-axis represents expression measurements of oligos that contain the two elements. (C–E) Comparison of oligos with either TATA, Inr, or TATA+Inr in three different promoter backgrounds. Presented P-values were computed by Wilcoxon rank-sum test (n = 16 in each group). (F–H) Testing the effect of the distance between the TATA and the Inr in three different backgrounds. We designed oligos in which we placed the Inr in its consensus position and systematically changed the location of the TATA (2- to 3-nt increments). Each blue dot represents the expression level at a single position. The consensus position of the TATA (−31) is denoted.
To test whether the activity of core promoter elements depends on the background sequence, we analyzed the effects on expression of the TATA and the Inr in three different backgrounds separately. Our results show that while for some backgrounds (RTRAF and RPLP0) expression increases when adding Inr to TATA-containing oligos (P < 0.01 and P < 0.05) (Fig. 4C,D), for others (HIV) adding an Inr does not increase expression beyond the effect of the TATA (P > 0.1) (Fig. 4E). Moreover, promoters for which adding Inr to the TATA leads to an increase in expression also present greater sensitivity to the distance between the two elements in general, with maximal expression achieved when placed in the consensus reported position (−31) (Fig. 4F,G; Supplemental Table S9). In contrast, the HIV promoter, for which adding Inr to the TATA had no significant effect, was more robust to changes in the TATA location, with similar expression levels for the majority of the positions tested (Fig. 4H).
In all three backgrounds, expression is higher when the TATA is placed around positions −10, −20, and −30 relative to the TSS than at positions −15 and −25 (Fig. 4F–H). This ∼10-bp periodicity, which matches the DNA double-helix geometry, implies that the stereospecific alignment between the TATA and the TSS is important for expression, as was previously described for transcription factors (Takahashi et al. 1986; Yu et al. 1997; Sharon et al. 2012; Weingarten-Gabbay and Segal 2014a). Periodicity was not observed in the CMV background (Supplemental Fig. S11), suggesting that the sequence in which the elements are embedded affects alignment-dependent interactions.
Together, our results show that the TATA and the Inr elements can act additively to enhance transcription at preferable distances that facilitate stereospecific alignment between the two elements.
Comprehensive activity screen for 133 TF binding sites and nucleosome disfavoring sequences
In addition to the core promoter elements, the recruitment of the PIC is regulated by specific TFs that bind the proximal promoter region. Computational and high-throughput experimental approaches had characterized binding specificity (Jolma et al. 2013) and mapped the positions of TF binding sites in the human genome (Xie et al. 2005; Gerstein et al. 2012; Wang et al. 2012). However, since the expression levels of TFs, their localization, and post-translational modification vary between cell types, we cannot determine which TF binding sites will affect expression and to what extent.
To directly survey the activity levels of TFs, we designed promoters in which we planted four copies of each of 133 binding sites for 70 different TFs in two different backgrounds (Fig. 5A; Supplemental Table S10). To test the effect of directionality, we placed the sites in either the forward or reverse orientation. We found positive activity for 63% and 58% binding sites in the ACTB and the CMV backgrounds, respectively, spanning a dynamic range of approximately 30-fold in expression (Fig. 5B). As expected, by comparing the activity levels of expressed and unexpressed TFs in K562 cells according to RNA-seq measurements from ENCODE (The ENCODE Project Consortium 2012), we find significant lower activity for unexpressed TFs (P < 10−12) (Fig. 5C). Expression levels in both orientations are highly correlated (R = 0.81, P < 10−20) (Fig. 5D), suggesting that TF-driven expression is not sensitive to the binding site directionality. Similarly, we find good agreement between expression measurements in the two tested backgrounds (R = 0.72, P < 10−20, Fig. 5E).
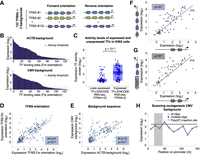
TF activity screen for 133 binding sites and the effect of nucleosome disfavoring sequence on expression. (A) Illustration of designed oligos for TF activity screen. One hundred thirty-three binding sites for 70 TFs were placed in four copies in either the forward or the reverse orientation in two backgrounds. (B) Expression measurements of oligos containing forward TF binding sites in two different backgrounds. Each bar represents a single binding site. Activity threshold determined by the empty vector is denoted. (C) TF activity measurements of expressed and unexpressed TFs as determined by ENCODE RNA-seq in K562 cells. Low activity is obtained for unexpressed TFs (P < 10−12, Wilcoxon rank-sum test). (D) Comparison between expression measurements of binding sites in two orientations. Each dot represents a pair of sequences for the same binding site placed in the forward or the reverse orientation (R = 0.81, P < 10−20, Pearson's correlation). (E) Comparison between expression measurements of binding sites in different backgrounds. Each dot represents a pair of sequences for the same binding site placed in the ACTB or the CMV backgrounds (R = 0.72, P < 10−20, Pearson's correlation). (F) Testing the effect on expression of adding two TF binding sites. Each dot represents a pair of designed promoters with either two or four sites for one of the 70 TFs tested in the CMV background. An increase in expression is observed for most TFs (P < 10−3, Wilcoxon signed-rank test). (G) Testing the effect on expression of nucleosome disfavoring sequence. A 25-mer poly(dA:dT) tract was added upstream to two binding sites for 70 TFs. An increase in expression is observed for most TFs (P < 10−6, Wilcoxon signed-rank test). (H) Systematic scanning mutagenesis to identify cis-regulatory elements in the CMV promoter. Eleven mutated oligos were designed; each contains a 14-nt window in which all nucleotides were mutated. Each dot represents expression of one mutated oligo. No elevation in expression is observed when mutating the sequences in which the poly(dA:dT) was inserted.
Previous studies from our laboratory demonstrated that transcription in yeast can be elevated either by increasing the number of TF binding sites or by adding poly(dA:dT) tracts that act as nucleosomes repelling sequences both in vivo and in vitro (Segal et al. 2006; Kaplan et al. 2009; Segal and Widom 2009; Raveh-Sadka et al. 2012; Sharon et al. 2012). To investigate these effects in human for a large number of factors, we designed promoters with two binding sites for 70 TFs in two backgrounds. We then placed either two additional binding sites or poly(dA:dT) tracts 25 bp in length upstream to the two existing sites. As expected, we find an increase in expression when adding two TF binding sites to the CMV background (P < 10−3) (Fig. 5F). Poly(dA:dT) tracts led to an increase in expression for most of the TFs tested, similar to what we reported for yeast promoters (P < 10−6) (Fig. 5G; Raveh-Sadka et al. 2012; Levo et al. 2017). To ensure that the obtained increase in expression is not a result of the destruction of a repressive sequence in the promoter background, we performed systematic mutagenesis to the CMV promoter, each time mutating a 14-nt region of its sequence. We found no increase in expression when introducing random mutations to the region in which we inserted the poly(dA:dT) tract (Fig. 5H). When we tested the ACTB promoter, we did not find a general increase in response to poly(dA:dT) tracts (Supplemental Fig. S12). However, the same background was also not affected by additional TF binding sites (Supplemental Fig. S12), suggesting that for some TFs the expression driven by two sites in the ACTB background is nearly saturated so that the contribution of additional elements cannot be accurately evaluated.
Together, our results, constituting the largest profiling of TF activity in human cells to date, demonstrate bidirectional activity and show that similar to what was shown in yeast, poly(dA:dT) tracts can increase expression in similar levels to TF binding sites at least in some promoters.
The effect of binding site numbers on expression is TF-specific
Proximal promoters and distal enhancers are enriched for multiple sites for the same factor, also known as homotypic clusters of TF binding sites (HCTs). Their conservation in vertebrates and invertebrates suggests that this is a general organization principle of cis-regulatory sequences (Gotea et al. 2010). Studies that examined the number of HCTs for different TF binding sites in the human genome found a wide range of behaviors, with some factors (e.g., SP1) forming a large number of HCTs, while others (e.g., cAMP-response element binding protein [CREB]) are rarely found in homotypic clusters (Gotea et al. 2010). This observation suggests that the effect on expression of multiple sites for the same factor depends on the identity of the TF, resulting in different relationships between binding site number and expression (Fig. 6A).
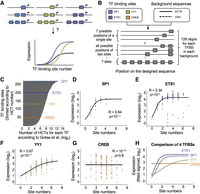
Systematic interrogation of the effect of homotypic TF binding site numbers on expression. (A) Illustration of different expression functions when adding homotypic binding sites for different TFs. (B) The design of 1024 synthetic oligos to systematically investigate the effect of site numbers on expression. Four different TF binding sites were planted in all possible combinations of one to seven sites in seven predefined positions within two different background sequences. (C) Shown is the number of homotypic clusters for TF binding sites (HCTs) of different TFs in the human genomes. Data were taken from Gotea et al. (2010). Each gray bar represents a single TF. Denoted are the four TFs chosen for the design of the synthetic oligos representing different numbers of HCTs. (D–G) Expression measurements of oligos with increasing number of sites for SP1 (D), ETS1 (E), YY1 (F), and CREB (G) in the ACTB background. Each dot represents a single oligo in the library. A logistic function was fitted (Methods), and the correlations between the expression measurements and the fitted values are shown for each TF. Missing data points in panel D are oligos with NaN value (less than 100 reads; see Methods). (H) A summary plot of the four expression curves that were computed in D through G for direct comparison between TFs.
To systematically interrogate the effect of homotypic site number on expression, we designed oligos in which we separately planted the sequences of four different TF binding sites in one to seven copies. To control for the effects of the binding site distance from the TSS, the distance between adjacent sites, and the immediate flanking sequence, we planted each TF binding site in all possible combinations of one to seven sites at seven predefined positions. We tested two backgrounds, resulting in a total of 1024 oligos (Fig. 6B; Supplemental Table S11). We selected four factors with different numbers of endogenous homotypic clusters as determined by a computational study that used known TF binding motifs and a hidden Markov model–based approach to detect HCTs in the human genome (Fig. 6C; Gotea et al. 2010). To evaluate the relationship between binding site number and expression, we fitted a logistic function to the expression measurements (see Methods) (Fig. 6D–G). By comparing the four TFs in the ACTB background, we find a striking agreement between the number of homotypic sites in the human genome and the obtained expression curves (Fig. 6C,H). Specifically, SP1 and ETS1, which have the highest number of homotypic sites of the four factors tested (3522 and 448, respectively), present the steepest increase (slopes of 1.67 and 1.91, respectively) and achieve the highest maximal expression levels (4.06 and 4.29, respectively; P < 10−11 and P < 10−5) (Fig. 6D,E,H). YY1, which has an intermediate number of homotypic sites (202), presents a moderate increase (slope = 0.62) and intermediate maximal expression levels (3.53; P < 10−17) (Fig. 6F,H). Finally, increasing the number of sites for CREB, which has the lowest number of homotypic sites (66), had no significant effect on expression (R = 0, P > 0.8) (Fig. 6G,H). By testing the expression curves in the CMV background, we find a similar trend, with three of the four factors preserving the same rank as in the ACTB background (Supplemental Fig. S13). Here too, we found that adding CREB sites does not increase expression (R = 0.17, P > 0.05).
Taken together, our findings demonstrate that the effect of homotypic sites on expression is factor-specific and that TFs that are naturally more prevalent in homotypic clusters in the genome also display higher dependency between binding site numbers and expression.
Discussion
Here we established a high-throughput experimental system to investigate thousands of designed promoters in a controlled genomic setting. By examining a large space of configurations and distances, we show how transcriptional regulatory elements combine to orchestrate a transcriptional output. We find that interactions between motifs, either core promoter elements or TF binding sites, are not universal and can vary between different backgrounds or factors. In turn, these differences can be translated into organizational principles of regulatory regions in the human genome.
A growing number of studies employ MPRAs to decipher gene expression regulation at the levels of transcription, translation, and mRNA stability (Zhao et al. 2014; Shalem et al. 2015; Ernst et al. 2016; Tewhey et al. 2016; Ulirsch et al. 2016; Weingarten-Gabbay et al. 2016). These types of experiments emphasize the need for accurate methodologies aiming at investigating designed sequences from a native genomic context. In this study, we developed a method for measuring thousands of designed oligos from a fixed location within the human genome with high efficiency and accuracy. Our method can readily be adapted to assay different types of regulatory elements, providing a valuable tool to interrogate gene expression. Site-specific integration followed by flow cytometry sorting provides single-cell information, allowing for systematic investigation of cell-to-cell variability that cannot be inferred from current MPRA methods, in which each cell is transfected with multiple different constructs. Thus, our method enables multidimensional investigation of the effect of sequence on expression at the population and single-cell levels, allowing us to infer both mean expression and noise in a single experiment.
Our findings shed additional light on the divergent nature of human promoters. The discovery of bidirectional transcription by genome-wide measurements of nested transcripts led to ongoing discussion on the existence and mechanisms underlying bidirectional promoters (Andersson et al. 2015; Duttke et al. 2015). In a recent study, Core et al. (2014) investigated the landscape and architecture of TSSs across the human genome and found that divergent transcription in both promoters and enhancers is facilitated by two distinct core promoters separated by ∼110 bp (Weingarten-Gabbay and Segal 2014b). Functional measurements of 300 promoters from the human genome (Trinklein et al. 2004) and 108 random genomic fragments (van Arensbergen et al. 2017) identified divergent promoter activity. However, since in both studies the assayed sequences were relatively long (up to 1 and 2 kb in Trinklein et al. 2004 and van Arensbergen et al. 2017, respectively), one cannot tell whether the activity observed represents true divergent sequences or two adjacent unidirectional core promoters. To address this question directly, we designed oligos to specifically match the core promoter region by taking 103 bp upstream of and 50 bp downstream from hundreds of PIC binding sites. Our functional measurements uncover that core promoters mostly drive unidirectional transcription. Moreover, in the model proposed by Core et al. (2014), a centered TF directs the PIC to initiate transcription from the two core promoters. In line with this model, we find high agreement between the activity levels of 133 TF binding sites when placed in the forward and the reverse orientations. Together, our study provides direct functional measurements supporting a model by which divergent promoter activity is driven by two distinct unidirectional core promoters sharing bidirectional TF binding sites.
Our systematic investigation of all possible combinations of six core promoter elements in various backgrounds reveals that while the TATA and the Inr increase expression, the BRE upstream and downstream elements lead to reduction in core promoter activity. BREu and BREd have been found to elicit both positive and negative effects on basal and TF-induced transcription (Evans et al. 2001; Chen and Manley 2003; Deng and Roberts 2005; Juven-Gershon et al. 2008; Kadonaga 2012). Considering that both BREs can act to stabilize the assembly of the PIC through interactions with the TFIIB general transcription factor (GTF), their negative effect on expression may be counterintuitive. However, it was suggested that while GTF–core promoter interactions can enhance the formation of the PIC, they might also impede the transition from initiation to promoter escape (Deng and Roberts 2005). Thus, sequence elements that increase the affinity between the initiation complex and the core promoter can have a negative impact on the transcriptional outcome. Although we find a positive relationship between GC content and promoter activity, our synthetic design, which is limited to sequences ∼200-nt long, did not facilitate the in-depth investigation of CpG islands, which are typically longer. These abundant genomic stretches of CG dinucleotides are associated with broad (dispersed) core promoters in which initiation can occur in multiple TSSs (Sandelin et al. 2007). Moreover, many core promoters lack any of the known motifs, and it is likely that additional elements will be discovered in the future. Thus, advances in technology that will allow the design of longer sequences as well as increasing the set of known elements will provide an exciting opportunity for additional studies for systematic investigation of both sharp and broad promoters classes.
TF binding sites can appear in homotypic and heterotypic clusters in the genome. An intriguing question is which of these organizational principles results in higher expression. A recent study that assayed 12 liver-specific transcription factors in homotypic and heterotypic clusters found that heterotypic elements are in general stronger than homotypic ones (Smith et al. 2013). However, since TFs differ in their DNA binding, trans-activation, and oligomerization domains, they may not adhere to one universal rule. Indeed, examining the human genome reveals that the tendency to appear in homotypic clusters is not uniform across TF binding sites (Gotea et al. 2010). Our systematic measurements of multiple homotypic sites for four distinct TFs uncover differences between their expression curves. Thus, TFs may employ different strategies to enhance transcription, and while some can “benefit” from homotypic sites, for others combining with a heterotypic site may result in higher expression. In addition, we find a striking agreement between the TF-specific expression curves resulting from multiple homotypic sites and the corresponding representation of a TF in homotypic clusters across the human genome. This finding suggests that intrinsic differences between TFs may be encoded in the genome and that we can use this information to increase our understanding of the various activation patterns of TFs. In order to determine the correlation between TF cooperativity and representation in homotypic clusters in the genome, the relationship between the number of sites and expression should be evaluated experimentally for additional factors beyond the four TFs tested here.
Genomic analyses of tumors using next-generation sequencing (NGS) have led to the identification of thousands of mutations, many of which reside within noncoding sequences. However, the effect of DNA sequence changes in regulatory regions remains elusive. Recent studies of breast cancer and melanoma have shown that the acquisition of mutations in the promoter sequences of four genes alter their expression by affecting TF binding sites and protein recruitment to the promoter regions (Horn et al. 2013; Rheinbay et al. 2017). Thus, deciphering the mapping between the promoters’ architecture and gene expression is key for understanding the transcriptional events underlying the development of cancer and additional genetic diseases. Our comprehensive characterization of human promoters, including the directionality of core promoters and TF binding sites, the portrayal of core promoter elements that positively and negatively regulate transcription, and the effect of element combinations and distances, adds new insights into rules of transcriptional regulation. In turn, these insights can facilitate the interpretation of the hundreds of DNA sequence changes associated with multiple diseases.
Methods
For additional methods, please see Supplemental Material.
Cell culture
K562 cells (CCL-243, ATCC) were cultured in tissue culture flasks (Nunc) in Iscove's medium (Biological Industries [BI]) supplemented with 10% fetal bovine serum (BI) and 1% penicillin and streptomycin (BI). H1299 human lung carcinoma cells with ecotropic receptor were cultured in RPMI 1640 medium (Gibco), supplemented with 10% fetal bovine serum (BI) and 1% penicillin and streptomycin (BI). Phoenix virus packaging cells were cultured in DMEM medium, supplemented with 2 mM l-glutamine, 10% fetal bovine serum (BI), and 1% penicillin and streptomycin (BI). Cells were kept at 37°C in a humidified atmosphere containing 5% CO2 and were frozen in complete media with 5%–7% DMSO (Sigma-Aldrich).
Plasmids
pZDonor AAVS1 was purchased from Sigma-Aldrich, as a part of the CompoZr Targeted Integration Kit–AAVS1, as were pZFN1 and pZFN2. pZDonor HindIII was a kind gift from Fyodor Urnov (Sangamo BioSciences). pPRIGp mChHA retroviral vector (Albagli-Curiel et al. 2007) was a kind gift from Patrick Martin (Université de Nice).
Construction of reporter master plasmids
A dual fluorophore master plasmid was constructed to allow cloning of the library as a proximal promoter of a single fluorophore while using another fluorophore for normalization. In order to minimize trans-activation between the eGFP-driving ACTB promoter, into which the library was cloned, and the EF1alpha promoter driving the mCherry control fluorophore, the master plasmid was designed to maximize the distance between the promoters. Thus, a sequence encoding two cassettes (each containing a promoter, fluorophore, and a terminator) placed back to back (with adjacent terminators) was synthesized by Biomatik (Canada) and cloned into the pZDonor plasmid. The eGFP cassette included a fragment of (−468,−122) of the human ACTB promoter (from genomic sequence NG_007992.1), followed by sites for AscI and RsrII restriction enzymes, a 5′ UTR and the chimeric intron from the pci-neo plasmid (Promega), eGFP gene, and the SV40 poly(A). A linker sequence of 25 bp was designed between the AscI and RsrII restriction enzyme sites (GGGTGTGTTGTTGGTGGGTTGGGTG) and was present instead of the library in the master plasmid control. The mCherry cassette included the EF1alpha promoter, mCherry, and the BGH poly(A).
Preparation of a dual-copy AAVS1 site K562 cell line
In order to reduce the number of possible AAVS1 integration sites from the three sites present in K562 cells, cells were nucleofected with ZFN mRNA and a pZDonor plasmid containing a HindIII site between the homology arms. Single cells were sorted by FACS and grown for up to a month to establish isogenic populations. Cells from the resulting populations were renucleofected with a fluorescent reporter to assess the number of possible genomic integrations. Cell lines exhibiting lower expression of the reporter were selected. In this manner, a cell line in which only two AAVS1 copies were present was retrieved and was used for all subsequent experiments.
Nucleofection of the plasmid library into K562 cells and site-specific integration into the AAVSI locus
The purified plasmid library was nucleofected into K562 cells and genomically integrated using the ZFN system for site-specific integration, with the CompoZr Targeted Integration Kit–AAVS1 Kit (Sigma-Aldrich). To ensure adequate library representation, 15 nucleofections with the purified plasmid library were carried out, each to 4 million cells. This number of cells was calculated to result in a thousand transfected cells per each sequence variant and at least 40 single integration events in average per variant. A master plasmid with no insert was also genomically integrated in the same manner. Nucleofections were performed using an Amaxa Cell Line Nucleofector Kit V (LONZA), program T-16. Cells were centrifuged and washed twice with 20 mL of Hank's Balanced Salt Solution (HBSS, Sigma-Aldrich), followed by resuspension in 100 µL room temperature solution V (Amaxa Cell Line Nucleofector Kit V). Next, the cells were mixed with 2.75 µg of donor plasmid and 0.6 µg of each in vitro transcribed ZFN mRNA just prior to nucleofection. A purified plasmid library was also nucleofected without the addition of ZFN to assess the background level of nonspecific integration and the time for plasmid evacuation. Nonnucleofected cells were taken after the washes in HBSS and seeded in 2 mL of precultured growth medium, serving as an additional control for FACS sorting.
Selecting for single integration by FACS sorting
Nucleofected K562 cells were grown for 15 d to ensure that nonintegrated plasmid DNA was eliminated, confirmed by the cells nucleofected without ZFNs. Cells were centrifuged, resuspended with PBS, and filtered using BD Falcon 12 × 75-mm tubes with cell-strainer cap (catalog no. 352235). Sorting was performed with BD FACSAria II special-order research product (SORP). To collect cells that integrated the reporter construct successfully in a single copy, we performed a preliminary calibration experiment to determine the mCherry gate representing single-integration population. We integrated a pZDonor plasmid containing an ACTB promoter upstream of the eGFP to K562 using ZFN-mediated site-specific integration and sorted this isogenic population according to different gates with increasing mCherry levels. Sorted cells were grown for additional 7 d and reanalyzed using flow cytometry (Supplemental Fig. S14). mCherry-expressing library cells corresponding to a single copy of the construct (∼4% of the population) were sorted using FACS (Supplemental Fig. S15A,B). The validity of this gate was verified by growing sorted cells for eight additional days and re-examining mCherry levels, verifying that no cells exhibited mCherry levels corresponding to a double integration (Supplemental Fig. S15C). A total of 7.5 million cells were collected in order to ensure adequate library representation. Master plasmid nucleofected cells were also sorted for single-copy integration.
Sorting single integration library into 16 expression bins
Following single integration sorting, the mCherry single integration population was grown for eight additional days before sorting into 16 bins according to the GFP/mCherry ratio. The bins were defined so they would span similar ranges of the ratio values, hence containing different percentages of the single integration population (from low expression to high: 2.5%, four bins of 8%, nine bins of 6.5%, 5.5%, 1%). We performed a single experiment of library sorting, and a total of 22 million cells were collected in order to ensure adequate library representation. The cells were grown further, and genomic DNA was purified from 5 million cells of each of the 16 bins using a DNeasy Blood and Tissue Kit (Qiagen) according to the manufacturer's protocol.
Aligning deep sequencing reads to the designed library
DNA was sequenced on Illumina NextSeq 500 sequencer. To determine the identity of each oligo after sequencing, we designed a unique 11-mer barcode upstream of the variable region. We obtained about 42 million reads for the entire library with a coverage of 100 or more reads for 91% of the designed oligos (14,375 of 15,753). As reference sequence for mapping, we constructed in silico an “artificial library chromosome” by concatenating all the sequences of the 15,753 designed oligos with spacers of 50 Ns. Single-end NextSeq reads in the length of 75 nt, respectively, were trimmed to 45 nt containing the common priming site and the unique oligo's barcode. Trimmed reads were aligned to the artificial library chromosome using NovoAlign aligner, and the number of reads for each designed oligo was counted in each sample.
Computing mean expression and noise for each designed oligo
Deep sequencing reads from each bin were mapped using the unique 11-bp barcode at the oligo 5′end. The distribution peak that contained the largest fraction of cells of each promoter was detected, and any cells outside of the peak were considered as technical noise. Here is a description of the procedure applied to each promoter expression distribution (an example of the process for a single promoter is shown in Supplemental Fig. S16): (1) Reads of each bin were normalized to match the fraction of the bin in the entire population; (2) expression bins that contained a fraction of cells smaller than a threshold were set to zero, and the threshold used in this work was 1/(#bins × 10) = 0.625%; (3) bin values were smoothed using MATLAB smooth() function with a span of three bins, and oligos with fewer than 100 reads were filtered and “NaN” values were assigned; and (4) we detected the peak that contains the largest fraction of reads and spans at least three adjacent bins. If obtained, additional smaller peaks were considered as technical noise as described before (Sharon et al. 2014). We used the chosen peak to compute both mean expression and standard deviation. Noise was quantified as the squared coefficient of variation (CV2), which is the variance divided by the square mean (Bar-Even et al. 2006).
Data access
Expression measurements of the designed oligonucleotides described in this study appear in the following Supplemental Tables: Supplemental Tables S2, S3, and S7 through S11. The raw sequencing data generated in this study have been submitted to the Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE118242.
Acknowledgments
We thank Eran Kotler, Martin Mikl, and Schraga Schwartz for critical comments on the manuscript. We thank Michal Levo, Leeat Keren, Yair Field, Hayden Metsky, and Steven Reilly for fruitful discussions. S.W.-G. is the recipient of the Clore PhD fellowship. E.S. is supported by the Crown Human Genome Center; the Else Kroener Fresenius Foundation; Donald L. Schwarz (Sherman Oaks, CA); Jack N. Halpern (New York, NY); Leesa Steinberg, Canada; and grants funded by the European Research Council (ERC 786344), the MRG-Grammar FET project (EU project 664918), and the Israel Science Foundation (ISF 161/16).
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.236075.118.
- Received February 14, 2018.
- Accepted December 5, 2018.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








