
Absolute nucleosome occupancy map for the Saccharomyces cerevisiae genome
- Elisa Oberbeckmann1,8,
- Michael Wolff2,8,
- Nils Krietenstein1,3,
- Mark Heron4,5,
- Jessica L. Ellins6,
- Andrea Schmid1,
- Stefan Krebs7,
- Helmut Blum7,
- Ulrich Gerland2 and
- Philipp Korber1
- 1Molecular Biology Division, Biomedical Center, Faculty of Medicine, Ludwig-Maximilians-Universität München, 82152 Planegg-Martinsried, Germany;
- 2Physik Department, Technische Universität München, 85748 Garching, Germany;
- 3Department of Biochemistry and Molecular Pharmacology, University of Massachusetts Medical School, Worcester, Massachusetts 01605, USA;
- 4Quantitative and Computational Biology, Max Planck Institute for Biophysical Chemistry, 37077 Göttingen, Germany;
- 5Gene Center, Faculty of Chemistry and Pharmacy, Ludwig-Maximilians-Universität München, 81377 Munich, Germany;
- 6Department of Biochemistry, University of Oxford, Oxford, OX1 3QU, United Kingdom;
- 7Laboratory of Functional Genome Analysis (LAFUGA), Gene Center, Faculty of Chemistry and Pharmacy, Ludwig-Maximilians-Universität München, 81377 Munich, Germany
-
↵8 These authors contributed equally to the work.
Abstract
Mapping of nucleosomes, the basic DNA packaging unit in eukaryotes, is fundamental for understanding genome regulation because nucleosomes modulate DNA access by their positioning along the genome. A cell-population nucleosome map requires two observables: nucleosome positions along the DNA (“Where?”) and nucleosome occupancies across the population (“In how many cells?”). All available genome-wide nucleosome mapping techniques are yield methods because they score either nucleosomal (e.g., MNase-seq, chemical cleavage-seq) or nonnucleosomal (e.g., ATAC-seq) DNA but lose track of the total DNA population for each genomic region. Therefore, they only provide nucleosome positions and maybe compare relative occupancies between positions, but cannot measure absolute nucleosome occupancy, which is the fraction of all DNA molecules occupied at a given position and time by a nucleosome. Here, we established two orthogonal and thereby cross-validating approaches to measure absolute nucleosome occupancy across the Saccharomyces cerevisiae genome via restriction enzymes and DNA methyltransferases. The resulting high-resolution (9-bp) map shows uniform absolute occupancies. Most nucleosome positions are occupied in most cells: 97% of all nucleosomes called by chemical cleavage-seq have a mean absolute occupancy of 90 ± 6% (±SD). Depending on nucleosome position calling procedures, there are 57,000 to 60,000 nucleosomes per yeast cell. The few low absolute occupancy nucleosomes do not correlate with highly transcribed gene bodies, but correlate with increased presence of the nucleosome-evicting chromatin structure remodeling (RSC) complex, and are enriched upstream of highly transcribed or regulated genes. Our work provides a quantitative method and reference frame in absolute terms for future chromatin studies.
It makes a fundamental difference whether a measurement yields values on a relative or absolute scale. In molecular biology, it is usually much easier to obtain relative values by comparing samples to references than to measure in absolute terms. For example, protein concentration is readily compared between cells by western blotting. However, the absolute number of protein molecules per cell is more difficult to obtain. Molecular biology generally suffers from the scarcity of quantitative data on absolute scales, which hinders a deeper understanding, modeling, and theoretical description of mechanisms and systems features.
Here, we amend such lack of absolute values for the basic packaging unit of eukaryotic genomes: the nucleosome. It is defined as a core of 147 bp of DNA spooled in 1.7 turns around a histone protein octamer (Luger et al. 1997) plus variable lengths of flanking linker DNA. For brevity and according to common usage, “nucleosome” refers to the nucleosome core in the following. The close interactions between DNA and histones in nucleosomes inhibit DNA access for many factors and thereby constitute an important level of regulation for all DNA-dependent processes, like transcription or DNA repair (Venkatesh and Workman 2015; Seeber et al. 2018). Therefore, the mapping of nucleosomes, the dynamics of their positioning and composition, as well as their roles in genome regulation are of paramount interest.
Several techniques map nucleosome positions along a genome (Jiang and Pugh 2009; Meyer and Liu 2014; Lieleg et al. 2015b). The most common tool is micrococcal nuclease (MNase), which digests nonnucleosomal DNA faster than nucleosomal DNA and yields mononucleosomes at a properly limited digestion degree (Rill and Van Holde 1973; Noll 1974). High-throughput sequencing of mononucleosomal DNA maps histone octamer DNA footprints (MNase-seq) (Albert et al. 2007). Because other DNA-bound factors may also inhibit MNase digestion and yield mononucleosome-sized DNA fragments (Chereji et al. 2017), additional criteria can ensure nucleosome specificity, for example, mononucleosome selection by anti-histone immunoprecipitation (MNase-anti-histone-chromatin immunoprecipitation-seq [MNnase-anti-histone-ChIP-seq]) (Albert et al. 2007; Wal and Pugh 2012). Alternatively, chemical cleavage (Flaus et al. 1996) is an MNase-independent and histone-specific method to map nucleosomes, especially its most recent version (Chereji et al. 2018). Cysteine residues are introduced in histones and coupled to copper-chelating phenanthroline so that incubation with hydrogen peroxide generates hydroxyl radicals that cleave defined DNA sites in the nucleosome. High-throughput sequencing of the resulting DNA fragments yields very precise genome-wide nucleosome maps (Brogaard et al. 2012; Moyle-Heyrman et al. 2013; Chereji et al. 2018). The flip side to measuring nucleosome positions is mapping linker DNA between nucleosome cores and wider nucleosome-free regions (NFRs), for example, by using a hyperactive transposase that inserts sequencing adapters into nucleosome-free DNA (assay for transposase-accessible chromatin [ATAC-seq]) (Buenrostro et al. 2013).
Although such techniques measure nucleosome positions or nonnucleosomal regions and may provide their relative occupancies, they all cannot measure absolute nucleosome occupancy. This quantity describes the fraction of a molecule/cell population where a certain base pair either is part of any nucleosome (Kaplan et al. 2010) or at a nucleosome center (Zhang et al. 2009; Lieleg et al. 2015b). The nucleosome center is also called dyad because of the pseudo-twofold nucleosome symmetry. In the following, we use the first definition because our approach scores the whole nucleosome footprint and not dyads. This fraction can vary between 0% (no molecule has any nucleosome covering this position) and 100% (all molecules have a nucleosome here). The aforementioned methods cannot measure this fraction because they are yield methods, meaning they only score either the nucleosomal or the nonnucleosomal amount at a time but lose track of the total population. A genomic position may yield more or less, for example, MNase-seq or ATAC-seq signal than another position, which is often interpreted as differential nucleosome occupancy (Jiang and Pugh 2009). However, this only refers to occupancy differences in relative terms. It remains unknown to which fraction of the total, that is, to which absolute occupancy such signals correspond.
Additionally, MNase-based methods suffer from further problems. First, MNase has some sequence bias (Dingwall et al. 1981; Hörz and Altenburger 1981) because it cleaves AT-rich DNA more readily, even within nucleosomes (Cockell et al. 1983; Caserta et al. 2009; Chereji et al. 2019a). Second, MNase digestion has to be limited, otherwise it also cleaves within nucleosomes. Thus, MNase-based methods do not operate at saturation. Standardization or normalization of digestion degrees is very challenging (Cole et al. 2011; Rizzo et al. 2012), but would be necessary because peak heights depend on the digestion degree (Weiner et al. 2010; DeGennaro et al. 2013; Chereji et al. 2019a). MNase must cut on both sides of the nucleosome to cut out a mononucleosome with a probability that depends on the digestion degree as well as on the sequence and length of the flanking DNA. Accordingly, long and AT-rich NFRs, which are typical for budding yeast promoters (Yuan et al. 2005), are frequently cut already at low digestion degrees leading to highest MNase-seq peaks for their flanking −1 and +1 nucleosomes, but are also the entry way for more efficient nucleosome digestion at higher digestion degrees, then leading to relatively lower flanking nucleosome peaks (Zhang et al. 2009; Stein et al. 2010; Weiner et al. 2010; Givens et al. 2012; DeGennaro et al. 2013; Flores et al. 2014). Only the combination of spike-in normalization controls and digestion degree titration allows quantitative relative, albeit still not absolute occupancy measurements by MNase (Chereji et al. 2019a).
Collectively, and as noted before (Rizzo et al. 2011; Ozonov and van Nimwegen 2013; Quintales et al. 2015), we are still blind to absolute nucleosome occupancies on the genome-scale, despite genome-wide mapping of nucleosome positions with increasing precision over the last 14 years (Yuan et al. 2005; Lieleg et al. 2015b).
Nonetheless, there are absolute nucleosome occupancy measurements at single loci based on the differential accessibility of nucleosomal versus nonnucleosomal DNA for restriction enzymes (REs) and DNA methyltransferases (DNMTs). Both enzymes will only cleave/methylate nonnucleosomal DNA, but not remove nucleosomal DNA. If both DNA types are monitored, the fraction of not cleaved/methylated molecules directly corresponds to the absolute occupancy. RE accessibility measurements by Southern blotting were pioneered, for example, at the budding yeast PHO5 and PHO8 promoters (Almer et al. 1986; Barbaric et al. 1992). DNA methylation footprinting was established using prokaryotic DNMTs that methylate either CpG or GpC sites (Jessen et al. 2004; Kilgore et al. 2007; Small et al. 2014).
To measure absolute occupancy, it is crucial that the RE- or DNMT-catalyzed reactions reach saturation, which requires that nucleosome dynamics are frozen. This is mostly true for ex vivo–prepared or in vitro–assembled chromatin under physiological buffer and temperature conditions (Korolev et al. 2007; Zhang et al. 2011), but not in vivo where ATP-dependent nucleosome remodeling enzymes (remodelers) reposition, disassemble, or restructure nucleosomes and generate transient DNA accessibility also within nucleosomes (Bartholomew 2014; Zhou et al. 2016).
Genome-wide methods using REs or DNMTs have not yet provided reliable absolute occupancies. Comparisons of RE accessibilities—NA-seq (Gargiulo et al. 2009) and RED-seq (Chen et al. 2014)—scored only cut fragments, for example, by ligating biotinylated adapters after RE digestion and sequencing only the streptavidin-immunoprecipitated DNA (RED-seq). DNMT approaches, for example, NOMe-seq (Kelly et al. 2012), were insufficient mainly for two reasons. First, genome sequencing coverage is often much too low, especially for metazoans, so that only coarsely grained absolute occupancy values could be discerned. Second, DNA methylation extent was either not saturating or chosen to match MNase-seq results. DNA methylation titration for establishing the NOMe-seq protocol (Kelly et al. 2012) led to a discrepancy at the human MLH1 locus where the GpC-specific DNMT M.CviPI methylated its cognate site even though MNase-seq gave a “nucleosome” signal here. The authors interpreted that the DNMT methylated within a nucleosome and accordingly chose less extensive methylation conditions, which were subsequently used by many in the field (Krebs et al. 2017; Levo et al. 2017).
Considering the aforementioned limitations of existing and especially MNase-based methods, we argue that a reliable and MNase-independent assessment of genome-wide absolute nucleosome occupancy is needed, and we hereby provide it for the Saccharomyces cerevisiae genome.
Results
ORE-seq: genome-wide absolute occupancy measurement by REs
As a first approach to measure genome-wide absolute occupancy at low resolution, we brought classical restriction enzyme accessibility assays (Almer et al. 1986; Gregory et al. 1999) to the genome level (Supplemental Methods) and called this method occupancy measurement via restriction enzymes and high-throughput sequencing (ORE-seq). Chromatin was prepared from logarithmically growing (Supplemental Fig. S1A) wild-type (WT) S. cerevisiae and digested with REs at different RE concentrations and for different incubation times to establish saturation (Fig. 1A, left). DNA purified after RE digest was sonicated and Illumina-sequenced in paired-end mode. Each fragment end was scored as generated by RE cut versus by sonication, primarily depending on whether its end is close to an RE site or not. Further, we corrected for fortuitous sonication breaks at RE sites, end resection at RE sites attributed to endogenous exonucleases, and deviations from calibration curves generated by analyzing mixtures of cut and uncut genomic DNA at defined ratios (Supplemental Methods).
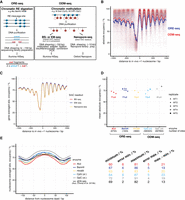
Genome-wide absolute occupancy measurement by restriction enzymes and DNA methyltransferases. (A) Method overview; for details, see text. Lollipops stand for DNA methylation sites: (open) unmethylated; (red fill “M”) methylated; (f.) fragments. (B) Composite plot of absolute occupancy ORE-seq and ODM-seq data averaged over all included samples and aligned at in vivo +1 nucleosome positions. Each dot represents the value of one genomic site, and the lines show the 10-bp bin mean occupancy of aligned sites. (C) As in B, but for WT5 replicate and the different 5mC readouts stated in A. (D) Absolute occupancy averaged over all sites (mean absolute occupancy) obtained by ORE-seq (Supplemental Table S3) or ODM-seq (Supplemental Table S4) for the indicated enzymes and biological replicates (WT1 to WT5) at saturation conditions. The number of sites implemented for each enzyme is indicated. (xl.) in vivo formaldehyde cross-linked. (E) Absolute occupancy values averaged in 10-bp bins around nucleosome dyads called from chemical cleavage-seq data (Chereji et al. 2018) and averaged over all replicates for the indicated enzymes. qDA-seq data are taken from Chereji et al. (2019b). On the right, absolute occupancy values and errors (mean over sites in the bin of the standard deviation among samples) are shown for the maxima and minima of each plot as well as the difference between maximum and minimum values for each enzyme.
The application of ORE-seq to biological chromatin replicates using different REs showed good reproducibility and clear saturation of RE digestions; that is, mean absolute occupancy values for each RE were within five percentage points for samples varying by different RE concentrations or incubation times (Supplemental Fig. S1B). As technical control, we exploited that each 6-bp HindIII site contains a 4-bp AluI site. Absolute occupancies measured at HindIII sites by high concentrations of either AluI or HindIII agreed well within 7% mean absolute difference for each site and 2% difference in mean sample occupancy averaged over all sites (mean absolute occupancy) (Supplemental Fig. S1C).
We selected absolute occupancy data for each RE site according to quality criteria, like saturation of digestion or sequencing coverage (Supplemental Methods), and combined them into a genome-wide ORE-seq map with low average resolution of approximately 870 bp (Fig. 1B).
ODM-seq: genome-wide absolute occupancy measurement by DNMTs
For increased resolution and as an orthogonal method, we established (Supplemental Methods) genome-wide absolute occupancy measurement in chromatin by differential cytosine methylation at position C-5 in CpG or GpC motifs and called it occupancy measurement via DNA methylation and high-throughput sequencing (ODM-seq) (Fig. 1A, right). Comparing DNA methylation of in vitro reconstituted nucleosomes using fly histones with ex vivo prepared yeast chromatin, we found that yeast nucleosomes were inherently unstable during prolonged incubation required for saturation of methylation unless magnesium was added or the chromatin was formaldehyde cross-linked (Supplemental Fig. S2A–E; Supplemental Methods). To avoid endogenous nucleases active in the presence of magnesium, we used cross-linked chromatin for ODM-seq (Supplemental Fig. S3; Supplemental Methods).
Methylated cytosines (5mC) were mostly detected by Illumina sequencing of sonication fragments after treatment with bisulfite (BS-seq), which converts only unmethylated cytosines to uracil and thereby changes the DNA sequence in a tractable way. Enzymatic (EM-seq, New England Biolabs) instead of bisulfite conversion in short or direct readout of 5mC in long DNA fragments by Oxford Nanopore sequencing resulted in equivalent occupancy maps (Fig. 1C). This controls against any systematic errors in our bisulfite sequencing and bioinformatics pipeline. Further, Oxford Nanopore sequencing excels in sequencing very long fragments (>10 kb) but did not yield completely unmethylated long DNA fragments. This argues against a contribution of DNA from unlysed cells, which would not have been accessible to REs or DNMTs and may have systematically distorted absolute occupancy measurements. Therefore, we conclude that known biases of bisulfite sequencing (Darst et al. 2010) did not affect our results.
Comparison of different methods and conditions with regard to absolute occupancy measurements
Now we had absolute occupancy measurements across the yeast genome for two independent methods (ORE-seq and ODM-seq) involving two different conditions (non-cross-linked chromatin in RE buffer with Mg2+ vs. cross-linked chromatin in DNMT buffer without Mg2+), five independent enzymes (AluI, BamHI, HindIII, M.SssI [CpG DNMT], M.CviPI [GpC DNMT]), five independent biological replicates (WT1 to WT5), two independent technical replicates for BamHI, and a comparison between purified in vitro chromatin and complex ex vivo chromatin for BamHI and the two DNMTs (Supplemental Fig. S2A). All these independent and partially orthogonal measurements yielded mean absolute occupancy values in the range of 71%–81% (Fig. 1D). Because there was no genome-wide precedent for such values, it was important that this multitude of approaches converged in a similar range and cross-validated each other.
Nonetheless, ORE-seq tended to yield lower mean occupancy values than ODM-seq (Fig. 1B,D) and CpG methylation yielded lower mean occupancy values than GpC methylation (Fig. 1D; Supplemental Fig. S2A,B). These differences were only in some cases significant, for example, GpC DNMT versus AluI or HindIII, and unlikely due to different conditions, methods, or replicates as values for BamHI and CpG DNMT overlapped across these differences (Fig. 1D). We considered that different enzymes may probe the same chromatin in different ways. Mean absolute occupancy values across the genome are influenced by the different distribution of cognate sites for each enzyme in nucleosomes versus nonnucleosomal regions (Supplemental Fig. S4A). To correct for such different site distributions, we used nucleosome positions mapped by chemical cleavage (Chereji et al. 2018) and plotted absolute occupancy values for each enzyme as averaged over 20-bp bins around the dyads of all called nucleosomes (Fig. 1E). Here, we also included AluI data from an independent approach called qDA-seq (Chereji et al. 2019b) that is conceptually equivalent to our ORE-seq and became available during the revision phase of our manuscript. This plot showed how each enzyme differently measured occupancy in and around nucleosomes, including linker regions, and the DNA at the entry and exit sites of the histone octamer that is known to transiently unwind (Polach and Widom 1995). For most enzyme pairwise comparisons, the differences for maximal and minimal occupancies, that is, at nucleosome dyads and in linkers, respectively, were within the error. Only GpC DNMT and our AluI data were just significantly different; that is, error bars almost touched. CpG DNMT, our AluI and HindIII, showed a larger min-max difference than BamHI, qDA-AluI, and GpC DNMT, which may mean that the former enzymes can access linker regions and entry/exit DNA more efficiently. However, these differences are within the mean standard deviation and barely significant. The average AluI occupancy values from us versus from the Clark group (Chereji et al. 2019b) agreed within 1% at dyads and differed by 9% in linkers, with almost touching error bars for the latter. When the data was gene averaged and +1 nucleosome-aligned, the qDA-AluI data showed a clear trend toward higher occupancies in linkers and NFRs (Supplemental Fig. S4B). This may be a result of different technical procedures or different biological conditions. For example, the Clark group (Chereji et al. 2019b) used a different strain background (W303), different media (synthetic complete), and G1-arrested cells (α-factor). However, their qDA-seq AluI data set of exponentially growing cells, that may be biologically closer to our logarithmic phase cells, showed the same trend (Supplemental Fig. S4B). This linker/NFR difference remains to be explained. Nonetheless, the majority of the RE and DNMT data cross-validated each other. Therefore, we were confident that we obtained an accurate measure for absolute occupancy, including the lower linker/NFR occupancy compared to the qDA-seq data.
Genome-wide absolute occupancy map and comparison with other nucleosome maps
Because the restriction enzymes were only important for cross-validation but did not contribute significantly to resolution (<1% compared to all DNMT sites) (Fig. 2A; Supplemental Table S1), we used just the ODM-seq absolute occupancy map with a mean resolution of 9 bp and an average error of 6% (mean over sites of standard deviation between samples) (Supplemental Fig. S4C, center and right; Supplemental Table S1) for further analyses. The +1 nucleosome-aligned composite plot of the ODM-seq map (Fig. 1B) was reminiscent of corresponding plots using MNase-seq, MNase-anti-histone-ChIP-seq (Zhang et al. 2011; True et al. 2016), or chemical cleavage-seq data (Fig. 2B), but the other methods show much more pronounced fluctuations in nucleosome peak heights and do not agree, for example, whether the relative occupancies for the +1 nucleosomes are on average higher or lower than those of the +2 nucleosomes (Fig. 2A,B). Our absolute occupancy map now provides meaningful peak heights in absolute terms and thereby resolves such questions; for example, the absolute occupancy for the +1, +2, and +3 nucleosomes is on average almost the same. The peak height of nucleosomes further downstream differs slightly if the complete downstream region is plotted or only up to the first transcription termination site (TTS) (Supplemental Fig. S4D) as a promoter NFR follows downstream from a TTS in many cases (Chereji et al. 2017, 2018). Of all compared methods, chemical cleavage-seq was closest to our data in terms of relative peak heights.
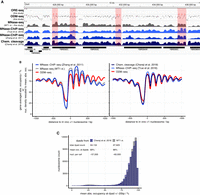
The ODM-seq absolute occupancy map. (A) Integrated Genome Viewer (IGV) browser shots comparing the indicated data sets (Supplemental Table S2) with our ORE-seq and ODM-seq absolute occupancy data. Regions in light red highlight pronounced differences in occupancy/signal between methods. (B) As in Figure 1C but for the indicated data sets. Because the external data do not provide absolute occupancy, we globally rescaled their signal to have the same genomic mean as the absolute occupancy map. Here and in following cases, nucleosome dyads of external data sets were extended to 147 bp. (C) Histogram of absolute occupancy at nucleosome positions called from the indicated data sets.
We measured generally high occupancy, with a median site occupancy of 84% (Supplemental Fig. S4C, left) but also a substantial fraction of sites with lower occupancies. We underscore that neither ORE-seq nor ODM-seq nor qDA-seq distinguish which kind of factor restricts DNA accessibility, that is, contributes to measured occupancy. To obtain not just an absolute occupancy but more specifically an absolute nucleosome occupancy map, our data must be combined with nucleosome-specific mapping data. We called nucleosome dyads either in our own matched MNase-seq data set for the cross-linked WT1 sample or used nucleosome dyad cluster medians (typical nucleosomes) called from chemical cleavage-seq (kindly provided by Razvan Chereji) (Chereji et al. 2018) and determined the absolute occupancy for each of the called dyads or dyad clusters, respectively, by averaging the occupancy of sites within ±20 bp of these calls. The corresponding histograms (Fig. 2C) for chemical cleavage-seq or MNase-seq show rather narrow distributions with means of 89% and 88%, respectively. Dyads with at least 70% absolute occupancy, which correspond to 97% (chemical cleavage) and 94% (MNase-seq) of all dyads that are mappable by ODM-seq have a mean absolute occupancy of 90% (chemical cleavage) and 91% (MNase-seq), both with standard deviation of 6%. The combination of nucleosome calls and absolute occupancy data allows calculating the absolute number of nucleosomes in a yeast cell as ≈57,000 or ≈60,000 nucleosomes per cell for chemical cleavage or MNase-seq calling, respectively. This difference is likely caused by method-specific limitations for scoring nucleosomes. MNase-seq is prone to score nonnucleosomal complexes in NFRs as “nucleosomes” (Chereji et al. 2017), which will lead to an overestimation of nucleosome number and a slight bump in the histogram around 25% occupancy (Fig. 2C). Conversely, high-resolution chemical mapping gives few such false positives but yields clusters that are not well-resolved, especially in gene bodies, that confound the calling algorithm. Therefore, some genic nucleosomes are not called in the latter data set and the number of nucleosomes is underestimated (R Chereji, pers. comm.). This encumbers the exact determination of the number of nucleosomes per cell. Nonetheless, this number likely is in the range of 58,000 ± 1000.
Detection of nonnucleosomal DNA-bound factors by ODM-seq
The more pronounced population of sites with low absolute occupancy around 25% in the histogram of absolute occupancy at CpG/GpC sites (Supplemental Fig. S4C, left) compared to the histogram of absolute occupancies at called nucleosome positions (Fig. 2C) stems mainly from sites in NFRs and linkers (Fig. 1B). NFRs are probably occupied by nonnucleosomal factors (Supplemental Fig. S5A). Indeed, our absolute occupancy map not only shows signals from nucleosomes, but also from DNA binding factors (Gutin et al. 2018) like the general regulatory factors (GRFs) Rap1, Abf1, Mcm1, and Cbf1, but only in few cases for Reb1 and the origin recognition complex subunit Orc1, which may be linked to the mappability of binding site motifs; for example, the Orc1 motif is very AT-rich and hardly mappable by DNMTs (Fig. 3A–D; Supplemental Fig. S5B). Such factor binding is hardly detected, as expected, by MNase-seq or by chemical cleavage (Fig. 3B, center and right). The GRF site-aligned composite plots of absolute occupancy (Fig. 3B, left) show the expected symmetrically aligned and regularly spaced nucleosomal arrays flanking Abf1 and Reb1 sites (Rossi et al. 2018), but much less regular arrays and lower occupancy immediately around Rap1 sites. This reflects that Rap1 sites often come in neighboring pairs, show a broader distribution relative to TSSs (Supplemental Fig. S5A), and occur in unusually wide promoter NFR regions, for example, of the highly expressed ribosomal protein (RP) genes (Knight et al. 2014; Reja et al. 2015).
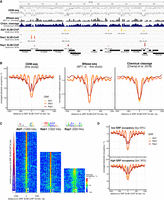
ODM-seq monitors not only absolute nucleosome but also absolute GRF occupancy. (A) IGV browser shot comparison of the indicated data sets (Supplemental Table S2). ODM-seq data are given both as individual (top) and as connected data points (second from top). (B) GRF site-aligned composite plots of absolute occupancy (left) or normalized signal (center and right) for the indicated GRFs and data sets. Signals are normalized to a mean of one. (C) GRF site-aligned heat maps of absolute occupancy sorted from top to bottom according to increasing absolute occupancy at GRF sites. The position weight matrix (Badis et al. 2008) and the number of binding sites detected by SLIM-ChIP for the indicated GRFs is given above the heat maps. White color denotes absence of signal (highlighted by white arrows). (D) As in B, left graph, but for genes of low and high GRF occupancy according to the SLIM-ChIP sorting in C.
Correlation of absolute occupancy with transcription rates
Because 97% of all nucleosomes called by chemical mapping have a mean absolute occupancy of 90 ± 6% (±SD) (Fig. 2C), the nucleosome occupancy landscape appeared rather flat and we did not expect strong correlations between absolute occupancies and biological features. Nonetheless, we asked which biological feature correlated with the few percent of nucleosomes that showed lower absolute occupancy.
Very high expression levels, like heat shock–induced genes, were reported to correlate with nucleosome loss over gene bodies (Zhao et al. 2005). We generated composite plots of absolute occupancy, chemical cleavage-seq, and MNase-anti-H3-ChIP-seq (Fig. 4A) as well as other mapping data (Zhang et al. 2011; Joo et al. 2017; Dronamraju et al. 2018; Chereji et al. 2019b) (Supplemental Fig. S6A) for gene quintiles of NET-seq data (nascent RNA bound to RNA polymerase) (Churchman and Weissman 2011), which measures transcription activity. Although MNase-anti-H3-ChIP-seq and ATAC-seq showed reduced signal for the most highly transcribed genes, this was much less the case for chemical cleavage-seq and hardly apparent for the ODM-seq and qDA-seq data. If genes were not binned but if their transcription rates measured by NET-seq or 4sU-seq (Xu et al. 2017) were individually correlated with the various occupancy mapping data (Fig. 4B; Supplemental Fig. S6B), an overall anti-correlation between transcription rates and occupancy was very poor for most and absent for methods that measure absolute occupancy and for chemical cleavage-seq.
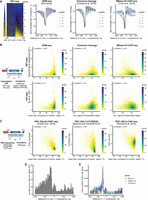
Correlation of absolute occupancy with biological features. (A, left) In vivo +1 nucleosome-aligned heat map of NET-seq data monitoring nascent RNA bound to RNA polymerase (Churchman and Weissman 2011) sorted from top to bottom by increasing signal over the gene body. (Right) As in Figure 2B but for the indicated data sets and genes subdivided according to quintiles of sorting in heat map on the left. (B) Correlation plots (color indicates number of occurrences) of transcription rate (NET-seq as in A or 4sU-seq [Xu et al. 2017]) against the absolute occupancy or coverage averaged over transcribed regions for the indicated data sets as in A. (C) As in B but correlation of absolute occupancy averaged over transcribed regions with RSC binding measured by the indicated methods. (D) +1 Nucleosome-aligned histogram (accumulated in 20-bp bins) of nucleosomes dyads (Chereji et al. 2018) with <70% absolute occupancy. (E) As in D but clustered by gene groups (Vinayachandran et al. 2018) as indicated. In brackets, mean number of low absolute occupancy nucleosomes per gene in 2-kb window around in vivo +1 nucleosome. Used data sets are listed in Supplemental Table S2.
Studies from the Pugh (Basehoar et al. 2004), Steinmetz (Xu et al. 2009), and Morillion (van Dijk et al. 2011) groups led to an instructive grouping of transcribed regions in yeast into RP genes, SAGA-, TFIID-dependent genes, cryptic unstable transcripts (CUTs), stable unannotated transcripts (SUTs), and Xrn1-dependent unstable transcripts (XUTs) (Vinayachandran et al. 2018). Our ODM-seq data did not show major differences over gene bodies between these groups (Supplemental Fig. S7A,B), in contrast to relative nucleosome occupancy measurements by MNase-ChIP-seq. Even if these occupancies were rescaled, the distribution of average occupancies across transcripts is too broad for these methods. The high expression levels of RP genes were confirmed by both 4sU-seq and NET-seq, but only 4sU-seq showed much higher transcription rates of SAGA- versus TFIID-dependent genes (Supplemental Fig. S7B).
In addition, we wondered if there was a correlation between the transcribed region length and absolute occupancy. Short units showed the whole range of absolute occupancies, but long units mostly had high absolute occupancy (Supplemental Fig. S6C).
Increased binding of RSC remodeling complex correlates with lower absolute occupancy
Because it was not RNA polymerase passage that reduced absolute occupancy in transcribed regions, we turned to the ATP-dependent chromatin remodeling complex RSC, which is the major nucleosome displacing activity in yeast (Cairns et al. 1996; Hartley and Madhani 2009). There was an inverse correlation between RSC signal detected over transcribed regions and their absolute occupancy (Fig. 4C). This was reproducible for different RSC data sets (Parnell et al. 2015; Kubik et al. 2018; Brahma and Henikoff 2019) with different degrees of correlation.
RSC mainly binds and depletes nucleosomes upstream of TSSs (Yen et al. 2012; Krietenstein et al. 2016). Accordingly, those nucleosome dyad clusters—as called by chemical cleavage, which is most reliable in these regions—that had lower than 70% absolute occupancy were mainly upstream of TSSs (Fig. 4D), where regulatory sites, like transcription factor binding sites, are enriched (Lee et al. 2007; Ozonov and van Nimwegen 2013) and mainly for RP and SAGA-dependent genes (Fig. 4E).
The enrichment of low occupancy nucleosomes upstream of TSSs explains in part that the absolute average occupancy upstream of and downstream from NFR minima is 70% and 79%, respectively (Fig. 1B). Nonetheless, this difference is also attributed to genes with wider than average NFRs (less steep upstream flank of the NFR trough in Fig. 1B), so that the alignment at the minimum in the +1 nucleosome-aligned composite plot used for calculating the aforementioned upstream and downstream average occupancies is not in the minimum for these genes. Note that the much lower site average (Fig. 1B, red line) upstream compared to downstream from the NFR is not explained by enrichment of low occupancy nucleosomes, but reflects that upstream nucleosomes are on average less regularly aligned to the +1 nucleosome position than downstream nucleosomes, so that the red line does not mainly average over nucleosome centers but also over linker regions. As shown by the individual data points, upstream nucleosomes also mostly have high absolute occupancy.
We conclude that absolute occupancy is mainly held constant for nucleosomes across the genome and particularly in transcribed regions, unless RSC depletes nucleosomes, which occurs mainly in regulatory regions upstream of TSSs.
Discussion
Here, we present the first genome-wide high-resolution map of absolute occupancy for a eukaryotic genome. Because there was no precedent, we established orthogonal methods, different analysis pipelines, and different experimental conditions that cross-validate each other. During revision of our manuscript, an independent low-resolution approach provided further validation for occupancy at nucleosome dyads (Chereji et al. 2019b). Absolute occupancy was measured as a mirror image of absolute DNA accessibility for REs and DNMTs and cannot distinguish what occupies the DNA. Therefore, we combined our measurements with data from more nucleosome-specific mapping techniques, like MNase-seq or chemical mapping-seq, to arrive at the first high-resolution absolute nucleosome occupancy map.
The main feature of this map is its uniform nucleosome occupancy at dyads. Of all nucleosomes called by chemical mapping, 97% have a mean absolute occupancy >70% with a mean of 90 ± 6% (±SD). This fits to single locus studies; for example, the absolute occupancy of the −2 nucleosome at the repressed yeast PHO5 promoter was estimated by RE accessibility as 90% (Almer et al. 1986). Nucleosomes are placed along the genome in an all-or-nothing manner without major occupancy differences between the −1, +1, +2, and +3 nucleosomes relative to the TSS. The few nucleosomes with lower absolute occupancy are enriched in regions upstream of TSSs, which fits to high histone turnover (Dion et al. 2007) and abundance of regulatory processes here. High absolute nucleosome occupancy seems to be conserved as qDA-seq in mouse hepatocytes (Chereji et al. 2019b), and quantitative mass spectrometry measurements of histones in Drosophila cells (Bonnet et al. 2019) reflected full coverage of the genome with nucleosomes at the species-specific nucleosome repeat length.
The uniformly high absolute nucleosome occupancy suggests that nucleosome depositioning operates as a highly effective default system. This is poorly defined but likely involves histone chaperones, specific histone modifications, like H3K56ac, and remodelers (Almouzni and Cedar 2016). In flies, nucleosomes are rapidly deposited in the wake of DNA replication (Ramachandran and Henikoff 2016) even in regions that are NFRs otherwise. In yeast, promoter NFRs are reestablished almost immediately after replication, maybe by RSC activity, and the kinetics of nucleosome repositioning over genes correlates with transcription levels (Fennessy and Owen-Hughes 2016; Vasseur et al. 2016). Many factors are involved in redepositing nucleosomes in the wake of RNA polymerase (Hennig and Fischer 2013; Smolle et al. 2013). Our finding that highly transcribed genes rarely exhibit low, but mostly exhibit high absolute nucleosome occupancy, argues that RNA polymerase passage, although it requires transient remodeling of nucleosomes (Farnung et al. 2018; Ehara et al. 2019), fosters high nucleosome occupancy, probably via concomitant recruitment of the nucleosome redeposition machinery. It is also compatible with bursty transcription (Haberle and Stark 2018). Conversely, it seems at odds with measurements, for example, by MNase-based methods that suggested an inverse relationship between transcription activity and nucleosome occupancy in gene bodies. MNase is usually not a reliable tool in this regard, as recently reiterated (Chereji et al. 2019a), and methods like DNase-seq or ATAC-seq exaggerate accessibility differences, which was explained recently (Chereji et al. 2019b). Our absolute occupancy measurements clarify this method-driven misconception. Nonetheless, anecdotal reports of nucleosome depletion over highly transcribed genes, like heat shock genes upon heat shock induction (Zhao et al. 2005), were corroborated by qDA-AluI absolute occupancy measurements for the case of a few genes induced by amino acid starvation (Chereji et al. 2019b). So, there may be special cases of highly induced genes that transiently lose nucleosomes over their gene bodies. In addition, regions transcribed by RNA polymerase I and III may be largely nucleosome depleted too, but our analyses focused on genes transcribed by RNA polymerase II.
The minimum of +1 nucleosome-aligned composite absolute occupancy of 29% at promoter NFRs (Fig. 1B) cannot be taken as evidence for higher nucleosome occupancy here than previously thought or for fragile nucleosomes (Kubik et al. 2015; Chereji et al. 2017) but rather reflects binding of nonnucleosomal factors, like transcription factors, GRFs or RNA polymerase. There remains the formal possibility that absolute occupancy, even at nucleosome positions determined by other methods, reflects a composite of nucleosome occupancy and, for example, occupancy by RNA polymerase. However, <1% of yeast genes have more than one RNA polymerase molecule bound (Pelechano et al. 2010), making this formal possibility less of a concern.
Low absolute occupancy correlates with the presence of RSC that is the major nucleosome-ejecting remodeling complex in yeast (Clapier et al. 2016) and particularly responsible for keeping NFRs nucleosome-free (Badis et al. 2008; Hartley and Madhani 2009; Wippo et al. 2011; Krietenstein et al. 2016; Brahma and Henikoff 2019). This correlation immediately suggests a mechanistic explanation for lower nucleosome occupancy, although it remains to be better understood what determines RSC-dependent nucleosome depletion.
Our methodology should be applicable to any chromatin preparation as long as nucleosome dynamics are frozen. We underscore that ORE-seq was necessary for our study as a validating approach but can be omitted in future applications. The application to chromatin of large genomes may be costly because high genome coverage (mean coverage >40-fold) is required and it remains to be tested if cross-linking will be required for stabilizing nucleosomes also in non-yeast species. For sure, care has to be taken to titrate DNA methylation into saturating conditions. The standard NOMe-seq conditions (Kelly et al. 2012) are likely insufficient, especially because DNA methylation by the GpC DNMT is more difficult to saturate.
In summary, DNA methylation under conditions of saturation, frozen nucleosome dynamics, and high sequencing coverage provides a measure for the long-missing chromatin quantity of absolute occupancy for nucleosomes or other factors. This will help distinguish if processes like DNA replication, DNA repair, or aging are associated with changes in nucleosome occupancy.
Methods
Yeast strains and media
The BY4741 strain (MATa his3Δ0 leu2Δ0 met15Δ0 ura3Δ0, Euroscarf) was grown to log phase (Supplemental Fig. S1A) in YPDA medium (1% w/v Bacto yeast extract, 2% w/v Bacto peptone, 2% w/v glucose, 0.1 g/L adenine, 1 g/L KH2PO4). Cross-linking was with 1% formaldehyde (final concentration) for 1, 5, or 20 min (WT5, other replicates only 20 min) at RT while shaking and quenched for 20 min with 125 (WT1, WT4) or 250 (WT5) mM glycine (final concentration).
Isolation of yeast nuclei
Yeast nuclei were prepared as described (Almer et al. 1986). In brief, cells were harvested by centrifugation, washed in water, resuspended in 2.8 mM EDTA, pH 8, 0.7 M 2-mercaptoethanol, incubated for 30 min at 30°C, washed in 1 M sorbitol, resuspended in 1 M sorbitol, 5 mM 2-mercaptoethanol and spheroplasted by incubation with 20 mg/mL freshly added Zymolyase 100T (MP Biochemicals) for 30 min at 30°C. Spheroplasts were washed with 1 M sorbitol and resuspended in lysis buffer (18% Ficoll, 20 mM KH2PO4, 1 mM MgCl2, 0.25 mM EGTA, 0.25 mM EDTA) followed by centrifugation at 22,550g for 30 min to collect chromatin (“nuclei”). Pellets were frozen in dry-ice/ethanol and stored at −80°C.
DNA methylation in chromatin
Nuclei pellets were washed in methylation buffer (20 mM HEPES-NaOH pH 7.5, 70 mM NaCl, 0.25 mM EDTA pH 8.0, 0.5 mM EGTA pH 8.0, 0.5% (v/v) glycerol, 1 mM DTT, 0.25 mM PMSF) (Darst et al. 2012). Per reaction, nuclei from approximately 0.1 g wet cell pellet were resuspended in 800 µL methylation buffer containing 640 µM freshly added S-adenosylmethionine (SAM). Two hundred units M.SssI or M.CviPI (both NEB) and 10 mM DTT were freshly added. Methylation reactions were dialyzed in Slide-A-Lyzer MINI Dialysis Devices 10 K MWCO (Thermo Fisher Scientific) against 15 mL methylation buffer with 200 µM freshly added SAM at 25°C for M.SssI or 37°C for M.CviPI. Next, 0.5–1 µg fully assembled pUC19-601-25mer plasmid (pFMP233) (Lowary and Widom 1998; Lieleg et al. 2015a) and Saccharomyces Genome Database (SGD) assembled Escherichia coli gDNA or SGD assembled E. coli plasmid library (limited Sau3A fragments of E. coli gDNA ligated into pJET 1.2 plasmid, Thermo Fisher Scientific) was spiked in before methylase addition. E. coli spike-in results were not further pursued owing to low coverage. Reactions were stopped by 0.5% SDS (final concentration), reversed cross-linked if required, and DNA was deproteinized by Proteinase K, phenol-chloroform extracted, and RNase A digested.
Restriction enzyme digestion of chromatin
Nuclei from approximately 0.1 g wet cell pellet were prewashed in methylation buffer, centrifuged, and resuspended either in 400 µL 1× CutSmart buffer (NEB) for BamHI-HF and AluI or in 1× NEB2.1 for HindIII. Digestions were started by RE addition, incubated for 30 or 120 min at 37°C, and stopped by 10 mM EDTA and 0.5% SDS (final concentration). DNA preparation was as above. For the cut-all cut method, Schizosaccharomyces pombe gDNA, fully digested with the corresponding RE, was spiked in at 5%–10% DNA mass of the final sample before phenol-chloroform extraction. After RNase A digestion, half of the sample was digested for a second time with 100 units of the corresponding RE and in the corresponding buffer for 1.5 h at 37°C. The reaction was stopped by 15 mM EDTA (final concentration). RE accessibility via Southern blot was done as previously described (Musladin et al. 2014).
Calibration samples for restriction enzyme digests
gDNA was purified from BY4741 cells using the Blood & Cell Culture DNA Midi Kit (Qiagen) and one aliquot completely digested with the respective RE. Mixed ratios of digested and not digested gDNA were treated as if they were DNA extracted after restriction enzyme digest of chromatin. The 0% cut calibration sample was an in vivo cross-linked nuclei preparation without RE addition.
DNA methylation and restriction enzyme digestion for in vitro–reconstituted chromatin
SGD chromatin was as in Krietenstein et al. (2012). For low or high nucleosome density, approximately 4 µg or 8 µg Drosophila embryo histone octamers, respectively, were assembled with 10 µg yeast plasmid library (Jones et al. 2008). DNA methylation and restriction enzyme digestion were done in the same buffer (20 mM HEPES-NaOH pH 7.5, 70 mM NaCl, 1.5 mM MgCl2, 0.5% [v/v] glycerol, 10 mM DTT) with the same units (20 units or 80 units) for 30 or 180 min at 37°C. For the latter, fresh enzyme (20 units) was refilled after 60 min, in case of DNA methylation, also fresh SAM. Reactions were stopped by 15 mM EDTA and 0.5% SDS (final concentration), followed by Proteinase K digestion and DNA purification. Methylated samples were directly used for library construction. RE digested samples were split after adding S. pombe gDNA spike-in, and one-half was digested a second time as described above.
Illumina sequencing library construction and sequencing
Purified DNA was sheared to ∼150 bp fragments (Covaris S220) and concentrated (NucleoSpin Gel and PCR Clean-up Kit, Macherey-Nagel). Then, 0.5–1 µg (determined by Qubit, Thermo Fisher Scientific) was used for library preparation: DNA end polishing (15 units T4 DNA Polymerase, 50 units T4 PNK, 5 units Klenow [NEB]) for 30 min at 20°C, DNA purification with AmPure XP beads (Beckman Coulter), A-tailing (15 units Klenow exo-[NEB]) for 30 min at 37°C, AmPure XP bead purification, adapter ligation (15 units T4 DNA Ligase [NEB], 75–150 pmol NEBNext Adapter or NEBNext Methylated Adapter for bisulfite conversion) for 20 min at 25°C, AmPure XP bead purification, PCR (only for half of the sample, 8 PCR cycles with NEBNext Multiplex Primers for Illumina and Phusion Polymerase [NEB]). Methylated samples were either bisulfite converted (Qiagen EpiTect Bisulfite Kit) and subjected to 12–14 PCR cycles (NEBNext Multiplex Primers for Illumina and Phusion U Polymerase, Thermo Fisher Scientific), or treated with the Enzymatic Methyl-Kit (NEB, E7120S). All samples were sequenced on an Illumina HiSeq 1500 in 50-bp paired-end mode.
Oxford Nanopore library construction and sequencing
For Nanopore sequencing, 1 µg of purified DNA was subjected to 1D native barcoding (Oxford Nanopore, SQK-LSK109). To get up to 1 Gb coverage per sample, up to five samples were loaded to a MinION flowcell (R9.4.1).
ORE-seq: cut and uncut fragment count and resection length
After demultiplexing (Girardot et al. 2016), sequenced reads were trimmed based on base-calling quality and mapped (Li and Durbin 2009) using the combined S. cerevisiae and S. pombe reference genome. Fragments longer than 500 bp or on rDNA loci were removed. At each cut site, fragment starts and ends were counted within a sample-specific window (green areas in Supplemental Fig. S1D; Supplemental Methods) to account for exonuclease resection and within the same window the mean resection length was calculated. Uncut fragments, that is, fragments covering, but not starting or ending at the cut site, were also counted at each cut site. Cut sites with neighbors closer than 200 bp were ignored completely and remaining cut sites with neighbors closer than 300 bp were analyzed only using the starting/ending counts not pointing toward the close neighbor.
ORE-seq: occupancy estimation (cut–uncut method)
Using the cut counts Ci and the uncut counts Ui at cut site i of the sample without a second RE digest, we calculated the effective cut and uncut counts which correct for cut fragment
ends caused by shearing and the loss of uncut fragments (detailed derivation in Supplemental Methods):
 in which w is the length of the count window, σ is the corrected ratio of all cut counts away from all cut sites, and all uncut fragment counts away from all cut sites and
the “uncut correction factor” γ. The estimated occupancy is then given by
in which w is the length of the count window, σ is the corrected ratio of all cut counts away from all cut sites, and all uncut fragment counts away from all cut sites and
the “uncut correction factor” γ. The estimated occupancy is then given by 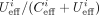 .
. 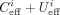 denotes the effective coverage and we ignored sites when its value was less than 40. For the uncorrected version, γ is one. In the corrected version, the value of γ was fitted using the calibration samples for AluI, BamHI, and HindIII, minimizing the deviation of the mean occupancy from
the prepared occupancy, and varies between 1.555 and 1.680 depending on the enzyme (details in Supplemental Fig. S1G, right, H; Supplemental Methods).
denotes the effective coverage and we ignored sites when its value was less than 40. For the uncorrected version, γ is one. In the corrected version, the value of γ was fitted using the calibration samples for AluI, BamHI, and HindIII, minimizing the deviation of the mean occupancy from
the prepared occupancy, and varies between 1.555 and 1.680 depending on the enzyme (details in Supplemental Fig. S1G, right, H; Supplemental Methods).
ODM-seq analysis for BS-seq and EM-seq data
We mapped the paired-end reads with BS-Seeker2 (version 2.1.8) (Guo et al. 2013) using Bowtie (Langmead et al. 2009) and trimmed the reads at the real fragment ends by 5 to 10 bp to obtain a constant conversion ratio along reads averaged over all reads, because the conversion ratio usually shows an increase or decrease at these ends owing to end repair. We ignored reads on the loci of rDNA genes and reads with an unconverted HCH motif, because HCH should be fully converted. The “anti-pattern” of the CpG/GpC DNA methyltransferases is the GCH/HCG pattern, respectively, and should also be fully converted. We discarded a sample if the average anti-pattern conversion ratio among all reads was less than 0.98. At each CpG/GpC methylation site, the occupancy is calculated as ratio of the converted reads over the number of all reads. We ignored methylation sites with a coverage less than 20.
ODM-seq analysis for Nanopore-seq data
We called the bases with Guppy (Oxford Nanopore Technologies, version 3.2.2) and mapped the reads with minimap2 (version 2.14-r892-dirty) (Li 2018). For methylation calling we used Nanopolish (version 0.11.0) (Simpson et al. 2017). At each CpG methylation site, the occupancy is calculated as ratio of unmethylated reads over the sum of methylated and unmethylated reads, ignoring reads where the site has been called “ambiguous” by Nanopolish. We ignored methylation sites with a coverage less than 20. Note that, currently, Nanopolish groups CpG sites within 10 bp into one site, thus having a lower resolution than BS-seq.
Calculation of ORE-seq and ODM-seq maps
For RE samples, very rare occupancy estimates outside the interval between 0 and 1 were truncated to 0 or 1. For the ORE-seq map (Fig. 1B), we averaged the occupancy values at the same sites in different RE samples with equal weights. Similarly, for the ODM-seq map (Fig. 1B), different bisulfite samples were averaged with equal weights. We also calculated individual enzyme maps in the same way (Supplemental Table S1).
Bioinformatics
BedGraph files were generated using the R (R Core Team 2018) package rtracklayer (Lawrence et al. 2013) and displayed with Integrative Genomics Viewer (IGV) (Robinson et al. 2011). +1 Nucleosome annotation was generated by calling nucleosomes within a 220-bp window downstream from transcription start sites (Xu et al. 2009) in our MNase-seq WT1 cross-linked data set using DANPOS (Chen et al. 2013). We used the R packages Biostrings (https://rdrr.io/bioc/Biostrings/), GenomicAlignments (Lawrence et al. 2013), and GenomicRanges (Lawrence et al. 2013) to load raw data files.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE132225. Source codes are deposited as Supplemental Code and at https://github.com/gerland-group/absolute-occupancy-analysis.
Acknowledgments
We thank Johannes Nübler for pointing us to the question of absolute nucleosome occupancy, Johannes Söding for letting M.H. help start the project, Michael Kladde for advice on DNA methylation of nucleosomes, Tobias Straub and Tamas Schauer for bioinformatics support, Björn Schwalb for sharing 4sU-seq data, Razvan Chereji for sharing chemical cleavage-seq nucleosome cluster calls, Nir Friedman for sharing SLIM-ChIP data, and Daan Verhagen and Felix Müller-Planitz for discussions on DNA methylation footprinting of nucleosomes. This work was funded by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) via the Collaborative Research Clusters SFB863 and SFB1064. M.W. is a member of the Graduate School of Quantitative Biosciences Munich (QBM). We thank the Amgen Foundation for funding J.L.E.’s stay in the Korber group as an Amgen Scholar.
Author contributions: Conceptualization and methodology, E.O., M.W., N.K., M.H., S.K., H.B., U.G., and P.K.; data curation, formal analysis, and software, E.O., M.W., and M.H.; visualization, E.O. and M.W.; funding acquisition, project administration, supervision, and resources, H.B., U.G. and P.K.; investigation and validation, E.O., N.K., A.S., J.L.E., S.K., and P.K.; writing (original draft), P.K.; writing (review and editing), E.O., M.W., N.K., M.H., U.G., and P.K.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.253419.119.
- Received June 7, 2019.
- Accepted October 31, 2019.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








