
Inherited DNA methylation primes the establishment of accessible chromatin during genome activation
- Guifen Liu1,2,3,
- Wen Wang1,3,
- Shengen Hu1,3,
- Xiangxiu Wang1 and
- Yong Zhang1
- 1Translational Medical Center for Stem Cell Therapy and Institute for Regenerative Medicine, Shanghai East Hospital, School of Life Science and Technology, Shanghai Key Laboratory of Signaling and Disease Research, Tongji University, Shanghai, 200092, China;
- 2Division of Translational Medicine, Institute for Advanced Study, Tongji University, Shanghai, 200092, China
-
↵3 These authors contributed equally to this work.
Abstract
For animals, epigenetic modifications can be globally or partially inherited from gametes after fertilization, and such information is required for proper transcriptional regulation, especially during the process of zygotic genome activation (ZGA). However, the mechanism underlying how the inherited epigenetic signatures affect transcriptional regulation during ZGA remains poorly understood. Here, we performed genome-wide profiling of chromatin accessibility during zebrafish ZGA, which is closely related to zygotic transcriptional regulation. We observed a clear trend toward a gradual increase in accessible chromatin during ZGA. Furthermore, accessible chromatin at the promoters displayed a sequential priority of emergence, and the locations of the accessible chromatin were precisely primed by the enrichment of unmethylated CpGs that were fully inherited from gametes. On the other hand, distal regions with high methylation levels that were inherited from the sperm facilitated the binding of DNA methylation-preferred transcription factors, such as Pou5f3 and Nanog, which contributed to the establishment of accessible chromatin at these loci. Our results demonstrate a model whereby inherited DNA methylation signatures from gametes prime the establishment of accessible chromatin during zebrafish ZGA through two distinct mechanisms.
Embryogenesis commences with the fusion of two terminally differentiated gametes that carry unique epigenetic signatures, and embryogenesis can efficiently activate epigenetic reprogramming to a transcriptionally permissive state (Burton and Torres-Padilla 2014; Zhou and Dean 2015). In mammals, chromatin modifications from gametes, including DNA methylation and histone modifications, are largely cleared after fertilization (Ooi and Henikoff 2007; Wu and Zhang 2010), but recent studies have shown that H3K4me3, H3K27me3, and DNA methylation can be partially retained during preimplantation embryogenesis (Hammoud et al. 2009; Borgel et al. 2010; Brykczynska et al. 2010; Tang et al. 2015; Dahl et al. 2016; Liu et al. 2016; Zhang et al. 2016; Zheng et al. 2016). In contrast to mammals, the DNA methylation pattern in the sperm is stably inherited during zebrafish embryogenesis (Jiang et al. 2013; Potok et al. 2013), and several important histone methylations are globally removed before zygotic genome activation (ZGA) (Vastenhouw et al. 2010; Lindeman et al. 2011). Although some chromatin modifications can be globally or partially inherited by progeny embryos from gametes, whether the inherited epigenetic signatures can facilitate the embryogenesis remains unclear. Several studies have demonstrated that the histone modifications in gametes are retained at the promoters of developmental genes and subsequently pattern the transcription of these genes (Hammoud et al. 2009; Wu et al. 2011; Ihara et al. 2014; Siklenka et al. 2015; Teperek et al. 2016). However, these findings contrast with evidence that shows that retention of histone modifications preferentially occurs at repeat elements and gene-poor genomic regions, which are usually distal to transcription start sites (TSSs) (Carone et al. 2014; Samans et al. 2014; Dahl et al. 2016; Liu et al. 2016; Zhang et al. 2016), highlighting the largely unexplored question of how inherited epigenetic signatures prime zygotic transcriptional regulation.
Chromatin accessibility is closely related to gene transcriptional regulation, and accessible chromatins are regarded as cis-regulatory elements, including promoters, enhancers, insulators, and locus control regions (Gross and Garrard 1988; Li et al. 1999; Gaszner and Felsenfeld 2006). Because accessible chromatins are usually bound by transcription factors (Li et al. 2007), the gene transcription regulation model can be inferred to some extent from chromatin accessibility data (Duren et al. 2017). One study showed that accessible chromatins contribute to ZGA in mice (Cho et al. 2002), and two recent studies have provided genome-wide accessible chromatin maps during ZGA in mice, which identify several transcription factors that are important for the establishment of accessible chromatin (Lu et al. 2016; Wu et al. 2016). However, to our best knowledge, the effects of inherited epigenetic signatures on the establishment of accessible chromatin during ZGA, which can reflect transcriptional regulation in the progeny, remain unclear.
In this study, we address the effects of inherited epigenetic signatures on the establishment of accessible chromatin during ZGA in zebrafish. In zebrafish, the major phase of ZGA occurs at the 10th cell cycle after fertilization (Wragg and Muller 2016). The fully sperm-inherited DNA methylation pattern (Jiang et al. 2013; Potok et al. 2013) and globally absent histone methylations before ZGA (Vastenhouw et al. 2010; Lindeman et al. 2011) largely exclude the priming effects of histone methylations in gametes on embryogenesis and thus provide an ideal in vivo system to investigate the mechanisms by which the inherited DNA methylation signatures can prime zygotic transcriptional regulation.
Results
Profiling accessible chromatin during ZGA
The early embryogenesis of zebrafish has two ZGA phases: a minor phase of ZGA that is initiated from the 64-cell stage, and a major phase that occurs at the 1k-cell stage (Wragg and Muller 2016). To investigate the effect of inherited epigenetic signatures on progeny transcriptional regulation during ZGA, we analyzed the establishment of accessible chromatin by ATAC-seq in the early embryos of zebrafish at five stages: the 64-cell (2 hpf), 256-cell (2.5 hpf), 1k-cell (3 hpf), oblong (3.7 hpf), and dome (4.5 hpf) stages (Supplemental Fig. S1A). Three biological replicates were generated for each stage, with very high reproducibility (Pearson correlation coefficient: 0.84–0.97) (Supplemental Table S1; Supplemental Fig. S1B), and we combined replicates from each stage for the following analysis. To profile the accessible chromatin (i.e., nucleosome-free loci), only ATAC-seq reads from fragments that were smaller than 100 bp were used for the following analysis.
Initiation of accessible chromatin during the minor phase of ZGA
By calculating the number of accessible chromatin regions in each developmental stage, we observed a clear trend toward a gradual increase in accessible chromatin during ZGA (Fig. 1A). At the 64-cell stage, only a few regions of accessible chromatin were detected (162 regions), including the miR-430 cluster (Fig. 1B; Supplemental Fig. S2A), which is consistent with a previous study showing that miR-430 genes are transcribed at the 64-cell stage (Heyn et al. 2014). Following initiation of the minor phase of ZGA, 398 regions were detected as accessible chromatin at the 256-cell stage, with 160 protein-coding genes that showed accessible chromatin at their promoters (TSS ± 2 kb) (Fig. 1C; Supplemental Fig. S2B). We next investigated the quantitative association between chromatin accessibility and transcription activity at the minor phase of ZGA and observed that genes with accessible chromatin at their promoters were closely related to transcription activity (see Supplemental Text; Fig. 1D,E; Supplemental Fig. S2C).
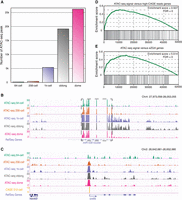
Establishment of accessible chromatin during the minor phase of ZGA. (A) The bar plot illustrates the number of accessible chromatin regions at each stage during ZGA. (B) The genome browser view shows the ATAC-seq signals around the miR-430 cluster regions, which are indicated by the dashed box. (C) The genome browser view highlights the ATAC-seq signals at the rps8a locus as a representative example of accessible chromatin that appeared at the 256-cell stage. (D) Gene Set Enrichment Analysis (GSEA) for the association between promoter accessibility and transcription activity. All genes were ranked according to the levels of chromatin accessibility at the promoters at the 256-cell stage. Genes with more than 500 CAGE reads around their TSSs at the 512-cell stage (TSS ± 50 bp; 811 genes) were treated as a gene set. Enrichment score and FDR were calculated by GSEA. (E) GSEA for the association between promoter accessibility and transcription activity. All genes were ranked according to the levels of chromatin accessibility at the promoters at the 256-cell stage. The reported early ZGA genes (993 genes) were treated as a gene set. Enrichment score and FDR were calculated by GSEA.
To confirm the lesser accessibility of chromatin before the major phase of ZGA, we performed a DNase I TUNEL assay, which is a direct measurement of chromatin accessibility (Jachowicz et al. 2017), in four developmental stages. The fluorescence signal was not detected in the 64-cell stage, and the signal was much weaker in the 256-cell stage than those in the 1k-cell or oblong stages (Supplemental Fig. S2D), which is consistent with our accessible chromatin profiles. Furthermore, for most accessible chromatin regions detected in the dome stage but not in the 256-cell stage, the patterns of nucleosome depletion were not observed at the 256-cell stage (Supplemental Fig. S2E), confirming the inaccessibility of those chromatin regions before the major phase of ZGA.
Burst of accessible chromatin during the major phase of ZGA
After the minor phase of ZGA, a burst of accessible chromatin was observed at the 1k-cell stage (5383 detected regions), which was accompanied by the initiation of the major phase of ZGA (Figs. 1A, 2A). After the 1k-cell stage, the number of detected accessible chromatin regions continuously increased at the oblong stage (19,160 regions) and dome stage (26,219 regions) (Fig. 1A). To investigate the feature of accessible chromatin regions during the major phase of ZGA, we examined the genomic distribution of the newly emerged accessible chromatin regions at the three stages. At the initiation of the major phase of ZGA, 63% of the accessible chromatin was located at the promoters, and the percentage decreased to 39% at the oblong stage and 29% at the dome stage (Fig. 2B), indicating a sequential priority of emergence of the accessible chromatin at the promoters during the major phase of ZGA. Clear nucleosome depletion patterns were observed for the newly emerged accessible promoters at each of the three stages (Supplemental Fig. S3). Gene Ontology (GO) analysis was performed on genes with newly accessible promoters at each of the three stages. The most enriched GO terms for all three gene sets were DNA binding (Fig. 2C), which was consistent with the activities of the transcription factors for zygotic gene expression (Lee et al. 2013) and early embryonic development (Hammonds et al. 2013). Our results show a burst of accessible promoters during the major phase of ZGA, raising questions regarding the mechanism that primes the establishment of these accessible promoters.
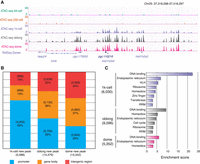
Establishment of accessible chromatin during the major phase of ZGA. (A) The genome browser view shows the ATAC-seq signals at the representative region as an example of accessible chromatin that appeared during the major phase of ZGA. (B) The bar plot shows the percentages of newly established ATAC-seq peaks that are in the promoter (TSS ± 2 kb), gene body, and intergenic region during the major phase of ZGA. (C) GO enrichment analysis of genes with newly accessible promoters at the major phase of ZGA. Enrichment scores were calculated by DAVID (Huang da et al. 2009a,b).
Unmethylated CpGs prime the emergence of accessible promoters
Because several important histone methylations undergo global erasure before ZGA and because the DNA methylation pattern is fully inherited from the sperm (Vastenhouw et al. 2010; Lindeman et al. 2011; Jiang et al. 2013; Potok et al. 2013), we further examined whether the inherited DNA methylation patterns had effects on the establishment of accessible chromatin during ZGA. Genes were divided into five groups based on the time of the appearance of accessible promoters, including the 256-cell newly accessible, 1k-cell newly accessible, oblong newly accessible, dome newly accessible, and inaccessible groups, and the average DNA methylation levels (at the 1k-cell stage) were calculated for each promoter separately. Genes in the inaccessible groups showed significantly higher DNA methylation levels at their promoters than the other genes. For the genes with accessible promoters during ZGA, the earlier the appearance of accessible promoters, the lower the DNA methylation level that was observed (Fig. 3A). Similar patterns were observed by using DNA methylation levels at other stages (Supplemental Fig. S4A–C). It was reported that unmethylated CpG-enriched regions have been observed at the promoters of developmental genes in zebrafish (Andersen et al. 2012; Jiang et al. 2013). We further observed that the larger number of unmethylated CpGs at the promoters was significantly associated with the earlier establishment of accessible promoters (Fig. 3B; Supplemental Fig. S4D–F). Compared with the above epigenetic features, DNA sequence features, such as CpG ratio and CpG density, showed much weaker discriminatory power to distinguish genes with accessible promoters during ZGA and those with inaccessible promoters (Supplemental Fig. S4G–I). For genes with accessible promoters during ZGA, the locations of the ATAC-seq peak summits, i.e., the regions with the local highest accessibility, were clearly associated with the local lowest DNA methylation levels and highest numbers of unmethylated CpGs (Fig. 3C; Supplemental Fig. S5). We then examined the extent to which the locations of the ATAC-seq peak summits at the promoters could be explained by the level of DNA methylation or the enrichment of unmethylated CpGs. With DNA methylation patterns at the 1k-cell stage, the local highest number of unmethylated CpGs precisely accounted for 69.5% of the locations of the ATAC-seq peak summits (Fig. 3D; Supplemental Fig. S6A), whereas the prediction power was 24.6% weaker for the local lowest DNA methylation level (Fig. 3D; Supplemental Fig. S6B; see Supplemental Methods). Our results show that accessible promoters that emerge during ZGA are clearly associated with DNA methylation patterns, especially the unmethylated CpG number, raising the assumption that the establishment of accessible promoters is primed by the number of unmethylated CpGs before ZGA.
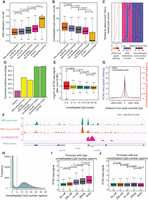
Unmethylated CpGs prime the emergence of accessible promoters. (A) The box plot shows the relationship between newly accessible promoters and DNA methylation levels (1k-cell stage). Earlier accessible promoters have lower DNA methylation levels. P-values were calculated by Wilcoxon test. (B) The box plot shows the relationship between newly accessible promoters and the largest unmethylated CpG numbers among 200-bp bins (1k-cell stage). Earlier accessible promoters have higher numbers of unmethylated CpGs. P-values were calculated by Wilcoxon test. (C) The heat map shows the relationship between the locations of the ATAC-seq peak, DNA methylation level (1k-cell stage), and unmethylated CpG number (1k-cell stage) at the promoters. The accessible promoters in C–E are newly emerged ones in the 1k-cell or oblong stages. (D) The bar plot shows the successful prediction percentage of the locations of the ATAC-seq peak summits at the promoters, based on the locally lowest DNA methylation level or the local highest number of unmethylated CpGs (128-cell and 1k-cell stages, respectively). Details of the prediction of the ATAC-seq peak summit locations were described in Supplemental Methods. (E) The box plot shows the log2-transformed fold change in the ATAC-seq signals at the 200-bp bins with largest numbers of unmethylated CpGs at each accessible promoter upon knockdown of cxxc1b. The accessible promoters were grouped based on the locally largest numbers of unmethylated CpGs. P-values were calculated by Wilcoxon test. (F) The genome browser view shows the ATAC-seq signals at the mrpl39 locus as a representative example of decreasing accessibility at the unmethylated CpG-enriched regions upon knockdown of cxxc1b. (G) The summit of the ATAC-seq signals overlaps with the highest number of unmethylated CpGs in the sperm at the promoters. The ATAC-seq peaks are newly emerged ones in 1k-cell or oblong stages. (H) All promoters are divided into two groups according to the locally largest numbers of unmethylated CpGs in sperm (cut-off of 5). (I) The box plot shows the ATAC-seq signal during ZGA at the promoters with a high locally largest number of unmethylated CpGs in sperm. P-values were calculated by Wilcoxon test. (J) The box plot shows the ATAC-seq signal during ZGA at the promoters with a low locally largest number of unmethylated CpGs in sperm. P-values were calculated by Wilcoxon test.
Some CXXC domain-containing factors specifically bind regions with enriched unmethylated CpGs (Thomson et al. 2010). To examine whether the CXXC domain-containing factors affect the establishment of accessible promoters with enriched unmethylated CpGs before ZGA, we screened the CXXC domain-containing factors (see Methods) in zebrafish and identified two factors (Cxxc1b and Kmt2a) that may participate in the potentially priming roles. We further knocked down the expression levels of two factors separately by injecting morpholino (MO) at the 1-cell stage and performed ATAC-seq at the oblong stage. Upon knockdown of cxxc1b, a significant decrease in chromatin accessibility was observed at the promoters with enriched unmethylated CpGs (Fig. 3E,F), whereas the kmt2a knockdown did not show significant differences (Supplemental Fig. S6C). The gene encoding the factor Cxxc1b is the homolog of mammalian CXXC1 in zebrafish. A study in mammalian cell lines revealed that the enriched unmethylated CpG influences the chromatin structure by recruiting CXXC1, which further recruits H3K4 methyltransferase (Thomson et al. 2010). H3K4me3 attracts specific PHD domain proteins, such as CHD1, which is essential for the establishment of accessible chromatin (Gaspar-Maia et al. 2009). Taken together, our results suggest that the enriched unmethylated CpG before ZGA can prime the establishment of accessible chromatin at promoters during ZGA and that Cxxc1b can mediate the transfer of DNA methylation patterns to the accessible chromatin landscape at promoters.
The DNA methylation pattern at the 1k-cell stage is almost identical to that of the sperm, rather than that of the oocyte (Jiang et al. 2013; Potok et al. 2013), suggesting that the accessible chromatin landscape at promoters during ZGA might be primed by paternal, instead of maternal, epigenetic inheritance. To examine this assumption, we compared the DNA methylation patterns in the sperm and oocyte at the accessible promoters during ZGA. The differences between the methylation levels at these promoters in the sperm and oocyte were much smaller than the methylation levels at the nearby genomic regions (4.6% versus 10.9%) (Supplemental Fig. S5A; Supplemental Fig. S6D), whereas the analysis of the number of unmethylated CpGs showed surprisingly identical patterns between the sperm and oocyte at these loci (Fig. 3G; Supplemental Fig. S5A; Supplemental Fig. S6E). We further divided all promoters into two groups based on the number of unmethylated CpGs in the sperm or oocytes, with five as the cut-off (Fig. 3H; Supplemental Fig. S6F), and found that the chromatin accessibility in the two groups of promoters showed distinct patterns during ZGA. Promoters with a high number of unmethylated CpGs showed an increase in accessibility, whereas the promoters with a low number of unmethylated CpGs were almost inaccessible during the ZGA process (Fig. 3I,J; Supplemental Fig. S6G,H). Our observations suggest that the patterns of unmethylated CpGs in the sperm and oocyte had equivalent priming effects on the establishment of accessible chromatin at the promoters during ZGA.
Transcription factors modulate the accessible distal regions with a high methylation level
In addition to the promoters, 10,506 distal regions were detected as accessible during the major phase of ZGA, and we examined whether the enrichment of unmethylated CpGs also primed the emergence of the accessible distal regions. Compared to the accessible promoters, the accessible distal regions showed a much higher level of DNA methylation at the 1k-cell stage (Fig. 4A), indicating that the emergence of accessible distal regions is unlikely to be primed by the enrichment of unmethylated CpGs. Several DNA methylation-preferred transcription factors can reportedly bind methylated DNA sequences (Yin et al. 2017). We ranked newly accessible distal regions at either the 1k-cell or oblong stages by their DNA methylation levels, and for each transcription factor DNA-binding motif, we performed an analysis that was similar to Gene Set Enrichment Analysis (GSEA) (Subramanian et al. 2005) by treating the accessible distal regions that contained the motif as the set. By setting FDR < 0.001 as the statistical threshold and by performing the ranking by calculating the enrichment score, the top list of transcription factor DNA-binding motifs that tended to associate with a high methylation state were identified, including factors with high RNA levels during ZGA, such as Pou5f3 (the homolog of POU5F1 in zebrafish), Nanog, and Eomesa (Fig. 4B). The factor Eomesa is associated with endoderm formation (Bjornson et al. 2005), whereas Pou5f3 and Nanog are essential for ZGA in zebrafish (Lee et al. 2013). Here, we investigated whether Pou5f3 and Nanog preferentially bound to regions with high DNA methylation (DNA methylation level >0.8) and subsequently affected the establishment of accessible chromatin at these loci. We observed the formation of a clear nucleosome depletion pattern at the Pou5f3 and Nanog binding sites during ZGA (Supplemental Fig. S7A,B), suggesting that both factors may contribute to the emergence of accessible chromatin at distal regions with high DNA methylation levels. To investigate the roles of Pou5f3 and Nanog in the establishment of accessible distal regions with high DNA methylation levels, we knocked down the expression of each gene by injecting MO at the 1-cell stage. We then performed ATAC-seq at the oblong stage and observed a significant decrease in chromatin accessibility at the binding sites of both factors (Fig. 4C–E; Supplemental Fig. S7C–E). Such a decrease in chromatin accessibility can be partially rescued when co-injecting MO and the mRNA of pou5f3 and nanog (Fig. 4C–E; Supplemental Fig. S7E). A recent study in a mammalian cell line reported that SMARCA4 (also known as BRG1), a chromatin remodeller, can be recruited by POU5F1 (also known as OCT4) and can contribute to the establishment of accessible chromatin (King and Klose 2017). To investigate whether Smarca4a (the homolog of SMARCA4 in zebrafish) participated in the establishment of accessible chromatin at the Pou5f3 or Nanog binding sites, we knocked down the expression of smarca4a and performed ATAC-seq at the oblong stage. A significant decrease in chromatin accessibility at the Pou5f3 or Nanog binding sites with high DNA methylation levels was observed (Supplemental Fig. S7F,G). Taken together, these results indicate that DNA methylation-preferred transcription factors, such as Pou5f3 and Nanog, combined with Smarca4a, contribute to the emergence of accessible distal regions with high DNA methylation levels during ZGA.
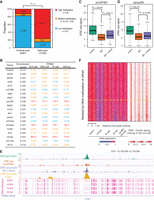
Transcription factors modulate accessible distal regions with high methylation levels. (A) The bar plot shows the proportion of newly accessible promoters and distal regions (at the 1k-cell and oblong stages) with high (>0.8), medium (>0.2 and ≤0.8), and low (≤0.2) DNA methylation levels (1k-cell stage). P-values were calculated by χ2 test. (B) The table shows the list of transcription factor DNA-binding motifs with the highest enrichment score, which is calculated by GSEA, and the enrichment score is associated with the enrichment of motifs at the high DNA methylation state. RNA levels of the transcription factors before and during ZGA are shown. RNA levels higher than 100 FPKM, between 1 and 100 FPKM, and less than 1 FPKM are indicated in red, blue, and orange, respectively. (C) The box plot shows the decrease in the ATAC-seq signals at the Pou5f3 binding sites (at distal regions with high DNA methylation levels) upon knockdown of pou5f3 and the partial recovery of the signals after co-injections with MO and the pou5f3 mRNA. P-values were calculated by Wilcoxon test. (D) The box plot shows the decrease in the ATAC-seq signals at the Nanog binding sites (at distal regions with high DNA methylation levels) upon knockdown of nanog and the partial recovery of the signals after co-injections with MO and the nanog mRNA. P-values were calculated by Wilcoxon test. (E) The genome browser view shows the ATAC-seq signals at the representative region as an example of decreasing accessibility at highly methylated distal regions upon knockdown of pou5f3 and the partial recovery after co-injections with MO and the pou5f3 mRNA. DNA methylation levels at sperm, oocyte, 1k-cell stage, and 24 hpf stage are also shown. (F) The heat map shows the relationship between the locations of the ATAC-seq peak (oblong stage), DNA methylation level (sperm, oocyte, 1k-cell stage, 24 hpf, and 24 hpf upon knockdown of tet) and ChIP-seq signals for Pou5f3 (5 hpf) and Nanog (high stage; 3.5 hpf) within ±1 kb that is centered on the distal accessible chromatin with a high DNA methylation level (1k-cell stage). Regions are ranked according to the DNA methylation level at 24 hpf. The yellow dotted line divides the regions into the upper group with maintained high DNA methylation levels at 24 hpf and the lower group with DNA demethylation at 24 hpf.
Because DNA methylation levels before ZGA have priming effects on the establishment of accessible distal regions that are mediated by DNA methylation-preferred transcription factors, we further examined the parental origin of these DNA methylation patterns. Unlike accessible promoters, the high DNA methylation levels observed at distal accessible regions were mainly inherited from the sperm, and these regions showed much lower DNA methylation levels in the oocyte, with 36.1% as medium or low methylation-level regions (DNA methylation level >0.2 and ≤0.8, and DNA methylation level ≤0.2, respectively) (Fig. 4F). Interestingly, although the above regions with medium or low methylation levels in the oocyte were fully methylated after fertilization, demethylation activity was observed at many of these regions at a later stage, such as 24 hpf (Fig. 4F; Supplemental Fig. S7H). Furthermore, the demethylated regions at 24 hpf showed significant overlap with the binding sites of Pou5f3 and Nanog during ZGA (Fig. 4F; Supplemental Fig. S7I,J). A study in a mammalian cell line reported that NANOG can physically interact with TET1 and TET2 (Costa et al. 2013), which are responsible for promoting DNA demethylation. Compared to wild-type sample, the binding sites of Pou5f3 and Nanog during ZGA showed a significant increase in DNA methylation levels at 24 hpf upon knockdown of tet (Fig. 4F; Supplemental Fig. S7K,L), indicating that Pou5f3 and Nanog may participate in Tet protein-mediated DNA demethylation at distal accessible chromatin. Taken together, these results indicate that the DNA methylation patterns that are inherited from sperm facilitate the binding of DNA methylation-preferred factors at distal regions during ZGA, and such factors may participate in the DNA demethylation activity, which in turn leads to oocyte-like DNA methylation patterns at these distal regions at later developmental stages.
Discussion
Here, we have provided two distinct mechanisms for how inherited epigenetic signatures can prime the global re-establishment of accessible chromatin during zebrafish embryogenesis, which is closely associated with the rebuilding of zygotic gene transcriptional regulation. The DNA methylation signatures in the promoters with enriched unmethylated CpGs in the sperm and oocyte are fully inherited after fertilization. The cluster of unmethylated CpGs can not only prime the formation of accessible promoters during ZGA but also precisely determine the loci with the highest accessibility at each accessible promoter; this determination can be mediated by CXXC domain-containing factors, such as Cxxc1b (Fig. 5A). For distal regions with high methylation levels in the sperm but with medium or low methylation levels in the oocyte, the maternal DNA methylation states are reprogrammed to paternally inherited signatures before ZGA (Jiang et al. 2013; Potok et al. 2013). During ZGA, such highly methylated distal regions facilitate the binding of DNA methylation-preferred factors, such as Pou5f3 and Nanog, recruit Smarca4a, and contribute to the formation of accessible chromatin at these regions. Interestingly, these distal accessible regions show oocyte-like DNA methylation patterns at later developmental stages, and DNA methylation-preferred factors, such as Nanog, may participate in Tet protein-mediated DNA demethylation activity at the binding sites (Fig. 5B).

Schematic model of the two mechanisms for the establishment of accessible chromatin in the promoter and distal regions. (A) Enrichment of unmethylated CpGs that are inherited from the sperm and oocyte primes the formation of accessible promoters during ZGA, which is mediated by CXXC domain-containing factors, such as Cxxc1b. (B) Paternally inherited DNA hypermethylation facilitates the binding of DNA methylation-preferred transcription factors, such as Pou5f3 and Nanog, and these factors recruit Smarca4a and contribute to the formation of accessible distal regions during ZGA. Such factors may participate in Tet-mediated demethylation and, in turn, lead to an oocyte-like DNA hypomethylation pattern after ZGA.
A recent in vitro study showed that hundreds of transcription factors prefer to bind methylated DNA (Yin et al. 2017). Consistent with this study, we observed that Pou5f3 and Nanog preferentially bind to their motifs on hypermethylated regions and contribute to the formation of accessible chromatin at these distal regions by recruiting chromatin remodeller Smarca4a during ZGA. To our best knowledge, this is the first study to report the functions of DNA methylation-preferred transcription factors in developing embryos. Compared to a previously defined putative enhancer list at 24 hpf (Lee et al. 2015), the demethylated regions at 24 hpf showed significant overlap with the list compared to all accessible distal regions during ZGA (35.7% versus 11.6%, χ2 test, P-value = 1.4 × 10−92), indicating that the demethylated regions are more likely to be considered as enhancers. Two recent studies on zebrafish reported that thousands of functional enhancers undergo demethylation during later developmental stages (Lee et al. 2015; Bogdanovic et al. 2016), and our study extended the findings of these studies by revealing that the enhancers with hypomethylated levels at the later stages are already accessible even before the demethylation activity.
The precise re-establishment of accessible chromatin during ZGA couples with the accurate control of the zygotic transcription program, and this study revealed the priming effects of inherited DNA methylation signatures from gametes in zebrafish. We do not exclude the possibility that other epigenetic signatures in gametes can also be transferred to the zygote and subsequently contribute to the establishment of zygotic transcription regulation; for example, the histone modification signatures in zebrafish sperm mark the embryonic developmental genes (Wu et al. 2011), and a recent study in Drosophila showed the role of maternally inherited H3K27me3 in regulating enhancer activation during ZGA (Zenk et al. 2017). Although several important histone modifications, including H3K4me3, H3K27me3, H3K36me3, and H3K9me3, are globally removed before zebrafish ZGA (Vastenhouw et al. 2010; Lindeman et al. 2011), residual methylation at these histones may also play a role during ZGA, and some unexamined histone modifications or histone variants may be inherited and have functions in this process. Besides, studying the inheritance of epigenetic signatures in zebrafish has many technical challenges, such as the potential off-target effects of MO, which cannot be fully excluded, and the limited materials of early embryos could be used in high-throughput experiments. Further studies are warranted to thoroughly dissect the inheritance of epigenetic signatures and their transgenerational effects during ZGA.
Methods
Zebrafish husbandry
Wild-type Tübingen-strain zebrafish were maintained under standard conditions (Westerfield 2000). Zebrafish embryos were obtained via natural mating of wild-type Tübingen-strain zebrafish (6–12 mo). Zebrafish care and experiments were approved by the Institutional Animal Care and Use Committee of Tongji University.
ATAC-seq
Embryos for ATAC-seq were harvested at five different developmental stages: 64-cell (2 hpf), 256-cell (2.5 hpf), 1k-cell (3 hpf), oblong (3.7 hpf), and dome (4.5 hpf). The ATAC-seq analyses of the zebrafish embryos involved a slightly modified approach that was based on the original method (Buenrostro et al. 2013); see Supplemental Methods. The libraries were sequenced by Illumina HiSeq X Ten sequencing. Public ATAC-seq data were collected from Bogdanovic et al. (2016) (Supplemental Table S2).
Sequenced reads were mapped to the zebrafish genome (zv9 assembly) using Bowtie 2 (version 2.2.3) with default parameters (Langmead and Salzberg 2012), and peak calling was performed using MACS (version 1.4.2 20120305) (Zhang et al. 2008) with the following parameters: -f BED −g 1.4 × 109 --keep-dup all --nomodel --shiftsize 25. Peaks with P-value ≤ 1 × 10−10 and fold ≥10 were kept. See Supplemental Methods for details.
DNase I TUNEL assay
Embryos were collected at the 64-cell, 256-cell, 1k-cell, and oblong stages. For each developmental stage, the numbers of cells used for the DNase I TUNEL assay were ∼1–2 × 104. The DNase I TUNEL assay was performed according to a previous study (Jachowicz et al. 2017) with some modifications. See Supplemental Methods for details.
MO injection, DNA construct, and mRNA injection
For MO injection, antisense MO oligos were synthesized by Gene Tools and resuspended in nuclease-free water. Unless otherwise stated, 1 nL of the MO solution was injected into the embryos at the 1-cell stage. All sequences were used to block translation. The concentrations of the MOs that were used in the injections were as follows: 0.75 pmol (cxxc1b), 1.0 pmol (kmt2a), 0.05 pmol (pou5f3), 0.05 pmol (nanog), and 0.25 pmol (smarca4a). See Supplemental Table S3 for MO sequence.
For the mRNA injection, the full-length cDNAs of pou5f3 and nanog were cloned by PCR into pCS2-gfp using the In-Fusion HD Cloning kit (Clontech, #639636) with BamHI (Takara, #1010A) sites. To prevent the binding of pou5f3 MO or nanog MO, mutations without amino acid changes were introduced within the MO target sites via PCR cloning (see Supplemental Table S4 for PCR primers). The mRNA was transcribed in vitro using the mMESSAGE mMACHINE SP6 kit (Ambion, #AM1340) after linearization of the constructs with KpnI (Takara, #1068A). To rescue the loss-of-function phenotype, 100 pg of the pou5f3 mRNA and 100 pg of the nanog mRNA were injected either individually or together into the MO-injected embryos at the 1-cell stage.
Screen for CXXC domain-containing factors
The list of CXXC domain-containing factors was compiled from a previous study (Blackledge et al. 2010). Only cxxc1b, dnmt1, mbd1b, and kmt2a were expressed during zebrafish ZGA. Factors Dnmt1 and Mbd1b play a repressive role (Jorgensen et al. 2004; Smith and Meissner 2013), which indicates that they are unlikely to participate in the establishment of open chromatin. After filtering, only Cxxc1b and Kmt2a were kept for experimental validation.
Motif scan
Motif scans were performed using FIMO (version 4.11.1) (Grant et al. 2011) against the JASPAR core 2016 vertebrates (Mathelier et al. 2016) and Cistrome (Mei et al. 2017) databases with the following parameters: –max-stored-scores 1000000. A motif with a P-value ≤ 1 × 10−5 was used for the analysis.
Data access
ATAC-seq raw and processed data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE101779.
Acknowledgments
We thank Ying Cao for the kind gifts of the Ctrl MO and plasmid (pCS2-gfp). This work was supported by the National Key Research and Development Program of China (2017YFA0102600, 2016YFA0100400), National Natural Science Foundation of China (31322031, 31571365, 31371288, 31601161), Program of Shanghai Academic Research Leader (17XD1403600), Shanghai Municipal Science and Technology Commission (17ZR1449500), and National Program for Support of Top-notch Young Professionals.
Author contributions: Y.Z. designed the study. G.L. performed the experiments, with the help of X.W. The data were analyzed by W.W. and S.H. The manuscript was written by G.L., W.W., S.H., and Y.Z.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.228833.117.
- Received August 9, 2017.
- Accepted May 23, 2018.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








