
Targeted deletion of a 170-kb cluster of LINE-1 repeats and implications for regional control
- Miguel L. Soares1,2,5,
- Carol A. Edwards1,5,
- Frances L. Dearden1,
- Sacri R. Ferrón1,6,
- Scott Curran1,7,
- Jennifer A. Corish1,
- Rebecca C. Rancourt1,8,
- Sarah E. Allen1,9,
- Marika Charalambous1,10,
- Malcolm A. Ferguson-Smith3,
- Willem Rens3,
- David J. Adams4 and
- Anne C. Ferguson-Smith1
- 1Department of Genetics, University of Cambridge, Cambridge CB2 3EH, United Kingdom;
- 2Departamento de Biomedicina, Unidade de Biologia Experimental, Faculdade de Medicina da Universidade do Porto, Porto; and i3S–Instituto de Investigação e Inovação em Saúde, Universidade do Porto, 4200-319 Porto, Portugal;
- 3Department of Veterinary Medicine, University of Cambridge, Cambridge CB3 0ES, United Kingdom;
- 4Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge CB10 1SA, United Kingdom
-
↵5 These authors contributed equally to this work.
Abstract
Approximately half the mammalian genome is composed of repetitive sequences, and accumulating evidence suggests that some may have an impact on genome function. Here, we characterized a large array class of repeats of long-interspersed elements (LINE-1). Although widely distributed in mammals, locations of such arrays are species specific. Using targeted deletion, we asked whether a 170-kb LINE-1 array located at a mouse imprinted domain might function as a modulator of local transcriptional control. The LINE-1 array is lamina associated in differentiated ES cells consistent with its AT-richness, and although imprinting occurs both proximally and distally to the array, active LINE-1 transcripts within the tract are biallelically expressed. Upon deletion of the array, no perturbation of imprinting was observed, and abnormal phenotypes were not detected in maternal or paternal heterozygous or homozygous mutant mice. The array does not shield nonimprinted genes in the vicinity from local imprinting control. Reduced neural expression of protein-coding genes observed upon paternal transmission of the deletion is likely due to the removal of a brain-specific enhancer embedded within the LINE array. Our findings suggest that presence of a 170-kb LINE-1 array reflects the tolerance of the site for repeat insertion rather than an important genomic function in normal development.
Interspersed DNA elements are a hallmark of eukaryotic genomes with more than 40% of the human and mouse genomes consisting of repeats derived from transposable elements (International Human Genome Sequencing Consortium 2001; Mouse Genome Sequencing Consortium 2002). The most abundant class is the long interspersed elements (LINEs), which are autonomous retrotransposons and thus transpose via an RNA intermediate. This “copy and paste” mechanism results in mainly truncated copies of LINEs in the genome that are incapable of further retrotransposition. LINE-1 (L1) is the youngest of the four families and the major LINE family in eutherian and marsupial mammals. There are thought to be approximately 3000 active L1s in the mouse genome (Goodier et al. 2001) and around 100 in the human (Brouha et al. 2003). In the mouse there are three families of active L1s: L1md_A, L1Md_Gf, and L1Md_Tf (DeBerardinis et al. 1998; Goodier et al. 2001). Members of two of the active families, Gf and Tf, are known to be transcribed from the inactive X Chromosome in female mice, where they have been suggested to facilitate X Chromosome inactivation (XCI) in regions which would otherwise escape inactivation (Chow et al. 2010).
LINEs are found throughout the eukaryotes (Malik et al. 1999) and have played a major evolutionary role in sculpting the genomic landscape. They can generate structural variation through insertion-mediated deletion, in which the insertion of an L1 element can result in the deletion of the adjacent genomic sequence (Symer et al. 2002; Gilbert et al. 2002, 2005) and via nonallelic homologous recombination between L1s that can create insertions, deletions, and inversions (Han et al. 2008; Cordaux and Batzer 2009). Active L1s have been shown to be a major source of variation within the human population (Beck et al. 2010; Huang et al. 2010; Iskow et al. 2010) and can modulate gene expression, for example, by alternative splicing (Belancio et al. 2006), disrupting transcription (Han et al. 2004), and generating alternative promoters (Speek 2001).
Although some insertions may be beneficial, most insertions in and around genes are likely to be deleterious and thus attract epigenetic silencing to prevent retrotransposition. In the majority of somatic tissues, L1 activity is suppressed by DNA methylation at the promoter (Yoder et al. 1997). The importance of epigenetic silencing is evident in mice with deletion of the DNA methylation cofactor Dnmt3l which fail to methylate L1s in the germ line. In males, this results in pachytene arrest (Bourc'his and Bestor 2004). It is also evident that small RNAs play an important role in the suppression of L1 retrotransposition in the mammalian germline (Girard et al. 2006; Watanabe et al. 2006; Yang and Kazazian Jr. 2006; Kuramochi-Miyagawa et al. 2008). Finally the APOBEC3 family of RNA editing proteins have been shown to inhibit L1 activity in human cells (Bogerd et al. 2006; Chen et al. 2006; Muckenfuss et al. 2006; Richardson et al. 2014). However, despite all these defense mechanisms, L1s are still active in some tissues. Low-level L1 expression does occur in the male and female germlines (Branciforte and Martin 1994; Ostertag and Kazazian Jr. 2001); however, little retrotransposition occurs. Instead, most L1 retrotransposition occurs early in embryogenesis leading to somatic mosaicism (Kano et al. 2009). L1 retrotransposition has also been observed in neuronal progenitor cells (Muotri et al. 2005) and in the adult hippocampus (Coufal et al. 2009; Muotri et al. 2009). Estimates for the number of L1 insertions per neuron in human range from less than 0.1 to 80 (Coufal et al. 2009; Baillie et al. 2011; Evrony et al. 2012), with a recent study, using single-cell retrotransposon capture, estimating 13.7 insertions per hippocampal neuron (Upton et al. 2015).
The distribution of L1 repeats in the genome is not uniform: They tend to be found in AT-rich, gene-poor domains (International Human Genome Sequencing Consortium 2001), and they have been shown to be enriched close to monoallelically expressed genes such as olfactory receptor genes (Allen et al. 2003). In human they are almost completely absent from HOX clusters, yet there is a region on the X Chromosome spanning ∼100 kb that contains 89% LINEs (International Human Genome Sequencing Consortium 2001). It is not known whether such dense arrays of L1 repeats perform specific functions such as acting as boundary elements or if they represent repeat “graveyards,” accumulating only in regions where they can be tolerated in the genome. Here, we have identified 66 L1-dense regions of >100 kb within the mouse genome. One of the identified regions is rodent specific and lies within the proximal end of the mouse Dlk1-Dio3 imprinted domain. Because LINEs have previously been shown to be enriched at imprinted regions (Allen et al. 2003), we have deleted this region and asked whether it is a “graveyard” for repeats or if it may have evolved a novel function within the rodent lineage. Here, we assess the consequences of the deletion on regional control of gene expression and imprinting.
Results
Large L1 arrays are associated with tandem gene clusters in the mammalian genome
To determine where in the mouse genome stretches of dense LINE content reside, regions containing >70% LINEs of >100 kb length were cataloged. Sixty-six regions were identified: 50 were autosomal, with 15 on the X Chromosome and 1 on the Y Chromosome (Fig. 1A; Supplemental Table S1). Each region was then analyzed for gene content: 38 regions were intergenic, 4 were intragenic (within known transcripts), 11 were gene-flanking, and 13 were found both within and flanking genes (Supplemental Fig. S1A; Supplemental Table S1). Thirty-four of the LINE-rich regions contain genes; of these, 20 contain at least one gene belonging to a tandemly repeated gene cluster. These tracts of high LINE content are enriched for tandemly repeated olfactory and vomeronasal receptor genes (seven and eight regions, respectively; P < 0.01 when compared with 500 randomly generated genomic regions by binomial test). High LINE repeat content has previously been shown to be associated with random monoallelic genes such as the olfactory and vomeronasal receptors (Allen et al. 2003). Possible reasons for this association include a L1-mediated duplication mechanism in the evolution of these clusters (Younger et al. 2001) or a regulatory or structural role for L1s in monoallelic expression (Allen et al. 2003).
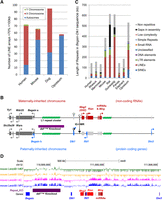
Distribution of LINE-rich arrays in mammalian genomes and identification of a large, intergenic LINE-rich repeat array in the mouse Dlk1-Dio3 imprinted gene cluster. (A) Chromosomal distribution of LINE-rich arrays of >70% LINE over >100 kb in the human, mouse, dog, and opossum genomes. In the mouse genome, LINE-rich arrays of >70% LINE extending over 100 kb are associated with vomeronasal and olfactory receptor genes. (B) The LINE-rich repeat cluster (green) is flanked by the Dlk1-Dio3 imprinted domain. Maternally expressed noncoding transcripts are shown in red, and paternally expressed protein coding genes are shown in blue. On the maternally inherited chromosome, noncoding RNAs Meg3 (Gtl2), Rtl1 anti-sense (AS), Rian, and microRNA-containing Mirg (red) are transcribed, whereas the protein-coding genes (light gray) are repressed. The paternally inherited chromosome expresses protein-coding Begain variant 1b, Dlk1, Rtl1, and Dio3 (blue), and silences the transcription of the noncoding RNAs (light gray). The imprinting control region (IG-DMR) and the Dlk1 DMR are unmethylated on the maternally inherited chromosome (white circles), and hypermethylated/partially methylated, respectively, on the paternally inherited chromosome (black and shady gray circles). Biallelic genes are shown in dark gray, and arrows denote the direction of transcription. (C) The total repeat coverage in the Begain-Dlk1 intergenic region in nine mammalian species illustrates the accumulation of LINE elements in the rodent lineage. Repeat content was ascertained using the RepeatMasker web server (Smit et al. 2015). (D) The mouse Dlk1-Dio3 intergenic LINE array between Begain and Dlk1 (purple bar) coincides with a facultative lamina-associated domain (fLAD) in astrocytes and neural precursor cells (Peric-Hupkes et al. 2010). Image taken from UCSC Genome Browser (Kent et al. 2002).
Because imprinted regions have previously been reported to be enriched for L1 repeats (Hutter et al. 2010) and three of the regions we identified reside in or flank imprinted domains (two in the Snrpn cluster and one in the Dlk1-Dio3 region), we assessed whether there was enrichment for large arrays of LINE elements. For this analysis, LINE arrays were defined as regions of >50 kb in length with >40% of LINE content, which is double the mouse genome average (∼20%). Twenty-one imprinted domains and 1 Mb of flanking sequence on either side were assessed for LINE arrays, but no significant enrichment was found either within or surrounding imprinted gene clusters (Supplemental Fig. S1B).
When the lineage specificity was ascertained for all clustered and unclustered L1 repeats in the mouse genome, the ratio between the number within Eutherian, Rodent, Muridae, and Mus elements is approximately 1:1:1:1 (the number of L1s in each lineage = 203,859; 190,500; 216,000, and 262,981, respectively). However, in the LINE arrays, the numbers are skewed toward the Muridae and Mus specific elements with total numbers of elements within all 66 regions being 372, 747, 2004, and 2800, respectively. These data indicate that the accumulation of L1s in these particular regions is Muridae specific. To ascertain if this is the case, similar LINE-rich arrays were identified in three other species: human, dog, and opossum. Seventy-two regions of >70% LINEs extending over 100 kb were identified in the human genome (Fig. 1A), the majority of which (56) are located on the X Chromosome. Only one autosomal region is conserved between mouse and human: that containing the cytochrome gene cluster. In the dog, 95 regions were identified: 32 autosomal and 63 on the X Chromosome. Eight of the 32 autosomal regions contain olfactory receptor genes. Of the 58 arrays identified in the opossum genome, only two are located on the X. This is in agreement with previous observations that the opossum X is depleted for LINEs (Mikkelsen et al. 2007).
Characterization of LINE-1 repeat array in the Dlk1-Dio3 imprinted domain
We identified 40 intergenic LINE-rich arrays in mouse. Of these, only one is entirely located between two genes falling within a known coordinately regulated gene cluster, hence allowing this region to serve as a paradigm for functional analysis. This is the 170-kb array located between the Begain and Dlk1 genes in the Dlk1-Dio3 imprinted domain. This domain encompasses >1 Mb on mouse Chromosome 12 and contains paternally expressed protein-coding genes (Dlk1, Rtl1, Dio3, and Begain transcript b) and maternally expressed noncoding regulatory RNA transcripts (Meg3 [also known as Gtl2], Rtl1as, and clusters of snoRNAs and miRNAs) (Fig. 1B). Imprinting is regulated via a long-range cis-acting mechanism controlled by a germ line–derived, intergenic, paternally methylated differentially methylated region (IG-DMR) located between Dlk1 and Meg3 (Gtl2) (Lin et al. 2003).
In order to assess the evolutionary context of this region in mammals, comparative sequence analysis was performed between the orthologous domains (start of Begain to the start of Dlk1) in nine mammalian species (Fig. 1C; Supplemental Table S2). In rodents, this region is almost three times as large as those of most nonrodent eutherians, in which the size is similar. The region in the rat and guinea pig is even more expanded than in the mouse (385,493 bp); however, there are a number of gaps in these reference genomes, and their true length is not known. The repeat content for the regions delineated by Begain and Dlk1 was ascertained using RepeatMasker (Fig. 1C; Supplemental Table S2; Smit et al. 2015). All three rodent species show an increase in L1 content compared with other mammals. Further analyses demonstrate that the expansion seen in the mouse domain is due to the accumulation of L1 elements within a small interval (Supplemental Fig. S2A). The closest common evolutionary conserved regions (ECRs), reported in the UCSC Genome Browser, that span the 170-kb repeat array in the mouse are 226,694 bp apart, whereas in human, the distance separating these ECRs is only 2270 bp (Kent et al. 2002; Karolchik et al. 2004). A similar insertion is also present in the rat and guinea pig.
The ∼227 kb mouse insertion contains 92.0% interspersed repeats, the majority of which are L1 elements (67.8%). Lineage-specificity, membership of different families, and their age and ability to transpose were ascertained for all 157 L1 repeats identified within the insertion (Supplemental Table S3). The L1Base 2 database (Penzkofer et al. 2016) and the L1Xplorer program (Penzkofer et al. 2005) were then used to identify full-length L1s in the insertion. Nine putative full-length L1s were identified in the interval in L1Base 2 and one further full-length element by L1Xplorer (Supplemental Fig. S3; Supplemental Table S4), but none were found to contain intact open reading frames. Both Orf1 and Orf2 of all these elements have gaps, frameshifts, or stop codons, and most have mutated poly(A) signals (Supplemental Table S4). Together these data indicate the elements in this region cannot autonomously retrotranspose. Nonetheless, it is conceivable that these elements could still produce RNA transcripts (see below).
The LINE-1-rich repeat array bordering the Dlk1-Dio3 imprinted cluster is nuclear lamina associated
LINE-1s have been shown to be enriched in nuclear lamina-associated domains (LADs) (Meuleman et al. 2013). Analysis of publicly available DamID profiles of LaminB association data confirms that the 227 kb LINE-rich insertion upstream of Dlk1 overlaps almost perfectly with a LAD in ES cell–derived neural precursor cells and astrocytes (Fig. 1D; Peric-Hupkes et al. 2010). This is a facultative LAD (fLAD) that is not present in the progenitor ES cell line or in mouse 3T3 embryonic fibroblasts. A further fLAD is present at the distal end of the imprinted domain running from the maternally expressed Rian gene to the paternally expressed Dio3. To determine whether association with LADs is a feature of all LINE arrays, the previously identified 66 regions of >70% LINE extending over 100 kb were intersected with LADs from the four cell lines, with a minimum of 10 kb overlap. Sixty-three of the 66 high-density L1 regions were found to overlap with a LAD in at least one of the cell lines, and 39 regions were constitutive LADs (cLAD) in all four lines (Supplemental Table S1).
Deletion of 260 kb of the Begain-Dlk1 LINE-1 repeat array
A large intergenic repeat cluster containing tandem repeat and retroelements has previously been reported between the Igf2/H19 and Kcnq1 imprinted domains; it has been proposed that this cluster may act as a target for epigenetic modifications that regulate imprinting (Shirohzu et al. 2004). To investigate whether the recently evolved, long, dense, lamina-associated LINE-1 array in the Dlk1/Dio3 domain might have an impact upon imprinting and gene regulation, it was subjected to targeted deletion by homologous recombination in embryonic stem (ES) cells using vectors from the Mutagenic Insertion and Chromosome Engineering Resource (MICER) (Adams et al. 2004). The targeted interval (Chr 12: 109096083–109354908) is located 27.8 kb downstream from Begain, at its 5′ end and 98.3 kb upstream of Dlk1 at its 3′ end and encompasses the entire 170-kb L1 array identified above (Fig. 1B). Cre-mediated recombination in ES cells produced a deletion of ∼260 kb of genomic sequence (Fig. 2A–F), which was confirmed cytogenetically by FISH (Fig. 2G). Chimeric mice bearing the deletion were generated and germ line transmission obtained: A mutant line (termed delL1rep) was then established on a C57BL/6J background.
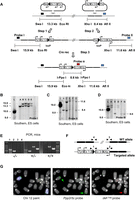
Targeting of 260 kb encompassing the LINE-rich repeat array: molecular and cytogenetic characterization of the delL1rep allele. (A) Complementary MICER vectors (Adams et al. 2004) were consecutively integrated into the genome by homologous recombination in embryonic stem (ES) cells. Ex 1–2 and ex 3–9 correspond to the split Hprt minigene exons in the 5′ and 3′ vectors, respectively. After expression of Cre, the loxP elements recombine excising the intervening sequence and reconstituting the Hprt mini-gene, which results in resistance to hypoxanthine/aminopterin/thymidine (hat). Mini-genes for the coat color markers agouti (Ag) and tyrosinase (Tyr) are shown. The location and direction of promoter elements is indicated by arrows. Probes used for Southern hybridizations are indicated by boxes. Positions of restriction sites and the sizes of WT and targeted alleles are also shown. (B–D) Southern hybridization blots of ES cells at Step 1 (B), Step 2 (C), and Step 3 (D). (L) ladder. (A) wild type; (B,C) 5′ vector-targeted; (D) double-targeted (5′ and 3′ vectors). (E–G) Distinct delL1rep (Cre-recombined) clones. (E) PCR genotyping of tail DNA of representative animals. (F) Schematic illustration of the respective assays. Primers pairs (1–4) used for screening are indicated by arrowheads. (G) Representative images of DNA FISH on metaphase spreads of mutant ES cells with BAC probes complementary to sequence within (delL1rep) and downstream (Ppp2r5c) from the deleted interval. The Ppp2r5c region (1 Mb downstream from the deletion) is identified in both Chromosome 12 homologs (green arrows), whereas the repeat region is seen in one homolog (red arrow) and is absent from the other (white arrowhead).
Both heterozygous (delL1rep/+ and +/delL1rep) and homozygous animals were viable and appeared phenotypically normal. No deviations from the expected Mendelian ratio were observed in any of the crosses, and the average litter size and number of litters from each cross agrees with published data for C57BL/6J (Verley et al. 1967), showing uncompromised viability and reproductive fitness (Supplemental Table S5). Postnatal survival rates measured for periods of up to 52 wk revealed no differences between delL1rep mutants and wild-type animals (Supplemental Table S5). There were also no significant differences in postnatal growth between wild types and delL1rep mutants (Supplemental Fig. S4).
LINE-1 elements from within the deleted repeat interval are transcribed biallelically
To assess LINE-1 transcription from within the repeat interval, two LINEs were shown to amplify from wild-type genomic DNA but not delL1rep homozygous mutants, indicating the amplicons were specific to the deleted region (Supplemental Fig. S5A, left). These two amplicons were in the 3′ UTR of a full-length Lx2 LINE (L1Base 2 ID 11235) (Supplemental Fig. S3) and the 5′ UTR of an L1Md_F2 element (L1Base 2 ID 11087) (Supplemental Fig. S3). Transcription was assessed by RT-PCR in fetal brain and liver, and in adult brain. A Lx2-3′ UTR (11235) transcript was present in all three tissues assayed, and L1Md_F2-5′ UTR (11087) was transcribed in fetal and adult brain only (Supplemental Fig. S5A, right). Despite being full length, the open reading frames for these elements are not intact; thus, the transcripts do not code for functional L1 proteins, and the RNA transcribed has unknown relevance.
The allelic expression of L1 transcripts within the cluster was ascertained using fetal brain cDNA from embryos with uniparental duplications of Chromosome 12 (matUPD12 and patUPD12) (Georgiades et al. 2000). Amplification of the two LINE elements was observed both in mat and patUPD12 cDNA, indicating that the transcripts are not imprinted in fetal brain (Supplemental Fig. S5B) despite the imprinting of genes on either side of the array in this tissue. We cannot exclude that these L1 elements may be imprinted elsewhere in the body or in specific areas in the brain.
Epigenetic control in the region is not affected by the absence of the LINE-rich array
To see if the LINE-rich array contributes to a landscape influencing the regulation of imprinted gene expression, we next examined the impact of delL1rep on imprinting at the Dlk1-Dio3 locus. Wild-type and heterozygous embryos with expressed polymorphisms on Chromosome 12 were obtained from reciprocal crosses between delL1rep heterozygous mutants and Mus musculus castaneus (Cas) or Mus musculus molossinus 12 (Mol 12), a congenic mouse line derived in our laboratory harboring a Mus molossinus Dlk1-Dio3 domain in a C57BL/6J background (Lin et al., 2003). Allele-specific expression of Begain b, Dlk1, Meg3 (Gtl2), Rtl1as, and Dio3 was ascertained by quantitative RT-PCR and pyrosequencing in fetal brain, liver, and placenta (Begain b and Rtl1as were only assessed in brain). All five transcripts were confirmed to be imprinted, retaining their original parent-of-origin specific expression (Supplemental Fig. S6). We also analyzed the allelic expression of Wars, Wdr25, and Begain a in the same tissues; each maintained their biallelic status (Supplemental Fig. S7). Hence deletion of the 170-kb L1 repeat cluster has no effect on imprinting at the region.
The allele-specific expression of all genes of the Dlk1-Dio3 imprinted cluster and that of Begain variant b is controlled by the methylation status of the IG-DMR. This region is unmethylated on the maternal allele, where the protein coding genes are repressed and the noncoding transcripts are expressed; it is hypermethylated on the paternally inherited chromosome resulting in the expression of the protein coding genes and the repression of the noncoding transcripts. The mechanism behind this regulatory action is unknown, but it results in the simultaneous and coordinated expression control of genes spread over 1249 kb. Methylation of the IG-DMR—assessed by pyrosequencing—was unchanged in delL1rep homozygotes, and maternal and paternal heterozygotes compared to wild-type samples (Supplemental Fig. S8). Together, these results indicate that the LINE-rich repetitive DNA tract is not involved in the establishment or maintenance of imprinting within the domain, nor is it required to shield the neighboring biallelic genes from the long-range epigenetic control exerted by the IG-DMR. The IG-DMR, however, appears to be unable to influence the allelic activity of the L1 transcripts within the cluster.
Paternal transmission of delL1rep results in temporal- and tissue-specific disruption of expression levels on the paternal chromosome through loss of an enhancer
To further explore the effects of the repeat deletion at the molecular level, we first evaluated gene expression in wild-type and mutant E16.5 embryos and placentas. Twelve genes flanking the deletion were assayed by real-time quantitative PCR in embryonic brain, liver, and placenta, where expression of the Chromosome 12 imprinted genes is abundant. Maternal transmission of delL1rep did not reveal detectable changes in gene expression in any of the three tissues (Fig. 3A, upper; Supplemental Fig. S9, middle). Normal levels of expression were also observed in liver and placenta in delL1rep mutants after paternal transmission and in homozygous mutants from heterozygote intercrosses, when compared to wild-type littermates (Supplemental Fig. S9). Strikingly, we found that on paternal transmission, delL1rep mutants exhibited significant down-regulation of the paternally expressed transcripts in the brain. Thus, the expression of Begain b, Dlk1, and Dio3 in fetal brain was significantly decreased by 65%, 32%, and 26%, respectively (Fig. 3A, middle). This effect, which was specific to only the imprinted, protein-coding transcripts on the paternal chromosome, was recapitulated in homozygous mutants. delL1rep homozygotes showed a comparable decrease in the expression of Begain b (63%), Dlk1 (28%), and Dio3 (28%) (Fig. 3A, bottom). Neither the biallelic nor the maternally expressed genes were found to have altered levels of expression in brain or in any of the tissues analyzed.
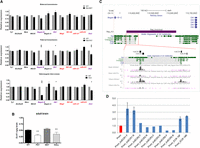
Paternal transmission of delL1rep results in temporal- and tissue-specific disruption of imprinted gene expression on the paternal chromosome. (A) Relative expression of five biallelic genes closest to the deleted interval (black) and of the transcripts within the neighboring imprinted cluster ([blue] paternally expressed; [red] maternally expressed) in fetal (E16.5) brain, as determined by RT-qPCR. (Upper) No significant differences in expression were observed upon maternal transmission of the delL1repallele (n = 12 wt, 14 mut; four litters). (Middle) Paternal inheritance of the delL1repallele elicited a significant down-regulation of all of the paternally expressed genes (n = 13 wt, 13 mut; four litters). (Lower) The effect of delL1repon the expression of Begain b, Dlk1, and Dio3 was closely recapitulated in homozygous mutants (n = 12 wt, 13 mut; six litters). Maternally expressed miR-127 and miR-433 are part of larger transcripts (Rtl1-AS and Mirg, respectively) but have their own promoters and can be independently transcribed (Song and Wang 2008). Data were normalized to Gap3dh expression and are shown relative to WT controls (=1). (*) P < 0.05; (***) P < 0.001 by two-tailed Student's t-test. (B) DLK1 ELISA performed on wild type (WT), paternal transmission of delL1rep (PAT), maternal transmission of delL1rep (MAT), and homozygous adult brains (NULL). DLK1 protein levels in PAT and NULL are significantly lower than wild type: (**) P < 0.01 by ANOVA with Bonferroni's post hoc test. Error bars indicate SEM. (C) The delL1rep region contains two evolutionary conserved regions (ECRs) as identified from the UCSC conservation track. ENCODE data for the enhancer-associated histone modifications H3K4me1 and H3K27ac in whole brain and olfactory bulb are also shown. The purple box denotes the deletion region; black boxes show the position of four conserved subregions that were used for luciferase assays. Image taken from UCSC Genome Browser (Kent et al. 2002). (D) koECRs 1, 2, 3, and 5 were cloned into pGL3-Promoter (Promega) in both orientations to test for enhancer activity. Constructs were transfected into E16.5 neurospheres. Graph shows the expression as measured in luminescence of luciferase relative to Renilla and normalized to the empty pGL3-Promoter vector (Prom). ECR19 was previously shown to have enhancer-like activity in ESCs and was shown to bind EP300 in brain (Visel et al. 2009). Statistics were calculated by the one-way ANOVA (and nonparametric) Friedman test to calculate the overall statistical significance (P-value), followed by Dunn's Multiple Comparison post-test to calculate the statistical significance (P-value) between specific samples. P < 0.05 was deemed as statistical significant. (Prom) pGL3-Promoter empty vector.
In the adult, in line with the results obtained with the embryo, maternal inheritance of the repeat deletion had no effect on the overall levels of gene expression in the mutant adult brain (Supplemental Fig. S10). In contrast, in paternal heterozygotes and homozygotes, Dlk1 showed a significant decrease of 31%–34% (Supplemental Fig. S10). ELISA performed on wild-type, heterozygous, and null adult brains showed a >30% decrease in DLK1 protein levels in the null and paternally inherited deletion but no significant change upon maternal transmission (Fig. 3B; Supplemental Table S6). Taken together, our results show that paternal transmission of delL1rep results in a tissue-specific reduction in expression of the imprinted genes on the paternally inherited chromosome.
Since functional elements within the genome evolve at slower rates than nonfunctional sequence, we scanned the repeat region for noncoding evolutionary conserved regions (ECRs). Genome alignments using the UCSC Genome Browser (Kent et al. 2002; Blanchette et al. 2004) identified two regions of conservation (Fig. 3C). These regions were then compared to whole-genome histone ENCODE ChIP-seq data (The ENCODE Project Consortium 2011) to determine if they overlapped established enhancer-associated histone signatures, such as H3 lysine 27 acetylation (H3K27ac) and H3 lysine 4 mono-methylation (H3K4me1), in neuronal lineage cells. One region, koECR1, was found to overlap with H3K4me1 in whole brain and olfactory bulb. KoECR2 and 3 also showed an H3K4me1 association in olfactory bulb (Fig. 3C).
These putative enhancers were tested for enhancer activity using in vitro luciferase reporter assays in neurosphere cells derived from the cortex of E16.5 brains. Enhancer-like activity (3.2- to 3.5-fold greater than empty vector) was observed from the koECR1 region in both orientations (Fig. 3D) consistent with positive control constructs generated using a known neural enhancer. The other three regions tested showed no enhancer-like activity in these cells, although they may have regulatory roles in other areas of the brain or tissues. However, no enhancer-like activity was observed from any of the regions in NIH-3T3 fibroblasts, mouse ES cells (E14Tg2a), or neural stem cells derived from 46C ES cell line (Supplemental Fig. S11; Ying et al. 2003). Dlk1 is expressed in fetal neurospheres (Ferrón et al. 2011), whereas Begain is not, suggesting koECR1 may function to enhance Dlk1 expression in these cells.
Together, our data indicate that the large dense array of LINE-1 elements residing within a coordinately controlled, epigenetically regulated imprinted domain, does not contribute to a landscape required for the genomic imprinting or transcriptional regulation of adjacent genes across a 1.25-Mb region.
Discussion
Dense arrays of LINEs accumulate in regions of the genome where they can be tolerated
Nearly half of the mammalian genome is composed of repetitive sequences, yet their influence on genome function is only now beginning to be considered, predominantly due to previous technical challenges associated with working with repetitive elements. Here, we characterized a class of large LINE repeat arrays and asked whether one such cluster of L1 repeats might function as a modulator of local control at a well-characterized imprinted cluster on mouse Chromosome 12.
It was previously suggested that coordinately regulated gene clusters are devoid of repeats because they are under the control of cis-acting elements that cannot tolerate their presence, for example, Hox clusters (International Human Genome Sequencing Consortium 2001; Simons et al. 2006). In agreement with this is the fact that the majority of the large LINE arrays identified were found in intergenic regions (38/66).
Dense LINE arrays were shown to be enriched in regions of tandemly duplicated olfactory and vomeronasal receptor genes in mice. The association between LINEs and random monoallelic genes, such as olfactory receptors, has been long established, and it is suggested that these LINEs may facilitate monoallelic expression (Allen et al. 2003). It has also been claimed that L1-mediated mechanisms may be responsible for the duplication of these genes (Younger et al. 2001). The large numbers of LINEs that flank these genes may be tolerated because they provide a mechanism by which the gene repertoire can be increased, allowing the animals to adapt more quickly to changing environments or indeed because they sit harmlessly outside regions required for the regulated allelic exclusion associated with such domains.
Similarly, imprinted gene clusters are coordinately regulated, reflecting both their cis-acting parental origin-specific imprinting control and their coregulation in common developmental or physiological pathways (Ferguson-Smith 2011; Charalambous et al. 2012). The existence of a large L1 repeat domain at the proximal end of the Dlk1-Dio3 imprinted domain raised questions as to whether it was performing some sort of topological role, or alternatively that the region provided a safe locale for a “graveyard” for such elements.
We deleted the intergenic LINE-rich array in the mouse Dlk1-Dio3 domain to ascertain the implications of the presence of a large repeat region on a large gene cluster, whose monoallelic expression is governed by a single cis-acting element. This repeat array was found to be rodent specific, and its existence within the Dlk1-Dio3 domain indicates that the gene cluster is able to tolerate such an accumulation without any detrimental effects. Clearly, the cis-acting function of the IG-DMR is unaffected by the presence of a repeat insertion since imprinting in the domain occurs in eutherian mammals with and without the LINE array (Smit et al. 2005; Hagan et al. 2009). Moreover, deletion of the LINE-rich region does not alter the imprinting status of any of the genes in the region. One possible function of such LINE-rich regions in the genome could be as boundary elements between differentially regulated genomic regions. This is not the case for the murine Dlk1-Dio3 LINE array since the IG-DMR is still able to exert its influence on the proximal Begain b gene to confer its paternal origin-specific expression in mouse despite the presence of the LINE-rich insertion.
LINE-1s have previously been found to be enriched in the regions surrounding imprinted and random monoallelic genes (Allen et al. 2003). This finding led to speculation that LINEs in autosomal monoallelically expressed regions were acting as “way stations” to aid the spreading of heterochromatin via a mechanism similar to that hypothesized for Xist in X inactivation at the time (Lyon 1998). However, this idea has been recently thrown into doubt because it is now believed that marsupials, who have LINE-poor X Chromosomes, also use a noncoding RNA (Rsx) to achieve XCI (Grant et al. 2012). In mice, the Dlk1-Dio3 domain extends from Begain through to Dio3 (Fig. 1B), and this is the same as in sheep (Smit et al. 2005), which do not contain a large LINE-1 cluster. The lack of conservation of this LINE-1 array indicates that it does not have a key role in conferring a particular chromatin state required for long-range control. Furthermore, the deletion of the mouse repeat has no effect on the imprinting status of any of the genes in the region or the methylation status of the IG-DMR, indicating that the repeat array has gained no role in the control of monoallelic gene expression in this imprinted cluster under normal developmental or physiological conditions.
Gain of function in gene regulation and chromosome architecture
Inheritance of delL1rep from either parent causes no gross phenotypic effects; however, there are changes to expression levels of paternally expressed genes within the region. These expression changes were only observed in embryonic and adult brain and suggest that any possible phenotype in the knockout mice may be behavioral. One likely explanation for reduction in paternally expressed transcripts upon paternal transmission is that brain-specific enhancers could have been removed along with the deletion. We identified a putative brain-specific enhancer on the basis of conservation and of an enhancer-associated epigenetic signature; moreover this element exhibited cell-type–specific enhancer activity in reporter assays in ex vivo neural cells. Alternatively, both LTRs and SINEs have previously been demonstrated to exhibit enhancer functions in some contexts (Hambor et al. 1993; Long et al. 1998), so it is also possible that the repeat elements themselves may have gained an enhancer function. This has previously been observed at the human lipoprotein, Lp(a) locus (Wade et al. 1997; Yang et al. 1998).
The delL1rep mutant displays no obvious physical phenotype. However, as L1 expression in the region was only observed in brain and changes to expression levels of local imprinted genes was only observed in neural tissues, it would be interesting to see whether there may be behavioral phenotypes in mutant mice, including whether the mutant mice react differently in stress conditions.
DamID data indicate that the repeat array is nuclear lamina-associated in neural precursor cells (Peric-Hupkes et al. 2010), and that these LINEs may contribute to the tethering of particular genomic regions to the nuclear periphery. In some contexts, nuclear lamina association has previously been shown to lead to reduction in gene expression (Finlan et al. 2008). It has recently been shown that in ES cells, the maternally inherited Dlk1-Dio3 chromosome, which is highly expressed for Meg3 (Gtl2), is preferentially located internally in the nucleus, and the paternally inherited locus from which little expression is observed is located peripherally (Kota et al. 2014). Perturbation of maternal gene expression by knocking down expression from the IG-DMR resulted in more peripheral localization, indicating that lack of expression itself may target a region to the periphery (Kota et al. 2014). Here, we show that upon maternal transmission of delL1rep, no significant changes in Meg3 (Gtl2) expression or any other genes in the domain was observed in any tissues analyzed. Furthermore, upon paternal transmission of delL1rep, the expression of both Dlk1 and Begain b is actually reduced in the brain, indicating that the lamina association in the region is not repressing gene expression, again supporting a causal role for a putative neural enhancer within the deleted interval. It is also of note that the Dlk1-Dio3 domain contains a second longer neuronal lineage LAD, suggesting that the region might remain tethered to the lamina even after deletion of the L1-rich array.
Together, our findings suggest that a 170-kb LINE-1 array, located between two paternally expressed imprinted genes within the Dlk1-Dio3 imprinted domain, reflects the tolerance of the site for repeat insertion rather than an important genomic or epigenetic function.
Methods
In silico analysis
For the whole-genome analyses, the following genome builds were used: Mouse, mm10; Human, hg38; Dog, canFam3; Opossum, monDom5. Whole-genome repeat annotations were downloaded from RepeatMasker (Smit et al. 2015). These annotations were uploaded into the SeqMonk program (developed by Simon Andrews at the Babraham Institute, https://www.bioinformatics.babraham.ac.uk/projects/seqmonk/). The percentage coverage by LINEs was ascertained for running windows of 100 kb with 10-kb steps. Next for each domain, the percentage covered by >70% LINEs >100 kb was ascertained. The genes in each region were obtained using the UCSC Table Browser (Karolchik et al. 2004). The coordinates of known LADs were taken from Peric-Hupkes et al. (2010) and intersected with the 68 regions with >70% LINE >100 kb using the Join utility in Galaxy (Giardine et al. 2005; Blankenberg et al. 2010; Goecks et al. 2010) for a minimum of 10 kb overlap. For the imprinted region analysis, running windows of 50 kb with 10-kb steps were used and regions of >40% LINE >50 kb were identified. Random sequences were generated using BEDTools (Quinlan and Hall 2010). Wilcoxon matched pair test was performed using GraphPad Prism version 5.00 for Windows, GraphPad Software (https://www.graphpad.com).
For Begain-Dlk1, repeat analysis in genomic coordinates and sequence were taken from the UCSC Genome Browser (http://genome.ucsc.edu/) (Kent et al. 2002). Repeat content was ascertained using RepeatMasker Web Server (Smit et al. 2015). Information on the lineage specificity of the LINE repeats was obtained from RepeatMasker (http://www.repeatmasker.org/) (Smit et al. 2015) and Repbase (http://www.girinst.org/repbase/).
Gene targeting in ES cells
The targeting vectors used to generate the allele with the deletion (delL1rep) were obtained from the Mutagenic Insertion and Chromosome Engineering Resource (MICER, http://www.sanger.ac.uk/resources/mouse/micer/) and have been described in detail previously (Adams et al. 2004). Ready-made MICER vectors MHPN49e02 (Chr 12: 110,325,911–110,334,290, NCBIM37) and MHPP67j17 (Chr 12: 110,593,112–110,597,765) were customized by the introduction of a I-PpoI restriction site in the tyrosinase 5′ flank region (878/880, MHPN) and in the vector backbone (6245/6249, MHPP). After digestion with PacI and SbfI, respectively, a double-stranded DNA oligonucleotide containing the I-PpoI restriction site with matching overhangs for each of the enzymes was ligated into the vectors by common procedures. I-PpoI (Promega) is a yeast-specific intron-encoded endonuclease from Physarum polycephalum that does not cut mammalian DNA or any of the commonly used plasmids or vectors.
Feeder-independent E14Tg2a ES cells cultured in KO-DMEM (Invitrogen) supplemented with 15% FBS (Hyclone), 1% beta-Mercaptoethanol, 1% L-glutamine/Pen-Strep solution, and ESGRO (LIF, Chemicon) supplement at 1000 units/mL, were electroporated with 50 µg of SmaI-linearized MHPN vector and selected with G418 (180 µg/mL) (Sigma). Targeting was determined by Southern blotting following digestion of genomic DNA with SwaI and EcoRI. Positive clones were subsequently electroporated with 50 µg of KpnI-linearized MHPP vector and selected with puromycin (3 µg/mL) (Sigma). Targeting was ascertained by Southern blotting following digestion of genomic DNA with XhoI and Afl II. Two double-targeted clones were transfected transiently with the Cre-expressing vector pOG231 (Addgene plasmid 17736) (O'Gorman et al. 1991) by electroporation and selected with hypoxanthine/aminopterin/thymidine (HAT) (Gibco). HAT-resistant colonies were genotyped by Southern blotting as described above and following double digestion with NdeI and I-PpoI. Successful deletion was confirmed by the presence of an 8.6-kb fragment resulting from the I-Ppo I digestion within the two neighboring targeting vectors, which demonstrated the removal of the intervening sequence. Euploidy was verified for five ES cell clones carrying the delL1rep allele by chromosome counting of 50 metaphase spreads per clone.
DNA fluorescence in situ hybridization (FISH)
Metaphase spreads of ES cells carrying the delL1rep allele were hybridized with two BAC clones from the 129S7/AB2.2 BAC repository (Sanger Institute). bMQ-177c10 (110,338,144–110,464,879) is located within the deleted interval; bMQ-98C5 (111,624,966–111,780,688) partially overlaps Ppp2r5c.
Mouse metaphase preparations were prepared from primary fibroblast cultures, grown in Dulbecco's modification of minimal essential medium (Gibco), enriched with 10% fetal bovine serum (Gibco), penicillin (100 units/mL) and streptomycin (100 mg/mL). Chromosome preparations were made following standard procedures that included 1 h of colcemid treatment (0.05 mg/mL) followed by 8 min of hypotonic treatment in 0.075 M KCl and fixation in 3:1 methanol/glacial acetic acid. Ten microliter metaphase preparation was dropped onto glass slides followed by 10 µL 3:1 methanol:acetic acid fixative in a 52°C water bath to achieve sufficient spreading. To remove excess cytoplasm, slides were dehydrated in 100% ethanol for 5 min and treated with 0.0025% pepsin in 10 mM HCl for 2 min. Slides were then rinsed in 2× SSC twice for 5 min followed by a wash with deionized water. Slides were then passed through an ethanol series of 70%, 90%, and 100% ethanol. Slides were then air dried and baked for 1 h at 65°C. BAC DNA was purified using the Sigma PhasePrep BAC DNA Kit (NA0100-1KT) and labeled with biotin or dUTP-Cy3 by a standard Nick Translation procedure using 1 µg of purified BAC DNA. Chromosome 12 Paint (Cambio) and approximately 150 ng BAC probe and 1 µg Mouse Hybloc DNA (Applied Genetics Laboratory MHB-0.5) were made up to 12 µL with hybridization buffer (50% deionized formamide, 10% dextran sulfate, 2×SSC, 0.5 M phosphate buffer, pH 7.3, and 1×Denhardt's solution) and denatured for 10 min at 75°C and preannealed for 30 min at 37°C. Metaphases were denatured for 1 min 40 sec at 67°C in 70% formamide in 2×SSC. Immediately after denaturing, slides were placed in ice cold 70% ethanol for 4 min. The ethanol series was repeated. BAC probes were applied to the slide and covered with a coverslip, sealed with fixogum, and incubated for two nights at 37°C in a humid chamber. Post-hybridization washes were performed at 42°C in 50% formamide, 2×SSC for 5 min twice, 2×SSC for 5 min twice, and 4×T (0.05% Triton X-100 in 4×SSC) for 4 min. Biotin-labeled probes were visualized using 1:500 Streptavidin-Cy3 (GE Healthcare) which was applied to the slides in 4×T covered with parafilm and incubated for 30 min at 37°C. Slides were rinsed in 4×T for 3 min, twice. Excess fluid was removed, and slides were mounted in DAPI II and sealed with nail varnish. Images were captured using a wide-field Leica DMRXA fluorescence microscope and a ×100 oil immersion objective with a numerical aperture of 1.4.
Generation of chimeric mice and germ line transmission of the delL1rep allele
All experimental procedures with live animals were conducted in accordance with UK Government Home Office Licensing regulations. CD-1 embryos were recovered at the four-cell stage from the oviducts of fertilized CD-1 females and cultured in KSOM medium up to the uncompacted eight-cell stage. Clumps of 6–15 mutant ES cells were placed in purpose-built wells covered with ES cell medium, in close contact with host morulae, which were allowed to develop to the blastocyst stage. Blastocysts were implanted into the uteri of pseudopregnant CD-1 females and the born chimeric animals were readily identified by the coat color, due to the expression of the Agouti minigene in the white CD-1 background. Chimeric males were mated to albino C57BL/6 females (Charles River) for germ line transmission of the delL1rep allele. Heterozygous mutant progeny were easily discriminated by the coat color (white-bellied chinchilla), the genotypes of which were confirmed by PCR and Southern blotting of genomic DNA isolated from tail biopsies at weaning (details below). A mutant line was then established by backcrossing the delL1rep allele onto the C57BL/6J background over 10 generations.
PCR and Southern blot genotyping
Genomic DNA isolated from tail biopsies by phenol/chloroform and from ear fragments with the Tissue-to-PCR kit (Fermentas) was PCR amplified using four sets of primers as follows: Pair 1, Fw CAAGCACTGGCTATGCATGT, Rv TGAACCCAGGAGGTTGAGAC, 60°C annealing, 40 cycles, 2 min 72°C extension; pair 2, Fw TTTAGAGCTTGACGGGGAAA, Rv CAAGAGCTCTGGGGTACTGG, 55°C annealing, 35 cycles, 30 sec 72°C extension; pair 3, Fw AGCGGATCCATCCTTTGAGT, Rv CAAGAGCTCTGGGGTACTGG, 65°C annealing, 34 cycles, 1 min 72°C extension; pair 4, Fw ACCAACCTCTCTGTGGCTGTG, Rv TCCACACGAGGACACCATGCC, 65°C annealing, 32 cycles, 30 sec 72°C extension. Primer pair 1 produces an amplicon of 589 bp spanning the recombination breakpoint on the mutant allele; pair 2 amplifies a fragment of 494 bp that spans the boundary between the MHPP backbone vector and the 3′ genomic arm on the mutant allele; pairs 3 and 4 amplify fragments of 832 and 647 bp, respectively, spanning genomic sequence of the wild-type allele that includes the 5′ and 3′ arms used for homologous recombination.
For Southern blot genotyping of ES cells and mice, genomic DNA was isolated by phenol/chloroform and digested as described. Probes for Southern hybridization were generated by PCR amplification using the following primers: 5′ flanking probe I, Fw TTAGGGGAGGGAAGGCTTTA, Rv CGGCATCGCTTATTGAAGAT, 60°C annealing, 311 bp; 3′ flanking probe II, AAGATAAGGACATGTTAGCC, Rv TCTGTGGAGCTTAATATCTG, 60°C annealing, 313 bp; Hprt internal probe III, Fw CTGGGTCAAGGGGAAAGAGT, Rv GAGGTGAGGTGGGAAAATCA, 60°C annealing, 704 bp. Probe I detects fragments of 13,331 and 15,875 bp from the wild-type and mutant allele, respectively, upon digestion with SwaI and EcoRI; probe II identifies fragments of 8441 and 11,648 bp from the wild-type and mutant allele, respectively, following digestion with XhoI and AflII; probe III reveals a fragment of 8653 bp from the mutant allele only, upon digestion with I-PpoI (Fig. 2).
Tissue collection, RNA extraction, and cDNA synthesis
Tissues from embryos and adult animals were snap-frozen in liquid nitrogen following quick dissection after uterine collection or humane sacrifice, respectively. Total RNA was isolated with TRIzol (Invitrogen) according to the manufacturer's instructions, quantified by spectrophotometry, and quality controlled by gel electrophoresis to assess the integrity of the ribosomal RNA and absence of degradation products.
Two micrograms of total RNA were treated with 1 unit of RQ1 DNase (Promega) to remove genomic DNA contaminants and were reverse transcribed using the RevertAid H Minus cDNA synthesis kit (Fermentas) with random primers. The resulting cDNA was diluted 1:20, aliquoted, and stored at −20°C for subsequent use.
To discriminate between Rtl1 and Rtl1 anti-sense (Rtl1-AS) transcripts, a strategy was adopted whereby M13-R and Mp-F “adaptor” sequences, which are not found in the mouse genome, were tethered to Rtl1-AS-specific primers. Strand-specific reverse transcription was then performed as described above with adaptor-containing primers 5′-TGTCAGGCAACCGTATTCACCGGAGTCCAGCGATGGTTCAC-3′, where the nucleotides in bold are the gene-specific sequence.
For the synthesis of miR-127 and miR-433 cDNA, reverse transcription was carried out in 6-µL reactions made up with 1 µg total RNA, 1× RT reaction buffer (Fermentas), 1 mM dNTPs (each), 1× RT microRNA primer (ABI), 100 units RevertAid H-minus reverse transcriptase (Fermentas), and 20 units RiboLock RNase inhibitor (Fermentas). The microRNA primers were used in multiplex in the RT reaction, which included the control small RNA snoRNA 202. The reaction mixture was covered with 10 µL mineral oil to prevent evaporation, incubated for 30 min at 16°C, for 30 min at 42°C, and for 5 min at 85°C to inactivate the enzymes. The cDNA was extracted following the addition of 44 µL distilled water and 20 µL chloroform, vigorous vortexing, and centrifugation at 1000 rcf.
Analysis of allelic expression
Wild-type and heterozygous embryos with expressed polymorphisms at Chromosome 12 were obtained from reciprocal crosses between delL1rep heterozygous mutants (C57BL/6 background) and Mus musculus castaneus (strain CAST/EiJ, abbreviated Cast) (Jackson laboratories) or Mus musculus molossinus 12 (strain B6.MOLF12A, abbreviated Mol 12) (Supplemental Table S7). The B6.MOLF12A congenic strain carries an 8 cM region of Chromosome 12 from the Mus musculus molossinus strain MOLF/Ei on a C57BL/6 genetic background. Allelic expression of eight transcripts was assessed by PCR amplification of fetal (E16.5) brain, liver, and placenta cDNA with biotinylated primers, followed by pyrosequencing. Primer sequences for PCR amplification of the analyzed polymorphisms and amplification conditions are shown in Supplemental Table S7. Pyrosequencing was carried out with up to 15 µL of PCR product. The biotinylated strand was purified using Strepdavidin Sepharose High Performance Beads (GE Healthcare), washed, denatured, and annealed to the pyrosequencing primer (0.3 µM) using PyroMark reagents (Qiagen), as described (Tost and Gut 2007). Pyrosequencing was performed on a PyroMark MD pyrosequencer (Biotage) using PyroMark Gold Qp6 SQA reagents (Roche), and the data were analyzed with the PyroMark MD version 1.0 software (Biotage).
Gene expression studies
Real-time quantitative PCR with SYBR Green was performed to measure the levels of gene expression in fetal brain, liver, and placenta, and in adult brain. Target gene expression was normalized to the expression of Gap3dh for brain, and of beta-2-microglobulin for liver and placenta. Thermocycling was performed on a DNA engine Opticon 2 thermocycler (MJ Research) in 12.5-µL reactions with SensiMix (Quantace), 400 nM of primers (except where noted) and 3 µL of cDNA synthesized and diluted as described above, in duplicate. A standard curve made up of doubling dilutions of pooled cDNA from the samples being assayed was run on each plate, and quantification was performed relative to the standard curve. All PCR products were subsequently checked for specificity by gel electrophoresis. Primer sequences and annealing temperatures are shown in Supplemental Table S8. All primers amplified with an estimated efficiency of 80%–110%.
The expression levels of miR-127 and miR-433 were measured by TaqMan assays. Ten-microliter reactions were made up according to manufacturer's instructions but scaled down: 1× TaqMan Universal PCR mastermix (ABI), 1× microRNAqPCR primer (ABI), and 2 µL diluted cDNA, synthesized as described. Thermocycling was performed on an Applied Biosystems 7500 Fast Real-Time PCR system with 10 min at 95°C and 40 cycles for 15 sec at 95°C and for 60 sec at 60°C. Data were normalized to snoRNA202 expression and quantified using the ΔΔCt method.
DLK1 Protein quantification by ELISA
Adult brains from wild-type, heterozygous, and homozygous delL1rep animals were homogenized in 1 mL RIPA buffer, and 25 µL were assayed as previously described (Charalambous et al. 2012).
Methylation analyses
Sodium bisulphite mutagenesis was carried out on 1 µg gDNA per sample using the two-step conversion protocol of the Imprint DNA Modification Kit (Sigma). Two samples with no template were included to confirm contamination had not occurred during bisulphite treatment. Five microliters of bisulphite-treated sample were PCR amplified with biotinylated primers (Forward 5′-GTGGTTTGTTATGGGTAAGTTT, Reverse 5′-Btn-CCCTTCCCTCACTCCAAAAATTAA, 54°C annealing for 36 cycles) and quality controlled by gel electrophoresis of 5 µL of each PCR reaction. A volume of up to 15 µL PCR product was then used for pyrosequencing, which was carried out as previously described (Tost and Gut 2007). The biotinylated strand was purified using Strepdavidin Sepharose High Performance Beads (GE Healthcare), washed, denatured, and annealed to the pyrosequencing primer (0.3 µM 5′-TGGTTTATTGTATATAATGT) using PyroMark reagents (Qiagen). Pyrosequencing was performed on a PyroMark MD pyrosequencer (Biotage) using PyroMark Gold Qp6 SQA reagents (Roche), and the data were analyzed with the Pyro Q-CpG 1.0.9 software (Biotage).
LINE-1 transcription studies
Primers for amplification of LINE-1 elements specific to the deleted interval on Chromosome 12 were designed by taking the alignment between the L1 of interest and the L1 consensus sequence from RepeatMasker (Smit et al. 2015) and identifying regions of low conservation. Primer pairs were designed to the low conservation regions using Primer3 (Koressaar and Remm 2007; Untergasser et al. 2012) then tested for successful amplification and specificity using genomic DNA of wild-type and delL1rep homozygous mice, respectively. Primer sequences and PCR conditions are as shown in Supplemental Table S9.
Transcription was examined by real-time PCR amplification of 3 µL cDNA from fetal brain and liver, and adult brain (prepared and diluted as described above), in a 12.5-µL reaction containing SensiMix (Quantace) and 200 nM primer. Reactions were carried out on a DNA engine Opticon 2 thermocycler (MJ Research) and subsequently checked by gel by electrophoresis. To assess the allele-specific mode of transcription, cDNA from fetal brain from embryos with uniparental duplications of Chromosome 12 (matUPD12 and patUPD12) (Georgiades et al. 2000) was used and amplified as above.
In vitro luciferase assays
DNA fragments were generated by PCR from mouse gDNA using primers designed with either two SacI or two NheI restriction sites at each end in order to clone both in sense and antisense orientation. Each 25-µL reaction contained 1× PCR Buffer (KOD Hot Start, Novagen), 300 µM dNTPs, 1 mM MgSO4, 0.5 units Hot Start KOD polymerase, 0.6 µM of each primer, and 50 ng of template. The cycling parameters were as follows: for 2 min at 94°C, 35 cycles for 15 seconds at 94°C, annealing temperature (specific for each primer) for 30 sec, 72°C for 1 min/kb of expected product, and a 5 min extension cycle at 72°C. PCR products were digested with appropriate restriction enzyme and cloned into pGL3 Promoter (Promega) in both orientations using standard molecular techniques. PCR primers and conditions are as shown in Supplemental Table S10.
Transfections were performed by electroporation (1500 V, 20 msec, 1 pulse), and 0.3 µg luciferase construct was cotransfected with 0.1 µg pSV40PK Renilla (Promega) into 2 × 105 neurospheres derived from E16.5 brains as described (Ferrón et al. 2007). Neurospheres were cultured in RHB basal media (Takara) supplemented with N2, Gentamycin, BSA, EGF, and FGF. A positive pGL3 Control vector (Promega) containing both the SV40 promoter and enhancer was transfected to test the assay conditions, while the pSV40PK Renilla vector alone demonstrated no firefly luciferase activity and acted as an internal control. Each construct was tested in triplicate per plate.
For mouse embryonic fibroblast cell line (NIH-3T3) and mouse embryonic stem cell line (E14Tg2a), cells were cultured under standard/normal tissue culture conditions. On the day prior to transfection, cells were plated into 48-well plates at a concentration of 1–2 × 104 cells per well, and 0.5 µg plasmid DNA was added to serum free media with 1 unit of transfection reagent TurboFect (Fermentas) in a 50-µL total reaction. The reaction was incubated at room temperature for 15–20 min then added dropwise to the media in the well. The media was changed 6 h after transfection, and the reaction was left for 48 h (for ES cells, media was changed daily). For neural stem cells derived from a mouse embryonic stem cell line, 46C (Ying et al. 2003) cells were cultured in RHB A complete media (StemCells) supplemented with EGF and FGF. 1.5–3 × 104 cells per well were seeded into 48 well plates. After 24 h, 0.5 µg DNA was added to 25 µL diluent and 1.6 µL Nanofectin (PAA) to 25 µL diluent, then combined. This reaction was then added dropwise to the media in each well. The media was changed 3–4 h after transfection.
After 48 h, cells were tested for their luciferase activity using the Dual-Luciferase Reporter (DLR) Assay System (Promega) according to the manufacturer's protocol. Each cell lysate was measured in duplicate on a single channel TD20/20 Luminometer (Turner Designs). The luminometer was programmed with a 2-sec pre-read delay, followed by a 10-sec measurement period. The firefly luciferase values were normalized to the Renilla values, and then each test construct was normalized to pGL3-Promoter. Statistics for the in vitro luciferase reporter assays were calculated by the one-way ANOVA (and nonparametric) Friedman test to calculate the overall statistical significance (P-value), followed by Dunn's Multiple Comparison post-test to the empty vector to calculate the statistical significance (P-value) between specific samples. P < 0.05 was deemed as statistically significant.
Statistical analyses
All statistical tests were performed using the GraphPad Prism Software Version 5.00 for Windows (GraphPad Software; https://www.graphpad.com) or R (R Core Team 2015) and are indicated in the respective section of the text or in figure legends. Figures show the mean, and error bars represent the standard error of the mean. Significance values and the number of samples analyzed (n) are indicated in the respective figure legends. P < 0.05 was deemed as statistically significant.
Acknowledgments
We thank members of the Ferguson-Smith laboratory for helpful discussions. We are particularly grateful to Mitsuteru Ito and Kazuki Yamasawa for advice and technical help with pyrosequencing, and to Elizabeth Radford, Joseph Gardner, and Dionne Gray for invaluable technical assistance. The work was funded by grants from the Medical Research Council (MR/J001597/1), Wellcome Trust (WT086838; WT095606RR), and Isaac Newton Trust. M.L.S. was the recipient of a postdoctoral fellowship (SFRH/BPD/29806/2006) from Fundação para a Ciência e Tecnologia, Portugal.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.221366.117.
-
Freely available online through the Genome Research Open Access option.
- Received February 4, 2017.
- Accepted January 10, 2018.
This article, published in Genome Research, is available under a Creative Commons License (Attribution 4.0 International), as described at http://creativecommons.org/licenses/by/4.0/.








