
Human cardiac cis-regulatory elements, their cognate transcription factors, and regulatory DNA sequence variants
- Dongwon Lee1,4,
- Ashish Kapoor1,5,
- Alexias Safi2,
- Lingyun Song2,
- Marc K. Halushka3,
- Gregory E. Crawford2 and
- Aravinda Chakravarti1,4
- 1McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, Maryland 21205, USA;
- 2Department of Pediatrics and Center for Genomic and Computational Biology, Duke University Medical Center, Durham, North Carolina 27708, USA;
- 3Department of Pathology, Johns Hopkins University School of Medicine, Baltimore, Maryland 21205, USA
Abstract
Cis-regulatory elements (CRE), short DNA sequences through which transcription factors (TFs) exert regulatory control on gene expression, are postulated to be the major sites of causal sequence variation underlying the genetics of complex traits and diseases. We present integrative analyses, combining high-throughput genomic and epigenomic data with sequence-based computations, to identify the causal transcriptional components in a given tissue. We use data on adult human hearts to demonstrate that (1) sequence-based predictions detect numerous, active, tissue-specific CREs missed by experimental observations, (2) learned sequence features identify the cognate TFs, (3) CRE variants are specifically associated with cardiac gene expression, and (4) a significant fraction of the heritability of exemplar cardiac traits (QT interval, blood pressure, pulse rate) is attributable to these variants. This general systems approach can thus identify candidate causal variants and the components of gene regulatory networks (GRN) to enable understanding of the mechanisms of complex disorders on a tissue- or cell-type basis.
The majority of sequence variation affecting inter-individual complex disease risk is common and noncoding, in contrast to the rare coding variation underlying Mendelian disorders (Chakravarti and Turner 2016). These findings are consistent with the intense natural selection on coding sequence variation and the much weaker selection on noncoding variation (Asthana et al. 2007). Most complex traits arise from multigenic effects with identified risk alleles having small effects so that no individual effect is either necessary or sufficient (Chakravarti and Turner 2016). Thus, there must be considerable redundancy in noncoding functions. The best candidates for these noncoding functions are cis-regulatory elements (CREs) of gene expression because they are the primary agents of regulatory control and affect disease risk through altered gene expression (Davidson 2010; Maurano et al. 2012; Phillips-Cremins and Corces 2013; Chatterjee et al. 2016).
Understanding the regulatory genomic architecture is, therefore, important for elucidating the etiologies of complex disorders, particularly at the tissue- and cell-type levels. This involves identifying the numbers of transcription factors (TFs) and enhancers engaged, their genomic distribution, and the feedback mechanisms required for expression control of each gene, namely, the components of its gene regulatory network (GRN) (Davidson 2010). Additionally, we need to quantify the effect of sequence variation within the GRN on gene expression. These questions can now be answered using high-throughput ChIP-seq (Barski et al. 2007; Johnson et al. 2007; Robertson et al. 2007), DNase-seq (Boyle et al. 2008; John et al. 2011), and ATAC-seq (Buenrostro et al. 2013) experimental data from the NIH ENCODE (The ENCODE Project Consortium 2012) and Roadmap Epigenomics Projects (Roadmap Epigenomics Consortium et al. 2015) on many cell types, tissues, and developmental times and states, in conjunction with human sequence variation (the 1000 Genomes Project; 1KGP) (The 1000 Genomes Project Consortium 2015) and its effects on gene expression across human tissues (the Genotype-Tissue Expression Project; GTEx) (The GTEx Consortium 2015). The fulcrum of a GRN are its enhancers (CREs); however, their complete experimental detection is not possible because CRE activity is affected by many factors (Misteli 2001; Degner et al. 2012; He et al. 2014): (1) Functional strength is variable across enhancers; (2) activity is stochastic across cells; (3) detected activity varies by experimental protocol and sample; and, (4) resolution of a CRE's genomic location from current data is approximate. In principle, ChIP-seq data are superior, but the general lack of TF-specific antibodies is a major roadblock to this approach. We solve these major impediments instead by using a generalizable genome sequence-based machine learning approach (Lee et al. 2011, 2015; Ghandi et al. 2014). This method learns the short genome sequence features that maximally discriminate sequences underlying CREs from random genome sequences. The learned models are then used to identify additional CREs with similar sequence features, including combinations of transcription factor binding sites that failed detection through experiments.
We focus here on identifying the GRN ‘parts list’ of the adult human heart and the effects of CRE genetic variation on an exemplary cardiac trait, the electrocardiographic QT interval (QTi), causal to sudden cardiac death and other conduction disorders (Tomaselli et al. 1994). We show the generalizability of our results by using these cardiac enhancers to explain phenotypic variation in systolic/diastolic blood pressure and pulse rate but not BMI. We focus on the heart because of the extensive genome-wide association study (GWAS) literature implicating CRE variation in many cardiovascular diseases and phenotypes (Arking et al. 2014; Kapoor et al. 2014; The CARDIoGRAMplusC4D Consortium 2015; Eppinga et al. 2016) but also because the QTi is likely affected by the cardiac system alone. A number of human cardiac CRE maps exist (Narlikar et al. 2010; May et al. 2012), with a recent study identifying distal heart enhancers based on H3K27ac and EP300/CREBBP profiles (Dickel et al. 2016). At least one study has also used DNase-seq and histone modification ChIP-seq data for heart tissues to identify functional regulatory variants associated with electrocardiogram traits (Wang et al. 2016). Our study improves on these maps by comprehensive identification of CREs. In addition, we identify their corresponding TFs and analyze the effects of genetic variants within these heart CREs for heart-related traits.
Results
Experimental detection of cardiac CREs is incomplete
A schematic overview of our approach is shown in Supplemental Figure S1. We first performed DNase-seq to identify open chromatin regions through DNase I hypersensitive sites (DHSs) as potential CREs in two adult human hearts (left ventricles) together with three publicly available heart DNase-seq data sets (Fig. 1A). Peak calling with MACS2 (Zhang et al. 2008) identified varying numbers of DHSs (50,000–110,000) across the five samples (Methods; Supplemental Fig. S2A). We defined DHSs as 600-bp regions centered at the identified summits. DHSs frequently cluster in small regions, and 22%–32% of extended DHSs overlap their neighbors, forming larger DHSs with multiple summits (Supplemental Fig. S2B). We next compared heart DHSs with other tissue DHSs using the top 50,000 DHSs from each sample and the Jaccard index (number of bases in the intersection over the union for each pair). These comparisons show higher similarity (∼50%) between the adult cardiac samples than with other tissues (∼30%), including fetal heart (Methods; Supplemental Fig. S2C). Thus, DHS maps do uncover tissue-relevant CREs, although many regions are open across many tissues. Nevertheless, technical and biological variation affects the adult cardiac DHS maps. To maximally detect CREs, we aggregated all DHSs across the five adult heart replicates and identified ∼160,000 distinct regions (“observed DHSs”) covering ∼4% of the genome. Using multiple samples is important because ∼40% of DHSs were detected only once across the replicates, while only ∼22% detected across all five (Fig. 1B).
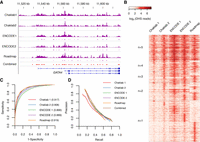
A machine learning algorithm (gkm-SVM) accurately predicts cis-regulatory elements. (A) An example of heart DNase-seq signals (raw data) and peaks (MACS2) at the GATA4 locus across multiple human samples. (B) A genome-wide heat map of DNase-seq read densities in 1000-bp windows centered at heart DHSs. Randomly sampled 1000 regions were used. Regions were grouped based on the configuration of the DHS peaks across the five samples with at least one observed DHS. (C) ROC curves of gkm-SVM models for five replicates against reserved test sets. (D) Comparisons of the fraction of DHSs overlapping predicted regions (precision) and fraction of predicted regions overlapping DHSs (recall).
Sequence-based models can predict additional CREs
To elucidate the sequence code for these cardiac CREs, we used the gapped k-mer support vector machines (SVM) method, gkm-SVM (Ghandi et al. 2014), widely accepted as one of the state-of-the-art sequence-based methods (Kreimer et al. 2017). Also, extracting predictive sequence features from gkm-SVM is easier than from other methods. We used an improved software LS-GKM (Lee 2016) that enabled training on larger data sets (∼100,000 DHSs) (Supplemental Fig. S3A). The gkm-SVM attained considerable accuracy in classifying CREs from non-CREs on reserved Chromosome 9 data (Methods; Fig. 1C). Of note, the choice of a specific chromosome did not affect the method's performance as its accuracy was unchanged with fivefold cross-validation (Supplemental Fig. S3B). Importantly, our model trained on a single data set can predict DHSs observed in the other data sets and systematically assigns higher scores to DHSs experimentally missed than random genomic regions, demonstrating that sequence-based predictions do indeed identify true DHSs (Supplemental Methods; Supplemental Fig. S4A).
The number of samples used for model building determines a model's accuracy. Our tests show that using all five samples identified an additional 5%–10% true DHSs than any single sample, although the maximum accuracy was rapidly attained (Supplemental Methods; Supplemental Fig. S4B). Further, the randomness of negative sets for training is also a major source of variation. To minimize this effect, we averaged 10 different models, trained on independently generated negative sets, and used the combined model for DHS prediction at balanced precision and recall rates (∼43%) (Methods; Fig. 1D).
Scanning the entire genome yielded an additional 88,026 distinct “predicted” heart DHSs after excluding regions that overlapped observed ones (Methods; Fig. 2A). Thus, these predicted DHSs do not suffer from overfitting by construction. For validations, we used several functional annotations to compare them to observed regions. First, we assessed sequence conservation against randomly permuted regions (Supplemental Fig. S5A): Both observed DHSs and, to a lesser extent, predicted DHSs significantly overlap genomic conserved elements (binomial test, P < 2.2 × 10−16) (Davydov et al. 2010). Second, we asked how frequently predicted DHSs were in open chromatin in other tissues. We defined two different DHS sets: (1) a universal set by combining DHSs from all ENCODE and Roadmap data sets, except from adult heart; and (2) a heart-related set of DHSs from cardiac-related samples only (Methods). Both observed and predicted DHSs significantly overlapped both DHS classes (Supplemental Fig. S5B,C): 94.7% of observed and 57.7% of predicted DHSs are open in heart-related tissues, with an additional 30.7% in other cell types (Fig. 2A). Third, we compared H3K27ac histone modification marks in heart tissues to show the same pattern: 52.3% of observed but only 10.9% of predicted DHSs overlapped H3K27ac marked regions (Supplemental Fig. S5D). Yet, we identified ∼7500 additional regions, under the stringent criteria that predicted regions overlap H3K27ac marks and are heart-related DHSs, not detected by DNase-seq alone. Predicted DHSs are largely restricted to specific tissues and cells (Fig. 2B; Supplemental Fig. S5E) while nearly half of observed DHSs are open across many different noncardiac cell types. Moreover, predicted DHSs show systematically weaker DHS signals than observed DHSs (Supplemental Fig. S5F).
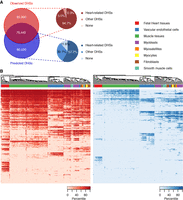
Learned sequence features predict additional CREs. (A) Venn diagram of observed and predicted DHSs and their proportions in noncardiac cell types. (B) Heat map of DHS signal intensities in heart-related cell types and tissues at observed (left) and predicted (right) DHSs. Randomly sampled 5000 regions from observed and predicted DHSs were used. Observed DHSs are mostly located in ubiquitously open chromatin regions in cardiac-relevant cells and tissues, while predicted DHSs show greater cell-type specificity.
Varying degrees of overlap with functional annotations led us to further investigate potential sources of variation in CRE identification. We divided predicted DHSs into four distinct categories based on overlap: heart H3K27ac histone modifications and heart-related DHSs (tier 1); heart-related DHSs only (tier 2); universal DHSs only (tier 3); and the remainder (tier 4). For each of these categories, we considered the (1) proportion of ambiguous bases, (2) average SNP (single nucleotide polymorphism) frequency per base (common and rare SNPs, 1% MAF as a threshold), (3) proportional overlap with H3K9me3 histone modifications in heart tissues, a representative heterochromatin mark (Nakayama et al. 2001), and (4) proportional overlap with FANTOM5 enhancers (Methods; Lizio et al. 2015). As a positive control, we included observed DHSs for comparison (Supplemental Table S1). First, the predicted DHSs in the lower tiers (3 and 4) have poorer mappability: Predicted DHSs in tier 4 have 2–3.5× more ambiguous bases than observed DHSs, depending on read length. Since the functional annotations (H3K27ac and heart-related DHSs) were also identified by sequencing, the variability in CRE detection can be explained at least in part by differential mappability. SNP frequencies also follow the expected pattern; higher SNP frequency in the lower tiers (2 & 3) imply that these predicted DHSs are more variable, making read mapping difficult. Predicted DHSs in tier 4 show the lowest SNP frequency because their extremely poor mappability reduces SNP identification (Nielsen et al. 2011). DHSs predicted in the lower tiers are more enriched in heterochromatin, suggesting that chromatin organization, a feature difficult to predict using sequence-based models only, influences CRE detection as well. Lastly, we used FANTOM5 enhancers (Lizio et al. 2015) as an orthogonal enhancer validation set and compared them to predicted DHSs. Significant proportions of predicted DHSs in tiers 1, 2, and 3 (12.3%, 5.9%, and 2.7%) were FANTOM5 enhancers (binomial test, P < 2.2 × 10−16 for all cases), but tier 4 had only 0.5% FANTOM5 enhancers (P < 0.97).
Another potential cause of variation in CRE detection lies in peak calling, with some predicted DHSs being missed due to being below our detection threshold. Thus, we recalled DHS peaks with relaxed thresholds (false discovery rate <0.1, 0.15, 0.2, and 0.25) and identified larger numbers of DHSs (193, 205, 242, and 280 k, respectively). We calculated the proportion of the original predicted DHSs overlapping these newly identified DHSs (Supplemental Table S2) to show that predicted DHSs, especially in the higher tiers, overlap many of these less stringent DHSs. Thus, a significant fraction of predicted DHSs are likely true. Taken together, our results suggest that sequence-based predictions of an additional ∼55% cardiac CREs are highly complementary to their experimental detection and necessary for obtaining comprehensive CRE maps.
Sequence-based models can also predict cardiac TFs
We next tested whether the sequence features of gkm-SVM allow identification of the cognate TFs through their TF binding sequences (TFBS) in the corresponding tissue/cell types (Lee et al. 2011; Gorkin et al. 2012). First, we used the distribution of all 11-mer weights in the SVM model and compared it to those that match TFBSs for known heart TFs. Indeed, TFBSs for CTCF and MEF2A as well as other known cardiac factors are significantly enriched in the top fifth percentile of the SVM weight distribution, while an exemplar noncardiac factor such as POU5F1 is not (Fig. 3A). Thus, to search for cardiac TFs, we used the Cis-BP database (Weirauch et al. 2014) augmented by 93 new position weight matrices (PWMs) of C2H2 Zinc-Finger (ZF) TFs (Schmitges et al. 2016), to show that 11-mers matching 54% of PWMs (473/868) associated with 506 TFs were enriched in the top fifth percentile of the distribution (Supplemental Data S1). As expected, 11-mers matching these predicted cardiac TFs systematically have larger SVM weights than those that don't (Supplemental Fig. S6). Since not all TFs are expressed in any given tissue, we further restricted attention to the 334 TFs expressed in heart left ventricles using gene expression profiles from the GTEx project (Supplemental Methods); Fisher's exact test confirmed that predicted TFs were significantly associated with cardiac gene expression (Fig. 3B).
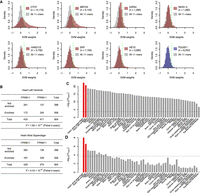
Learned sequence features identify cardiac TFs. (A) SVM weight distributions of PWM-matched 11-mers for the top two scoring TFs (CTCF and MEF2A), five well-known cardiac-specific TFs (GATA4, NKX2-5, HAND1, HAND2, SRF, and HEY2), and a cardiac-irrelevant TF (POU5F1). The fifth percentile is shown as a green line. The enrichment is defined as the fraction of TFBS-matching 11-mers in the top fifth percentile of all 11-mers compared to the expected fraction (5%). (B) 2 × 2 contingency tables comparing TFs enriched in heart DHSs with TFs expressed in heart left ventricles (top) and atrial appendages (bottom). (C,D) −log10(P) of one-sided Fisher's exact test for every tissue tested using all TFs (C) and after removing commonly expressed TFs (D); the two tissues from adult heart (atrial appendage and left ventricle) are highlighted in red with the Bonferroni-corrected P-value threshold (0.05) shown as a dashed line.
These results are physiologically relevant since left ventricles and atrial appendages from adult hearts were the two most significant tissues among the 53 GTEx tissue gene expression profiles we examined (Fig. 3C). Many other tissues also showed significant association because many TFs are expressed across multiple tissues. Thus, enrichment tests after removing 353 commonly (>90% of tissues) expressed TFs dramatically reduced the significance for every tissue except for the two heart tissues (Fig. 3D). Thus, cardiac CRE functions are activated by a large number of both commonly expressed and cardiac-specific TFs. Our analyses revealed 78 selectively expressed and CRE-enriched TFs in heart tissues, of which 29 are C2H2 ZFs with diverse binding specificities, with 44 others belonging to seven other major classes; 16 basic-helix-loop-helix (bHLH), nine Nuclear Receptor (NR), five Sox, four Homeodomain (HD), four basic leucine zipper (bZIP), three GATA, and three ETS transcription factors (Fig. 4). Many expressed TFs in the same family have similar sequence specificities, explaining some part of the expected functional redundancy.
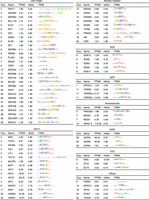
A list of 78 TFs enriched in heart DHSs as well as selectively expressed in cardiac tissues. The 78 TFs were grouped based on their DNA binding domain families. These PWMs were also clustered based on their sequence specificity. For each TF, cluster number (Clus), name, gene expression (FPKM) in left ventricle, fold enrichment (nfolds), and PWM are shown, respectively.
We validated these predicted cardiac TFs also using ENCODE ChIP-seq data (Supplemental Methods) based on two classes: potential cardiac TF ChIP-seq data (n = 1054 for 176 TFs) and the remainder (n = 313 for 126 TFs). For each, we calculated the overlap between ChIP-seq peaks and our observed and predicted DHSs to show that candidate cardiac TF-bound regions overlap heart DHSs significantly more often than noncardiac TFs (P < 2.2 × 10−16 for observed DHSs, P < 2.84 × 10−16 for predicted DHSs, with one-tailed two-sample Kolmogorov-Smirnov tests) (Supplemental Fig. S7).
Common cardiac DHSs are mostly promoters and CTCF sites
Many DHSs are accessible across diverse cell types (Xi et al. 2007; Song et al. 2011; Thurman et al. 2012). These common DHSs are typically enriched in transcription start sites (TSSs) or CTCF binding sites or both, with the remainder enriched in cell-type–specific distal enhancers. To discriminate the cardiac DHSs, we defined common DHSs as regions that are open in ≥30% of all ENCODE/Roadmap tissue samples (Supplemental Methods; Lee et al. 2015). Consistent with previous observations (Xi et al. 2007; Song et al. 2011; Thurman et al. 2012), ∼47% of observed heart DHSs are common DHSs, of which ∼63% overlap TSSs or CTCF bound regions (Supplemental Fig. S8). To understand the sequence features of these classes of DHSs we separately trained a gkm-SVM model after removing these common DHSs. In this heart-specific model, consistent with the CTCF ChIP-seq peak overlap analysis, most of the predictive 11-mers that showed a major decrease in SVM scores (Z-score differences > 6) were CTCF binding sites. In the top five, motifs showing a major decrease were additionally ZBTB4, PLAG1, SP4, and KLF10 binding sites, known to be promoter-binding TFs (Supplemental Fig. S8C,D). These analyses provide a principled, statistical way to distinguish TSSs, CTCF binding sites, and distal enhancers from DHS data, in a tissue- or cell-type–specific manner.
Predicted cardiac regulatory variants affect chromatin accessibility and gene expression
Our sequence-based model allows systematic predictions of whether a DNA sequence variant within a CRE is likely to affect its function, thus making detection of (cardiac) regulatory variants possible. To assess this function, we used the deltaSVM method (Lee et al. 2015; Beer 2017; Kreimer et al. 2017) to first assess a small set of high-confidence cardiac regulatory variants associated with allele-biased DHSs (Methods). We tested two different gkm-SVM models, one trained on all heart DHSs (“generic model”) and the other on cardiac-specific DHSs after removing common DHSs (“specific model”): deltaSVM scores from both models were significantly correlated with their allele-biased chromatin accessibility (Fig. 5A; Supplemental Fig. S9A). Both models achieved comparable precision—46% and 52% at 40% recall—using 4× larger sets of control variants with no allele bias (Fig. 5B). Although their performance is similar, they do not predict the same variants since ∼30% of variants are unique to each model (Supplemental Fig. S9B). This result is consistent with the k-mer weight distribution differences between the models (Supplemental Fig. S8C) and suggests that the specific model is better at detecting cardiac-specific TFBSs by ignoring CTCF and general promoter TFBSs.
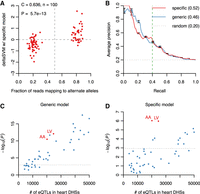
Identification of common, cardiac-specific regulatory sequence variants. (A) deltaSVM scores from the cardiac-specific model as compared to allele-biased chromatin accessibility. (C) Pearson correlation coefficient, (n) number of variants, (P) t-distribution P-value. (B) Precision-recall curves of deltaSVM scores of allele-biased DHSs against 4× larger control SNP sets; the dashed green line indicates the recall rate 40%. Error bars are the standard errors calculated from 10 independently sampled control SNP sets. (C,D) Statistical significance (P) of deltaSVM SNPs from the generic (C) and specific (D) cardiac models as compared to eQTLs using the χ2 test; the two heart tissues are highlighted. (AA) Heart atrial appendages, (LV) heart left ventricles. The Bonferroni-corrected P-value threshold (0.05) is shown as a dashed line.
These results prompted the question of whether deltaSVM predicted variants affected local gene expression. We identified all high scoring variants within heart DHSs (Supplemental Data S2) and compared them to GTEx expression quantitative trait loci (eQTLs) from 44 different tissues (Methods): deltaSVM variants predicted by the generic model are significantly associated with eQTLs in many tissues (Fig. 5C). This significance is strongly correlated with the number of eQTLs in CREs, which is also correlated with the sample size used for gene expression studies (The GTEx Consortium 2015). On the other hand, variants predicted by the specific model are mostly associated with eQTLs from heart tissues, although their overall statistical significance is lower (Fig. 5D). We concluded that, despite the pleiotropy of many regulatory variants, the specific model does identify cardiac-specific regulatory variants with increased specificity but at the cost of missing some regulatory variants common to many tissues. Further, inclusion of predicted DHSs enhanced detection by increasing the statistical significance of eQTL associations (Supplemental Fig. S9C,D).
Predicted cardiac regulatory variants explain cardiac phenotypes
We hypothesize that these predicted causal variants, within observed and predicted CREs explain a significant fraction of cardiac phenotype heritability, which we tested using QTi GWAS (Arking et al. 2014), an intermediate trait involved in long QT syndrome and sudden cardiac death (Tomaselli et al. 1994). We used our published QTi meta-analysis on 76,061 European ancestry subjects and ∼2.7 million SNPs and performed Q-Q analysis. First, variants within heart CREs (Fig. 6A, red dots) are significantly enriched in QTi GWAS SNPs in comparison to all common SNPs (black dots). A subset of these heart CRE variants, predicted to be causal by deltaSVM, especially by the specific model (green dots), are further enriched in QTi-associated variants (Fig. 6A).
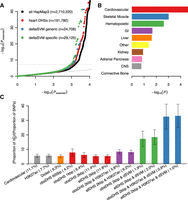
Cardiac regulatory variants explain QT-interval heritability. (A) Q-Q plots of QTi GWAS results using different subsets of common genome-wide sequence variants (numbers of variants in parentheses); the dashed gray line indicates the genome-wide significance threshold (P < 5 × 10−8). (B) Comparison of enrichment P-values between 10 predefined cell-type group functional annotations. (C) Enrichment values, estimated as the fraction of the heritability (hG2) explained by variants over the fraction of the SNPs, for various heart DHS-based annotations. (Cardiovascular) the predefined cell-type group functional annotation for cardiovascular tissues, (H3K27ac) H3K27ac marks in heart, (Dickel) heart CRE map from Dickel et al. (2016), (obsDHS 600 bp) observed DHSs, (obsDHS 1 kb/2 kb) observed DHSs with 1-kb/2-kb extension, (allDHS 2 kb) observed and predicted DHSs with 2-kb extension, (dSVM) deltaSVM predicted variants. A combination of multiple annotations indicates intersection of them. For example, “obsDHS 2 kbp & dSVM” means a set of variants predicted by deltaSVM and overlapping observed DHSs with 2-kb extension. The proportion of SNPs are in parentheses; error bars denote standard errors estimated by a block Jackknife method; no enrichment is shown as a dashed line.
Taken together, these variants contribute substantially to QTi heritability, as shown for some functional annotations of the genome relative to other complex phenotypes (Yang et al. 2011). To quantify this contribution, we used linkage disequilibrium (LD) score regression methods on GWAS summary statistics (Bulik-Sullivan et al. 2015; Finucane et al. 2015) to evaluate the heritability contribution from CRE causal variants (Supplemental Methods). All common autosomal variants (1KGP SNPs with MAF > 5% in European ancestry subjects) explained 11.2% of QTi variation. Using predefined cell-type group functional annotations (Finucane et al. 2015), the cardiovascular cell-type group contributed to the most significant enrichment as compared to other tissues (Fig. 6B); this cell-type group includes lung tissues, so that the specificity for QTi may be diluted. Consequently, we tested our heart DHS-based annotations by comparing the enrichment under various definitions of causality (Fig. 6C). We first evaluated different DHS lengths, discovering that a higher heritability (61%) could be explained by extending DHSs to 2-kb lengths, although higher enrichment (7.9×) was achieved at the original definition of 600 bp. We surmise that narrower DHSs truncate some CREs, missing true variants. As a comparison, we also evaluated a recently published cardiac CRE map based on EP300 and H3K27ac bound regions from multiple cardiac developmental stages (Dickel et al. 2016). This enrichment is comparable to that achieved by our 2-kb heart DHSs (5.4× vs. 5.3×; one-tailed Z-score P < 0.51). The average size of Dickel's cardiac CREs is bigger than our 2-kb heart DHSs (3211 bp versus 2467 bp), but the number of elements is smaller (82,119 versus 133,102). The majority of Dickel's CREs (49,506 out of 82,119, or 60%) overlap our DHSs, but many regions are still detected by just one approach, suggesting that each method detects somewhat different types of CREs. Next, adding H3K27ac marks to DHSs further increases (from 5.7× to 8.5×) the enrichment by effectively filtering out less informative DHSs. While the difference between these two enrichments is only marginally significant (one-tailed Z-score P < 0.06), H3K27ac marks alone capture more variants than H3K27ac overlapping DHS variants (7.7% vs. 5.8%) but explains a smaller heritability fraction (44.3% vs. 48.8%) (Fig. 6C; Supplemental Fig. S10). Thus, some H3K27ac peaks do not capture CRE variants, consistent with the fact that H3K27ac peaks are typically located at CRE boundaries. Adding deltaSVM predictions greatly increased enrichment but explained a smaller fraction of heritability, likely due to false negatives in deltaSVM predictions. Nonetheless, our heart DHS-based annotations, combined with deltaSVM predictions and H3K27ac marks, achieved a highly significant 33.1× enrichment. Tier 1 predicted DHSs consistently increase the explained heritability by 5%–10% (i.e., 1∼3% additional heritability) without compromising enrichment (Fig. 6C; Supplemental Fig. S10). These results confirm that regulatory sequence variation is the major source of QTi phenotypic variation and that we can identify a majority of such causal variants. Of note, including predicted DHSs with weaker functional support (Tiers 2, 3, and 4) decreased overall SNP heritability (Supplemental Table S3). This is probably due to the assumption in the LD score regression method that effect sizes are normally distributed. Under this assumption, adding a large number of SNPs with very small effect sizes can introduce a downward bias. It is also consistent with our observation that many predicted DHSs are weaker compared to observed ones, making them less contributory to the phenotype.
To demonstrate our method's broad applicability, we also analyzed three additional cardiac phenotypes (systolic blood pressure, diastolic blood pressure, and pulse rate) and one noncardiac phenotype (BMI) using the UK Biobank project public data (Methods; Sudlow et al. 2015). Similar to the QTi result, predicted cardiac regulatory variants significantly contributed to all three cardiac-relevant phenotypes but not BMI (Supplemental Fig. S11). The heritability of blood pressure explained by the cardiac variants is consistently less than that for QTi and pulse rate, suggesting that regulatory variants active in other tissues, such as kidney and blood vessels (Hoffmann et al. 2017), also contribute to blood pressure phenotypes.
The heart CRE map we generated and the deltaSVM predictions of causality for specific variants within these CREs can be used to probe each of the known QTi GWAS loci in much greater detail (Supplemental Data S3). The QTi GWAS meta-analysis (Arking et al. 2014) identified 35 independent loci with genome-wide significant (P < 5 × 10−8) common variants. By restricting attention to significant SNPs and their LD proxies (r2 > 0.9), we identified 149 variants as potentially regulatory (Supplemental Table S4). Of these, only ∼50% (n = 72) are highly associated (r2 > 0.6) with one of the 67 sentinel SNPs (cf. some of the 35 loci have multiple independent sentinel SNPs). On the other hand, 108 variants have alternative measures of high association (|D′| > 0.9) suggesting that many index SNPs may tag multiple regulatory variants within their haplotypes. Note that these regulatory variants are not uniformly distributed across the associated loci: 75% are located within eight loci (PLN, NOS1AP, ELP6, LITAF, CNOT1, SCN5A, LAPTM4B, and KCNH2). These variants are significantly enriched in GTEx eQTLs as well: 104 of 149 variants are eQTLs in at least one tissue (2.7× enrichment; binomial test, P < 3.6 × 10−30), of which 57 are eQTLs in the two heart tissues (7.5× enrichment; P < 2.2 × 10−34), implying that these specific variants affect the QTi phenotype by perturbing gene expression in the heart.
As an independent validation of deltaSVM predictions, we finally analyzed a small number of GWAS variants obtained from a recently published study (Wang et al. 2016). In this report, 18 putative functional SNPs were selected based on subthreshold GWAS significance (5 × 10−8 < P < 1 × 10−4) for the QTi and QRS duration, additionally supported by other functional annotations (histone modification marks, DHSs, or eQTLs). These were tested by luciferase assays in human iPSC-derived cardiomyocytes. Of these, 14 were found within our heart DHSs, of which five were predicted to be potentially regulatory by deltaSVM. All of these deltaSVM-predicted SNPs showed differential enhancer activities by the luciferase assay (100% specificity), but the study also identified five additional regulatory SNPs (50% false negative rate) (Supplemental Table S5). Although the small sample size explains the lack of statistical significance (Fisher's exact test P < 0.125), the result is consistent with our predictions.
Discussion
This study shows that sequence-based models can allow the systematic detection of specific noncoding CREs, the TFs that engage them, and sequence variants that affect their binding to regulate target gene expression. These models also allow prediction of the functional effects of noncoding sequence variation within these CREs on human phenotypes, as shown here for cardiac traits. Although much more progress is required, improved epigenomic and genomic data sets, data at cell-type resolution, more refined sequence-based models, yet more sophisticated machine learning algorithms, can lead to reading and assessing the entire human genome sequence of thousands of individuals comprehensively. These advances have important implications for understanding the role of both regulatory and structural variation in both Mendelian and complex disease. In the short run, as our results on the QT interval, blood pressure, and pulse rate demonstrate, we have specific variants with strong a posteriori evidence of regulatory effects on specific genes that regulate these phenotypes. These predictions are specific because they fail to predict noncardiac phenotypes (BMI). Thus, high-throughput functional tests of specific variants in specific CREs modulated by specific TFs and affecting specific target cardiac genes are likely to be fruitful. In turn, our cardiac model with genome-wide QT interval or other cardiac trait data can also enable prediction of additional genes, not merely loci, which modulate these traits. Eventually, the specific pattern of use of cardiac enhancers and their target genes across many traits is likely to teach us many new facets of cardiac physiology.
The research described here is integrative, general, and broadly applicable to all cell types and tissues. In time, such regulatory CRE maps, their cognate TFs, and sequence variants affecting CRE activity can be routinely constructed for a wide variety of cell types and tissues. The comparative analyses of such data will teach us a great deal about what is common and what is specific regulation for each cell type and the GRNs within them. Recently, Pritchard and colleagues have advanced the hypothesis that most complex traits and diseases are omnigenic, arising from the perturbations of thousands of genes by neighboring sequence variants: Although some of these genes have a physiologic role, many (most?) others merely happen to be expressed in disease/trait-relevant cell types (Boyle et al. 2017). They attribute this behavior to GRNs that are so interconnected that numerous ‘trait-irrelevant’ genes affect the functions of a much smaller set of core (‘trait-relevant’) genes. The comparative analyses of the models we describe will be essential for distinguishing the core from the peripheral trait genes as well as for assessing the effects of different tissues on a given disease.
Methods
Heart DNase-seq data sets
We collected two human heart left ventricle (LV) samples and performed DNase-seq experiments as previously described (Supplemental Methods; Song and Crawford 2010). In addition to the DNase-seq data sets we generated, three additional heart DNase-seq data sets were obtained from the ENCODE and the Roadmap Epigenomics projects using the mapped reads provided by the consortia (GSM1027322, ENCFF000SPN, and ENCFF000SPP). To identify DHSs, we processed the mapped DNase-seq reads (hg19) using MACS2 (Zhang et al. 2008) with the following parameters “-g hs –nomodel –shift -50 –extsize 100”. Six-hundred basepair regions centered at the identified summits were used to define DHSs. The identified DHSs from all five samples were then merged to observe a total of 164,235 distinct regions. For pairwise comparisons, we selected the top 50,000 regions based on the MACS2 P-values from each sample and calculated the Jaccard index for each pair. For comparative analyses, we chose four adult tissues (ovary, pancreas, psoas muscle, and small intestine) as well as five fetal tissues (adrenal gland, brain, heart, kidney, and spinal cord), all available from the Roadmap project.
Genome-wide prediction of cis-regulatory elements using gkm-SVM
For each of the heart DHS data sets, we trained the gkm-SVM models as previously described, with some modifications, and systematically evaluated its ability to predict new DHSs missed by experiments (Supplemental Methods). To identify the best model, we evaluated the combined model as well as the five individual ones by varying SVM thresholds. The combined model, which averages the SVM scores over the five heart data sets, consistently outperformed other models and was chosen as the best model for subsequent genome-wide CRE prediction. For predicting CREs, we scored the whole human genome (hg19) for every 600-bp interval with a 100-bp sliding window. We applied a SVM score cut-off (>0.9) that balanced precision and recall values estimated from the test set (precision = recall = 0.43). In this study we used hg19 as a reference genome and not GRCh38 because realigning reads to the new reference does not substantially alter hg19-based versus GRCh38-based gkm-SVM models (Supplemental Methods).
Genomic properties of detected DHS elements
For each heart DHS set (observed and predicted), we calculated the proportion of regions overlapping three different genomic annotations: a set of conserved regions, and two different sets of open chromatin regions—a universal DHS set and a heart-related DHS set. We used GERP++ evolutionarily constrained elements (Davydov et al. 2010) as conserved regions, and defined an overlap if at least 50 bp of the GERP++ element(s) was contained. For open chromatin regions, we downloaded publicly available DNase-seq data sets from the ENCODE UCSC Genome Browser (https://genome.ucsc.edu/encode/) and the Roadmap Epigenomics project website (www.roadmapepigenomics.org) and identified DHS peaks as described above. We defined the universal DHS set by merging all DHSs from 797 DNase-seq data sets (counting replicates separately, when available) except for the three heart DNase-seq data sets. To reduce false positive DHSs, we excluded regions that were detected only once across all data sets, resulting in 967,724 unique DHSs covering ∼45% of the genome. To define a cardiac-relevant DHS set, we only used samples derived from muscles, blood vessels, and fetal hearts (n = 126), resulting in 605,466 heart-related elements covering ∼19% of the genome. We defined an overlap by at least 300 bp. For histone modification marks of enhancers, we used H3K27ac ChIP-seq peaks and defined 118,597 distinct cardiac H3K27ac marked regions (Supplemental Methods). As a negative control, we created 100 independent sets for each of the heart DHS sets (observed and predicted) by randomizing their genomic positions. We then calculated the average proportion of regions overlapping genomic annotations and performed binomial tests using the average as a parameter p of the null distribution.
To investigate the potential source of variation in CRE identification, we evaluated additional genomic properties of the predicted DHSs: mappability, heterochromatin DNA, and SNP frequencies (Supplemental Methods). To further validate our predictions, we used CAGE-based enhancers from the FANTOM5 (Functional ANnoTation Of the Mammalian genome) project as an independent set (Lizio et al. 2015) and calculated the proportion of the predicted DHSs overlapping these elements (>1-bp overlap).
Predictive sequence feature analysis
To determine potential cardiac TFs, we identified TFs whose binding sites have systematically higher gkm-SVM weights. Specifically, we first scored all nonredundant 11-mers (N = 411/2 = 2,097,152) using gkm-SVM. We averaged these SVM scores over the five data sets to reduce the variance between biological replicates and normalized them to zero means. In parallel, we also identified 868 distinct motifs associated with 904 human TFs (Supplemental Data S4 and S5; Supplemental Methods). For each of these 868 motifs, we found all 11-mers matching the motif, determined by FIMO (Bailey et al. 2009; Grant et al. 2011) with default setting and tested whether they were enriched in the top 5% of the 11-mer score distribution. To resolve the issue that FIMO cannot align k-mers to PWMs longer than the k-mers, we generated sets of shorter PWMs of lengths between 8 and 11 bp tiling across full-length PWMs longer than 8 bp. We then defined “a hit” when a given 11-mer matched any of these PWMs. We tested the null hypothesis that the expected number of the motif matching 11-mers found in the top 5% scoring 11-mers is equal to 5% of all matching 11-mers, using a Poisson distribution, and identified PWMs with the Bonferroni-corrected P < 0.01. We note that our motif identification is robust with respect to the choice of k-mers: Identical analysis using 10- and 12-mers yielded 446 and 514 motifs, respectively, among which 436 (96.1%) and 466 (94.6%) overlap the original 473 motifs with 11-mers.
Allele-biased heart DHSs
To evaluate deltaSVM predictions, we identified DNA sequence variants that directly affected chromatin accessibility in cardiac tissues by using allele-biased mapping of DNase-seq reads (Supplemental Methods). In each of the 17 heart DNase-seq data sets (fetal and adult combined), we identified all heterozygous regions with at least 10 reads and calculated P-values of allele-biased DHSs using QuASAR (Harvey et al. 2015). We combined P-values using Fisher's method and identified 100 high-confidence allele-biased DHS SNPs (P < 0.001 and observed in at least three samples with the same direction of effect). For precision-recall analysis, we also identified a 4× larger control set of SNPs that do not exhibit allele-specific alignment (combined P > 0.9 and observed in at least four samples) by random sampling. To estimate standard errors, we generated 10 independent control SNP sets.
deltaSVM analysis of variants within CREs
We adapted the deltaSVM method (Lee et al. 2015) to predict the regulatory effect of variants within observed and predicted DHSs. We identified ∼610,000 common variants with at least 1% MAF in European ancestry 1KGP subjects residing within cardiac CREs: Of these, ∼420,000 and ∼190,000 were in observed and predicted CREs, respectively. Next, we extracted 21-bp sequences centered at each of these CRE variants from the reference genome (hg19) and calculated the SVM scores of both alleles using the heart DHS gkm-SVM models. The final deltaSVM score is the average of the differences in these scores from five different heart gkm-SVM models. These were repeated using gkm-SVM models trained on heart-restricted DHSs. Approximately 15% of these variants have potential cardiac regulatory effects, given a deltaSVM cut-off that yields 40% recall when predicting allele-biased heart DHS SNPs. Note that all of the deltaSVM significant SNPs predicted in this study are, by definition, restricted to the heart CREs. To investigate the relationship between deltaSVM predicted causal variants and their effect on gene expression, we checked for their overlap with GTEx eQTLs (V6P), restricting attention to eQTLs within ±50 kbp of the associated genes, to increase specificity. Using the variants in the cardiac DHSs, we tested for associations using the χ2 test with the binary variables of whether the variants have significant deltaSVM scores or not and whether they are eQTLs in a given tissue or not.
Data access
All sequencing reads from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo) under accession number GSE104989. Supplemental data and custom scripts are available in the GitHub repository at https://github.com/Dongwon-Lee/heart_cre_map and in the Supplemental Material.
Acknowledgments
We thank Dr. Jody Hooper for assistance in obtaining autopsy hearts and Dr. Dan Arking for providing summary statistics from his published meta-analysis of the QT-interval GWAS. This study has benefited greatly from advice and discussions with Dr. Michael A. Beer, as well as constructive comments from the Chakravarti and Crawford laboratories. The research reported here was supported by the computational resources of the Maryland Advanced Research Computing Center (MARCC) and National Institutes of Health grants GM104469, HL086694, and HL128782.
Author contributions: D.L., A.K., and A.C. conceived and designed the study; M.K.H. collected the adult heart tissues; A.S., L.S., and G.E.C. performed DNase-seq experiments; D.L. conducted all computational analyses; and, D.L. and A.C. wrote the manuscript. All authors were involved in manuscript revision.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.234633.118.
-
Freely available online through the Genome Research Open Access option.
- Received January 11, 2018.
- Accepted August 16, 2018.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








