
Capturing in vivo RNA transcriptional dynamics from the malaria parasite Plasmodium falciparum
- 1Department of Biochemistry and Molecular Biology and Huck Center for Malaria Research, Pennsylvania State University, University Park, Pennsylvania 16802, USA;
- 2Department of Chemistry, Pennsylvania State University, University Park, Pennsylvania 16802, USA
- Corresponding author: manuel{at}psu.edu
Abstract
To capture the transcriptional dynamics within proliferating cells, methods to differentiate nascent transcription from preexisting mRNAs are desired. One approach is to label newly synthesized mRNA transcripts in vivo through the incorporation of modified pyrimidines. However, the human malaria parasite, Plasmodium falciparum, is incapable of pyrimidine salvage for mRNA biogenesis. To capture cellular mRNA dynamics during Plasmodium development, we engineered parasites that can salvage pyrimidines through the expression of a single bifunctional yeast fusion gene, cytosine deaminase/uracil phosphoribosyltransferase (FCU). We show that expression of FCU allows for the direct incorporation of thiol-modified pyrimidines into nascent mRNAs. Using developmental stage-specific promoters to express FCU-GFP enables the biosynthetic capture and in-depth analysis of mRNA dynamics from subpopulations of cells undergoing differentiation. We demonstrate the utility of this method by examining the transcriptional dynamics of the sexual gametocyte stage transition, a process that is essential to malaria transmission between hosts. Using the pfs16 gametocyte-specific promoter to express FCU-GFP in 3D7 parasites, we found that sexual stage commitment is governed by transcriptional reprogramming and stabilization of a subset of essential gametocyte transcripts. We also measured mRNA dynamics in F12 gametocyte-deficient parasites and demonstrate that the transcriptional program required for sexual commitment and maturation is initiated but likely aborted due to the absence of the PfAP2-G transcriptional regulator and a lack of gametocyte-specific mRNA stabilization. Biosynthetic labeling of Plasmodium mRNAs is incredibly versatile, can be used to measure transcriptional dynamics at any stage of parasite development, and will allow for future applications to comprehensively measure RNA-protein interactions in the malaria parasite.
Plasmodium falciparum, the causative agent of malaria, has a complex life cycle that includes development in multiple tissues within the human host and the female Anopheles mosquito vector (Bannister and Mitchell 2003). A mosquito initiates infection by injecting Plasmodium sporozoites into a human, which migrate to the liver where they undergo asexual replication (schizogony) forming thousands of merozoites. When merozoites are released from the liver, they initiate the blood stage of infection. The intraerythrocytic development cycle (IDC) consists of continuous 48-h cycles of maturation and cell division, during which up to 32 new daughter cells are formed and released in each cycle. These clonal progeny invade uninfected red blood cells and repeat the asexual replication process, exponentially increasing the population size. During the IDC, a small proportion of asexual parasites transition to the sexual stage and differentiate into female or male gametocytes (Guttery et al. 2015). Gametocytogenesis is a stochastic process that is obligatory for parasite transmission, as asexual forms cannot propagate within the mosquito vector (Josling and Llinás 2015). These gamete precursors are morphologically and functionally distinct from their asexual blood stage counterparts, which is reflected in their cellular development, metabolism, and patterns of gene expression (Liu et al. 2011).
Gene expression in eukaryotic systems is comprised of many levels of regulation including chromatin modification, active transcription mediated by trans-acting factors, and post-transcriptional mRNA turnover or stabilization (Lelli et al. 2012). Similarly, Plasmodium differentiation and growth within various cell types and hosts involves complex regulatory mechanisms that govern both transcriptional and post-transcriptional processes (Hughes et al. 2010; Cui et al. 2015; Vembar et al. 2016). Development during the IDC is driven by a coordinated transcriptional cascade with the majority of genes expressed in a “just-in-time” fashion (Bozdech et al. 2003; Le Roch et al. 2003), although the mechanisms that underlie the coordination and specificity of active transcription remain largely uncharacterized (Hughes et al. 2010; Painter et al. 2011). Thus far, only a single transcription factor family, the 27-member Apicomplexan AP2 (ApiAP2) proteins, has emerged as transcriptional regulators with functions across all developmental stages (Balaji et al. 2005; Painter et al. 2011; Iwanaga et al. 2012; Josling and Llinás 2015; Yuda et al. 2015). Given the task of regulating roughly 5500 Plasmodium genes with such a small repertoire of transcription factors, it has been proposed that mRNA dynamics in Plasmodium parasites are greatly influenced by post-transcriptional regulatory mechanisms (Hughes et al. 2010; Bunnik et al. 2016; Vembar et al. 2016).
Evidence for post-transcriptional regulation in Plasmodium spp. includes several studies that have reported a significant delay between transcription and translation (Le Roch et al. 2004; Hall et al. 2005; Foth et al. 2008, 2011), and recent studies have demonstrated ribosomal influence on the timing of mRNA translation (Bunnik et al. 2013; Caro et al. 2014). Despite these insights, the proteins that regulate these processes are largely unknown. Surprisingly, although bioinformatic analyses have suggested that between 4% and 10% of the Plasmodium genome encodes RNA-binding proteins (RBPs), only a handful have been characterized to date (Reddy et al. 2015; Bunnik et al. 2016). Evolutionarily conserved post-transcriptional regulatory factors, such as DOZI (DDX-6 class DEAD box RNA helicase), CITH (Sm-like factor homolog of CAR-I and Trailer Hitch), and a pumilio family protein (PUF2), play critical roles in the translational repression (TR) of essential genes throughout Plasmodium sexual development (Cui et al. 2015; Vembar et al. 2016). Other important RBPs are the Alba-domain containing proteins (PfAlba1-4) which interact with RNA throughout the parasite's life cycle and are found associated with TR-complexes (Chêne et al. 2012; Vembar et al. 2015). Although the role of Plasmodium RBPs in gene regulation is a growing area of interest, techniques to accurately measure mRNA dynamics and identify post-transcriptional regulatory factors in Plasmodium parasites lag far behind those available for other eukaryotic systems.
In recent years, the ability to incorporate thiol-modified pyrimidines into nascent mRNA transcripts from human cells (Cleary et al. 2005), mice (Kenzelmann et al. 2007; Gay et al. 2013), Drosophila (Miller et al. 2009), and yeast (Miller et al. 2011; Munchel et al. 2011; Neymotin et al. 2014) has afforded an effective tool to evaluate mRNA dynamics of diverse cellular populations, both spatially (Miller et al. 2009; Gay et al. 2013) and temporally (Miller et al. 2011; Munchel et al. 2011). However, thiol-modified mRNA capture is dependent upon pyrimidine salvage, and the evolutionary lineage of Plasmodium has lost this biochemical capacity (Hyde 2007). Genetic supplementation of pyrimidine salvage enzymes in Plasmodium would restore this metabolic pathway, thereby enabling methods that utilize biosynthetically labeled RNA (Friedel and Dolken 2009).
To probe transcriptional dynamics in P. falciparum, we have developed a customizable approach to capture active transcription and mRNA stabilization in the parasite. Our method is made possible by the exogenous expression of two enzymes involved in pyrimidine salvage: the Saccharomyces cerevisiae gene fusion of cytosine deaminase and uracil phosphoribosyltransferase (FCU). With an active pyrimidine salvage pathway, P. falciparum can readily uptake biosynthetically modified pyrimidines and incorporate these into nascent transcripts, which can be tracked at various time points or in specific cells throughout parasite development. These transcripts can subsequently be captured and analyzed with common comprehensive transcriptomic approaches such as DNA microarrays or RNA-seq. This mRNA capture method from FCU-expressing parasites can be used to temporally profile the transcriptional dynamics of the developing Plasmodium parasite at all stages of development.
Results
Engineering a functional pyrimidine salvage pathway into Plasmodium
The measurement of transcriptional dynamics is greatly facilitated by the exogenous incorporation of modified pyrimidines, which allows for the biosynthetic labeling, capture, and subsequent analysis of newly transcribed RNAs (Fig. 1A). Plasmodium parasites are naturally unable to incorporate pyrimidine nucleoside precursors via salvage and instead rely on de novo pyrimidine biosynthesis (Reyes et al. 1982). To enable parasites to salvage pyrimidines, we genetically modified the P.f. 3D7 laboratory strain to exogenously express a functional S. cerevisiae fusion protein of cytosine deaminase (FCY1) and uracil phosphoribosyltransferase (FUR1) (Erbs et al. 2000), with a C-terminal GFP-tag (FCU-GFP) under the control of a constitutively active P.f. calmodulin promoter (cam, pf3d7_1434200) (Fig. 1B; Crabb and Cowman 1996). Transgenic 3D7cam parasites, but not wild-type parasites, readily incorporate the thiolated pyrimidine precursor 4-TU in a concentration dependent manner (3D7) (Fig. 1C).
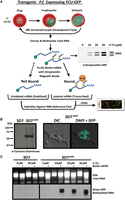
Engineering P. falciparum to salvage pyrimidines and generate thiol-modified RNAs. (A) Schematic of 4-TU biosynthetic mRNA capture method. Transgenic P. falciparum expressing a fusion gene containing cytosine deaminase/uracil phosphoribosyltransferase tagged with GFP (FCU-GFP) under the control of the calmodulin (cam) promoter (3D7cam) enables 4-TU salvage and incorporation into RNA. Thiolated-RNA can be biotinylated and detected by Northern blot or affinity purified by streptavidin magnetic beads for analysis by DNA microarray (or RNA-seq). (B) Expression of FCU-GFP in 3D7cam parasites was verified by Western blot when probed with anti-yeast cytosine deaminase and by live fluorescence microscopy: (green) GFP; (blue) nuclear DNA stained with DAPI. (C) Both wild-type and 3D7cam parasites were grown for 12 h in the presence of increasing 4-TU concentrations. The specificity of RNA thiol-incorporation and biotinylation was assessed by running 2 µg of each RNA sample with and without EZ-link Biotin-HPDP incubation (top). Total RNA was transferred to a nylon membrane and probed with streptavidin-HRP to detect biotinylated RNAs (bottom).
To ensure that cyclical progression through the 48-h IDC was not detrimentally affected by the incorporation of thiol-modified pyrimidines, 3D7cam parasites were incubated in the presence of increasing concentrations of 4-TU (0, 20, 40, 80, 160 µM), while monitoring parasitemia for 72 h (Supplemental Fig. S1A). Based on these data, we selected 40 µM 4-TU for further experimentation, because concentrations at or below this value had no effect on parasite growth (Supplemental Fig. S1A). Global transcriptional profiling by DNA microarray analysis of a complete 48-h cycle of 3D7cam grown in 40 µM 4-TU further indicated that developmental progression is unperturbed (Supplemental Fig. S1B). To ensure that the uptake and incorporation of 4-TU was consistent throughout development, pulses of 4-TU (40 µM) were administered at the ring (10 h), trophozoite (24 h), and schizont stages (38 h) for varying lengths of time (0, 1, 2, and 4 h) (Supplemental Fig. S1C). Our results indicated that 3D7cam parasites were fully capable of transporting and incorporating 4-TU into the total RNA pool throughout the IDC. Nascent transcripts that incorporated 4-TU were readily isolated from total RNA through covalent biotinylation of the thiol-labeled RNAs, followed by affinity purification (Fig. 1A; Supplemental Fig. S1D).
To verify robust incorporation of 4-TU into the pyrimidine pool, we determined the efficiency of the transgenic pyrimidine salvage pathway versus endogenous de novo pyrimidine synthesis (Fig. 2A). Using liquid chromatography mass spectrometry (LC-MS), we directly measured the incorporation of 15N2-uracil into the uracil monophosphate (UMP) pyrimidine pool. The results clearly show that 3D7cam parasites expressing FCU-GFP incorporated heavy-labeled uracil (15N2-UMP) into 50% of the pyrimidine pool in 10 min (Fig. 2A,B). We next tested whether 3D7cam parasites could grow in the absence of de novo biosynthesis by using the antimalarial drug atovaquone to inhibit the cytochrome bc1 complex in the mitochondrial electron transport chain (mtETC). Atovaquone treatment blocks ubiquinone regeneration, which in turn leads to an inhibition of dihydroorotate dehydrogenase (DHODH), an essential enzyme in pyrimidine de novo biosynthesis requiring ubiquinone (Fig. 2A; Painter et al. 2007). 3D7cam parasites were grown in the presence of atovaquone (10xIC50) for 3 h, followed by supplementation of the medium with 15N2-uracil and 13C-bicarbonate (13C-HCO3−). We found that these parasites readily salvaged the exogenously supplied pyrimidine (15N2-UMP), whereas de novo pyrimidine synthesis was disrupted by atovaquone (13C-UMP) (Fig. 2A,C). As expected, 3D7cam parasites were able to bypass the effect of atovaquone when uracil was supplied as a pyrimidine precursor to the growth medium (Fig. 2D). Therefore, the introduction of the FCU enzyme resulted in parasites that rapidly and efficiently salvage pyrimidine precursors and can proliferate independent of de novo pyrimidine biosynthesis.
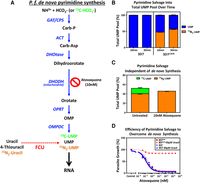
Efficiency of pyrimidine salvage in FCU-GFP expressing P. falciparum. (A) In Plasmodium parasites, pyrimidines are metabolized de novo from bicarbonate and ammonia through a series of enzymatic reactions (blue arrows) resulting in the generation of UMP, which can be incorporated into nascent RNA (black arrow). FCU-GFP allows for salvage of the pyrimidine precursor uracil (red dashed arrow) to UMP, which can then be incorporated into RNA. Inhibition of de novo pyrimidine synthesis is achieved via treatment with the mitochondrial inhibitor atovaquone. (B) 4-TU is efficiently taken up and incorporated into the pyrimidine pool by P.f. 3D7cam parasites grown in the presence of 40 µM 15N2-uracil for 10 and 30 min. Cellular metabolites were detected via LC-MS, and the proportion of total UMP pool that is unlabeled (blue) and labeled with 15N2 (orange) was calculated (n = 2 ± SD performed in triplicate). (C) Flux through de novo pyrimidine synthesis and salvage was measured in treated and untreated parasites by LC-MS after the addition of 13C-bicarbonate (13C-HCO3-) and 15N2-uracil to the culture medium for 30 min. The percentage of UMP derived from de novo synthesis (13C-UMP) and salvage (15N2-UMP) was determined in the presence or absence of the inhibitor atovaquone (10 nM) (n = 2 ± SD performed in triplicate). (D) Early ring stage P. falciparum 3D7 and 3D7cam were exposed to titrating concentrations of atovaquone with or without supplementation of 40 µM uracil to the medium. Parasite survival was determined using a traditional 48-h SYBR-green growth inhibition assay and plotted as an average of three technical replicates (±SD).
Stage-specific expression of pyrimidine salvage allows for biosynthetic labeling of parasites committed to sexual development
The 3D7cam parasite line ubiquitously expresses functional pyrimidine salvage enzymes in all cells from a relatively strong constitutive promoter. To demonstrate the utility and universality of the FCU-GFP mediated biosynthetic mRNA capture methodology in P. falciparum, we characterized the mRNA dynamics of a subpopulation of cells undergoing sexual commitment (gametocytogenesis). Capturing the transcriptional program of early gametocytes is difficult because of the low conversion frequency as well as technical challenges associated with isolating and distinguishing early stage gametocytes from asexual trophozoites (Sinden 1982; Silvestrini et al. 2010). To circumvent these challenges, we generated parasite lines engineered to express FCU-GFP using the established gametocyte-specific pfs16 (pf3d7_0406200) promoter (3D7pfs16), which is only actively transcribed and expressed in a subset of parasites committed to gametocytogenesis (Fig. 3A,B; Supplemental Fig. S2A,E; Dechering et al. 1999; Pradel 2007; Eksi et al. 2008, 2012; Adjalley et al. 2011). As anticipated, 3D7pfs16 parasites have a reduced number of GFP positive cells compared to the 3D7cam parasite population (Fig. 3A; Supplemental Fig. S2B). Direct quantification of 4-TU incorporation into cellular RNA from 3D7cam versus 3D7pfs16 parasites also demonstrated a significant reduction in the level of thiolated RNA in 3D7pfs16 parasites (Fig. 3C; Supplemental Fig. S2C). As observed in the 3D7cam parasites, mRNA abundance profiles of 3D7pfs16 throughout the IDC were unperturbed when grown in the presence of 4-TU (Supplemental Fig. S1B). Therefore, we conclude that FCU-GFP is expressed from the pfs16 promoter only in the subpopulation of parasites undergoing sexual differentiation (Fig. 3A).
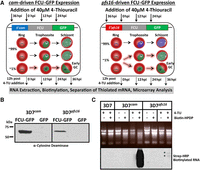
Stage-specific pyrimidine salvage for detection of early gametocyte transcription. To measure mRNA from a subpopulation of cells, we expressed FCU-GFP under the control of the gametocyte-specific promoter pfs16 in the 3D7 parasite line (3D7pfs16). (A) Schematic representation of the experimental design including the timing of 4-TU incubation (black arrows) and RNA extraction (dashed black arrows), plasmids transfected into P. falciparum strains, and a depiction of the highly synchronous cell populations that express cam- and pfs16-fcu-gfp throughout the 48-h IDC. All parasites express FCU-GFP from the constitutive cam promoter (green) regardless of developmental stage (left, 100% GFP+). Only a small proportion (∼1% GFP+) of parasites becoming committed (dashed highlighted arrows) during the IDC, and those that have entered gametocytogenesis in the previous cycle and are sexually committed rings express FCU-GFP from the pfs16 promoter (green), whereas asexual parasites do not (right, ∼99% GFP−). (B) FCU-GFP protein from uninduced asexual cultures of 3D7cam and 3D7pfs16 was detected by Western blot probed with anti-yeast cytosine deaminase. (C) Detection of the subpopulation of FCU-mediated thiol-tagged RNA was carried out by incubating highly synchronized P.f. 3D7, 3D7cam, and 3D7pfs16 in the presence or absence (top) of 40 µM 4-TU for 12 h. Total RNA was extracted, biotinylated, and assayed by Northern blot probed with Streptavidin-HRP (bottom), demonstrating that thiol tagging occurs at a much lower level in 3D7pfs16 (*) than in 3D7cam, representing the minor sexual-stage parasite subpopulation.
To capture the transcriptional dynamics associated with early gametocyte formation in P. falciparum, we compared the gametocyte producing 3D7 parasite line to the nongametocyte-producing F12 clone of 3D7 (Alano et al. 1995). The F12 parasite line is developmentally blocked due to a loss-of-function nonsense mutation in the gene encoding the PfAP2-G transcriptional regulator (pf3d7_1222600). Parasites carrying this mutation show reduced levels of gametocyte-specific gene markers (Silvestrini et al. 2005) and cannot produce mature gametocytes (Supplemental Fig. S2D; Kafsack et al. 2014). Therefore, any significant differences detected in the transcriptional dynamics of 3D7 and F12 would be attributable to gametocyte commitment and development. To do this, the F12 parasite strain was modified to express FCU-GFP driven by either the cam (F12cam) or pfs16 (F12pfs16) promoters (Fig. 3A). Expression of FCU-GFP within each strain was verified, and all strains actively incorporate pyrimidines through salvage (Fig. 3B,C; Supplemental Fig. S2A–F), enabling the temporal detection of differences in the mRNA dynamics during commitment to gametocytogenesis for both strains. Interestingly, the activity of the pfs16-promoter is independent of a parasite's ability to form mature gametocytes (Supplemental Fig. S2D), as F12pfs16 parasites incorporate 4-TU through salvage albeit at significantly reduced levels compared to 3D7pfs16 (Supplemental Fig. S2F).
Stage-specific biosynthetic capture in various strains reveals asexual vs. gametocyte-associated mRNA dynamics
To measure the mRNA dynamics in early gametocytes, we performed biosynthetic mRNA capture (Fig. 1A) from synchronized 3D7cam, 3D7pfs16, F12cam, and F12pfs16 parasites at four time points throughout the 48-h IDC (Fig. 3A), beginning with the ring stage of development. These highly synchronous parasites were separated into four equal fractions, which were incubated with 40 µM 4-TU for a duration of 12 h beginning at 36, 0, 12, or 24 h post-invasion (hpi) (Fig. 3A). To enable the direct comparison of mRNA dynamics in the cam- versus pfs16-fcu-gfp expressing parasites, 12-h incubations were used to ensure sufficient 4-TU incorporation into the small population of GFP-positive 3D7pfs16 and F12pfs16 (Supplemental Fig. S2B) parasites that are committing to gametocytogenesis (Fig. 3C; Supplemental Fig. S2F). The first 12 h incubation was initiated at 36 hpi, the point at which differentiation of asexual schizogony and stage I gametocyte development occurs (Bruce et al. 1990; Eksi et al. 2012), with the last starting at 36 hpi of the subsequent IDC. Following each 12-h exposure, total RNA was extracted and biotinylated to separate thiol-labeled mRNAs from the unlabeled mRNAs via streptavidin-magnetic beads (Figs. 1A, 3A), enabling the capture of Plasmodium mRNA dynamics in a single experiment. Throughout, we define 4-TU labeled RNA as nascently transcribed, and preexisting unlabeled RNA that is not turned over during the 12 h following the addition of 4-TU is stabilized (Dolken et al. 2008).
To identify differences in the mRNA dynamics of parasites committed to gametocytogenesis versus asexually replicating parasites, purified streptavidin-bound and -unbound mRNAs from the four strains were analyzed using DNA microarrays at each of the four time points (Fig. 3A). Microarray data from streptavidin-bound nascent mRNAs are referred to as “transcription” (containing newly incorporated 4-TU) (Fig. 4B), and the remaining unbound mRNAs (existing before 4-TU labeling) are referred to as “stabilization” (Fig. 4C). All genes identified by DNA microarray analysis were K10 means clustered based on the 3D7 strain data (Fig. 4B,C; Supplemental Table S1). An examination of the nascent mRNAs captured from 3D7cam and F12cam parasites demonstrates developmentally regulated gene expression of the P. falciparum transcriptome (Fig. 4B), similar to that previously reported from total mRNA abundance measurements (Bozdech et al. 2003; Le Roch et al. 2003; Llinás et al. 2006; Otto et al. 2010). Similarly, stabilized transcript dynamics are also stage-specific, although they follow a different pattern (Fig. 4C), demonstrating that nascent mRNA transcription and stabilization are independently regulated. Because the cam promoter is ubiquitously expressed in both asexual and sexual stage parasites, mRNAs from 3D7cam and F12cam parasites represent genes that are actively transcribed or stabilized in all parasites tested (Fig. 4A; Supplemental Fig. S3A). Conversely, mRNAs from 3D7pfs16 and F12pfs16 parasites only represent the transcriptional dynamics of the subpopulation that are committing to gametocytogenesis (Fig. 4A; Supplemental Fig. S3A).
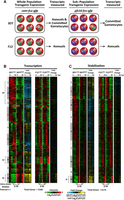
Biosynthetic mRNA capture of whole-genome thiol-labeled and nonlabeled mRNAs during asexual and sexual development. (A) Expression of FCU-GFP from the cam promoter occurs in the parasite strains 3D7 and F12 regardless of developmental stage. 3D7 produces gametocytes, and the transcriptional dynamics in 3D7cam are representative of both asexual (A) and committed gametocytes (cG). Expression of FCU-GFP from the pfs16 promoter in 3D7pfs16 captures mRNA dynamics only in parasites committed to gametocytogenesis (cG). F12 is not able to produce mature gametocytes, and mRNA dynamics occurring prior to gametocytogenesis can be measured regardless of the promoter used. (B) Thiol-tagged RNA was separated from the total pool by streptavidin magnetic purification. mRNAs eluted from the beads are 4-TU labeled (representing “transcription”); (C) unbound mRNAs were present before the addition of 4-TU (representing “stabilization”). Each column represents the Log2(Cy3/Cy5) ratio for every gene in the sample detected above background for mRNAs transcribed (5168 genes) or stabilized (5175 genes) at 0, 12, 24, or 36 h post-invasion (hpi). Ratios for each gene were K10 means clustered (1–10) and ordered according to their peak value starting at 0 hpi. The median Pearson's r for each gene over time between strains is shown below the respective time-course. The fold change in transcription and stabilization for each gene (Log2[Cy3/Cy5] ratio of 3D7pfs16/3D7cam and F12pfs16/F12cam) was calculated and represented to aid in determining enrichment in the gametocyte-specific population. (*) Clusters highlighted in the text.
When comparing the “transcription” time course data sets for 3D7cam and F12cam (Fig. 4B), we found a strong correlation (median Pearson's r = 0.71, data not shown), suggesting minimal inter-strain transcriptional differences when using the calmodulin promoter to drive the expression of FCU. However, when we calculated the intra-strain median Pearson's correlation at each of the IDC time-points between cam-fcu-gfp (asexual cells plus committed gametocytes)- and pfs16-fcu-gfp (committed gametocytes only)-expressing parasites for both 3D7 and F12 (S3B), these correlations directly reflect the strain's ability to produce gametocytes. F12, which cannot produce gametocytes, has an almost perfect correlation for both newly transcribed and stabilized mRNAs, irrespective of the activity of either FCU-GFP promoter (transcription median Pearson's r = 0.95, stabilization median Pearson's r = 0.94) (Fig. 4B,C; Supplemental Fig. S3B, bottom). In contrast, 3D7 has a significantly reduced intra-strain correlation (transcription median Pearson's r = 0.39, stabilization median Pearson's r = 0.35) (Fig. 4B,C; Supplemental Fig. S3B, bottom). From these data, we concluded that there is a significantly altered gene regulatory program between 3D7cam and 3D7pfs16 parasites that reflects the parasite's ability to commit to gametocytogenesis.
Upon closer examination of the 3D7 parasite strain data, we found several striking examples that reflect differences in transcription between pfs16- and cam-fcu-gfp-expressing parasites. For example, Clusters 1–3 represent a large number of genes that are significantly down-regulated (or no longer transcribed) in pfs16-expressing 3D7 cells (3D7pfs16). Of particular note is Cluster 8 (Fig. 4B; Supplemental Table S1), which contains 320 genes that are highly transcribed in both cam-fcu-gfp-expressing strains and includes genes involved in merozoite egress and invasion (enrichment of GO:004409, entry into host, P-value = 9.69 × 10−10) (Supplemental Fig. S4; Supplemental Table S1; Cowman et al. 2012). These same transcripts are largely undetectable in pfs16-expressing 3D7 cells (3D7pfs16) (Fig. 4B, Cluster 8). For example, the gene encoding for merozoite surface protein 1 (msp1, pf3d7_0930300), which is essential for asexual parasite invasion of erythrocytes, is transcribed at 36 hpi in both F12cam and F12pfs16 (Supplemental Fig. S5A). However, msp1 is only transcribed in 3D7cam (Supplemental Fig. S5A), presumably because sexually committed 3D7pfs16 parasites no longer require robust nascent transcription of this gene for reinvasion of erythrocytes. This suggests that we are measuring the transcriptional dynamics of a distinct subpopulation of parasites that no longer requires active transcription of invasion ligands as might be expected for gametocytes, which develop for 10–12 d within the same red blood cell. Conversely, Cluster 10 (Fig. 4B; Supplemental Table S1) contains 492 genes that are highly transcribed in 3D7pfs16; many of these genes have been previously reported to be expressed in gametocytes. For example, the gametocyte-specific gene pf11-1 (pf3d7_1038400) (Scherf et al. 1992) is highly transcribed in 3D7pfs16 at 36 hpi, but not in F12pfs16 (Supplemental Fig. S5A). Together, these data demonstrate that we are capturing nascent transcription from the sexual stage subpopulation of parasites.
Biosynthetic mRNA capture detects gametocyte-specific transcription and stabilization during sexual commitment
One of the goals of using our biosynthetic pfs16-FCU-GFP-based capture method was to detect the mRNA dynamics during early gametocytogenesis. To identify these genes, we calculated the fold change in the transcribed or stabilized signal intensities between the pfs16- and cam-fcu-gfp-expressing 3D7 and F12 parasite strains (Fig. 4B,C). The fold changes between the 3D7cam and 3D7pfs16 microarray data clearly show significant variation in the patterns of mRNA dynamics for virtually all transcripts measured (Fig. 4B,C, fold change). These ratios reveal that a large number of genes are no longer expressed in 3D7 gametocytes (e.g., transcription Clusters 1–3 and 8) (Fig. 4B), whereas other gene sets are strongly transcribed and stabilized (e.g., transcription and stabilization Cluster 10) (Fig. 4B,C). Using these data, we defined a subset of 808 genes whose fold change ratio is higher in either the transcribed or stabilized data between 3D7pfs16 and 3D7cam (Log2 fold change >1, 95th percentile) (Fig. 5A; Supplemental Table S2). We hypothesize that these genes are likely to be involved in gametocyte commitment and development. A comparison of these 808 genes with published gametocyte data sets revealed that many have been detected previously in gametocytes (672 of 808) by RNA-seq (Stage II), DNA microarray (Stage II–IV), or proteomic (Stage I–V) analyses (Fig. 5A; Eksi et al. 2005, 2012; Silvestrini et al. 2005, 2010; Young et al. 2005; Mair et al. 2010; López-Barragán et al. 2011; Brancucci et al. 2014; Tao et al. 2014). However, we found 136 genes that, to our knowledge, have not been previously associated with the gametocyte stage. Interestingly, among these genes, we found 35 rRNAs, 24 tRNAs, and 16 noncoding small RNAs, suggesting that the regulation of these RNAs may also play a role in the sexual differentiation of Plasmodium parasites (Supplemental Table S2). Although the majority of differences between 3D7cam and 3D7pfs16 are reflected by an increase in transcription (Fig. 5A, Clusters 2–4), a smaller subset of mRNAs are stabilized (Fig. 5A, Cluster 1 and 5). These clusters of stabilized transcripts include a number of well-established gametocyte markers, but surprisingly also contain invasion genes such as msp2 (pf3d7_ 0206800), rhoph3 (pf3d7_0905400), and the gene encoding the merozoite TRAP-like protein (mtrap, pf3d7_1028700) (Fig. 5A; Supplemental Table S2; Baum et al. 2006; Cowman et al. 2012). This finding supports recent studies that have identified an essential role for MTRAP in mature gametocytes (Bargieri et al. 2016; Kehrer et al. 2016). Our results demonstrate the specificity and robustness of mRNA biosynthetic capture for measuring the mRNA dynamics from the gametocyte parasite subpopulation.
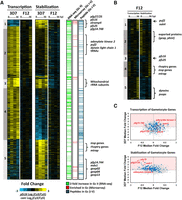
Dynamics of putative gametocyte-specific genes. (A) Genome-wide gametocyte transcription and stability was calculated by determining the fold change in Log2(Cy3/Cy5) ratio of 3D7pfs16/3D7cam. A total of 808 genes were identified as gametocyte-associated (≥1 log2 fold change, 95% percentile ranking) and ordered by K5 means clustering based on 3D7 (Supplemental Table S1). The gametocyte-associated genes identified were compared to previously published RNA sequencing analysis (green) (López-Barragán et al. 2011), DNA microarray (identified in ≥50% of analyses) (red) (Eksi et al. 2005, 2012; Silvestrini et al. 2005; Young et al. 2005; Mair et al. 2010; Brancucci et al. 2014; Pelle et al. 2015), and mass spectrometry identification of sexual-stage peptides (blue) (Silvestrini et al. 2010; Lindner et al. 2013; Tao et al. 2014). Genes characteristic of gametocytogenesis and newly identified gametocyte-associated genes are noted adjacent to the clusters in which they are enriched. (B) A subset of 431 of 808 genes identified as gametocyte-associated were significantly enriched in F12 (≥0.5 log2 fold change, 95% percentile ranking) and were ordered by K5 means clustering. Genes characteristic of both asexual invasion and gametocytogenesis are labeled. (C) Dot plots representing the median fold change (pfs16/cam) of each gene in both 3D7 and F12 transcription and stabilization arrays. Highlighted in blue are gametocyte genes frequently detected (95th percentile) in previously published transcriptomic and proteomic analyses (Supplemental Tables S2, S3). Example gametocyte gene markers, pfs16, pfg27/25, pf14.744, adenylate kinase, and mdv1 are noted in red.
Biosynthetic mRNA capture in F12 reveals initiation of a gametocyte-associated transcriptional program
Examining the same 808 genes differentially expressed in committed versus noncommitted 3D7 parasites, we found that the “transcription” and “stabilization” patterns between F12cam and F12pfs16 parasite populations are largely identical as seen by the overall low fold changes in transcription and stabilization (Supplemental Fig 5A). For example, Cluster 4 is comprised of genes whose patterns of transcription are invariant between F12cam and F12pfs16, but are highly altered in 3D7pfs16 gametocytes (Fig. 5A). In the 808 gametocyte-associated genes identified in 3D7, we found a subset of 431 genes that are differentially transcribed or stabilized (>0.5 log2 fold change, 95th percentile) in at least one F12 time point (Fig. 5B). However, in contrast to 3D7pfs16, transcription or stabilization in F12pfs16 parasites is generally not maintained throughout the IDC. This is interesting, because F12 parasites cannot produce gametocytes, and yet we measure active transcription or stabilization of gametocyte-associated genes. These genes include several well-established markers of gametocytogenesis such as pfpuf2, pfs25, and pfs16 (Fig. 5B; Supplemental Fig. S5A,B; Josling and Llinás 2015) as well the transcription and stabilization of genes involved in merozoite invasion (Fig. 5B, Cluster 4 and 5; Cowman et al. 2012). Interestingly, in the gametocyte-less F12 parasites, we also detect the transcription of a number of recently identified P. falciparum male and female gametocyte markers (Lasonder et al. 2016), such as dynein-encoding genes, NIMA related kinase 4 (nek4, pf3d7_0719200), Pfgabc2 (pf3d7_1426500), and p48/45 6-cysteine protein (pf3d7_0208900) (Supplemental Table S2; Supplemental Fig. S5A). Proteomic analysis has shown that these sex-specific gene products are not detected before Stage V of gametocyte development (Supplemental Table S2; Silvestrini et al. 2010), suggesting these mRNAs may be translationally repressed. These data support previous studies demonstrating that male and female sex determination occurs very early on during commitment (Silvestrini et al. 2000; Smith et al. 2000) and that this likely precedes PfAP2-G activity since it is nonfunctional in F12.
Although many gametocyte genes are nascently transcribed in the pfs16-fcu-gfp-expressing 3D7 and F12 parasite lines, a number of known gametocyte markers are stabilized only in 3D7pfs16 parasites, but not in F12pfs16 (blue dots Fig. 5C; Supplemental Fig. S5B). Therefore, it is likely that in F12, the lack of stabilization of these transcripts, perhaps due to a missing RNA-binding protein, may result in an inability to commit to gametocytogenesis. Interestingly, Pfgexp05 (pf3d7_0936600), which was identified as the earliest marker of committed gametocytes (Tiburcio et al. 2015), is actively transcribed during the ring-stage in all strains except 3D7pfs16 (Supplemental Fig. S5A), supporting the notion that early gametocyte genes are transcribed independently of the PfAP2-G transcriptional regulator. Therefore, these results suggest that F12 parasites are attempting to commit to gametocytogenesis, but appear to abort prematurely and resume asexual developmental progression possibly due to the lack of stabilization of essential gametocyte-specific transcripts.
Gametocyte mRNA dynamics support metabolic rewiring and membrane restructuring
Gametocytogenesis is associated with drastic alterations in the morphology, surface-adhesion properties, and metabolic functions of the parasite (Liu et al. 2011). To determine whether such alterations are predicted by the gametocyte-associated mRNA dynamics we measured, we performed GO-term and KEGG-pathway enrichment analysis on the five gene clusters differentially regulated in pfs16-FCU-GFP expressing parasites (Fig. 5A; Supplemental Fig. S6). We found an enrichment of genes involved in membrane structural integrity and transport (fatty acid metabolism, Clusters 1 and 2; membrane ultrastructure, Clusters 1, 2, and 5; and metabolite transport, Clusters 2 and 5) (Fig. 5A; Supplemental Fig. S6A,B) indicative of the parasite's preparation to undergo morphological change. Cluster 3 contains genes that are highly transcribed from mtDNA and stabilized post-transcriptionally, including ribosomal subunits and cytochrome c oxidase III (Fig. 5A). Genes encoding additional enzymatic subunits of the mitochondrial electron transport pathway and mitochondrial-associated metabolic pathways are enriched in Cluster 4 (Fig. 5A; Supplemental Fig. S6A), supporting an increase in the mitochondrial metabolic functions of gametocytes (MacRae et al. 2013; Ke et al. 2015). Interestingly, a significant proportion of nuclear encoded tRNAs (24/45) are highly transcribed during this developmental transition (Fig. 5A). Although little is known regarding the role of tRNAs in Plasmodium beyond translation, a wide variety of alternative functions, such as the promotion of cell survival, energy metabolism, heme biosynthesis, and as insulators between euchromatic and heterochromatic regions of the genome, have been identified in other eukaryotes (Kirchner and Ignatova 2015). The coordinated up-regulation of Plasmodium tRNAs warrants further investigation to define their role during gametocytogenesis.
Gametocyte-associated cis-regulatory motif enrichment reveals multilayered regulation of mRNA dynamics
To predict whether gametocyte mRNA transcription or stabilization is guided by specific DNA/RNA-binding proteins, we performed a DNA sequence motif enrichment analysis on the 808 gametocyte-associated genes from Figure 5 (Supplemental Table S2). We found that the DNA motif AGACA was enriched in the 5′ UTR (within 1 kb upstream of the start codon) of a significant proportion (371 of 808 genes, Z-score = 191.0, P-value = 1.33 × 10−55) of these genes (Supplemental Fig. S7). This motif was associated with gametocyte-specific genes in prior bioinformatic analyses of the Plasmodium genome and transcriptome (Wu et al. 2008; Young et al. 2008; Russell et al. 2015); however, it has not been associated with any specific transcription factors such as the ApiAP2 DNA-binding proteins (Campbell et al. 2010; Russell et al. 2015). Interestingly, the AP2-G associated motif GTAC was also identified in our search but at a lower Z-score (24.1) (Supplemental Fig. S7; Campbell et al. 2010; Kafsack et al. 2014; Sinha et al. 2014). This suggests that PfAP2-G may regulate a small subset of gametocyte-related genes, and additional regulators are likely required. In the 3′ UTR of the same gametocyte-associated genes, we detect enrichment of a UGUR motif, which resembles the RNA sequence motif recognized by the PfPUF2 RNA-binding protein (Supplemental Fig. S7; Cui et al. 2002; Miao et al. 2010, 2013), indicating that post-transcriptional regulatory mechanisms play a role in gametocytogenesis. The identification of independent sequence motifs associated with the early gametocyte stage suggests a need for further characterization of the complex gene regulatory network that coordinates mRNA dynamics during gametocyte commitment and maturation.
Discussion
This study presents a new method to capture and probe transcriptional and post-transcriptional mRNA dynamics in a cell-specific manner at distinct stages of Plasmodium development. We have shown that parasites expressing FCU-GFP can readily salvage pyrimidine precursors such as 4-TU, which allows for direct incorporation of this labeled nucleotide into newly transcribed mRNA. Biosynthetic mRNA capture with 4-TU can be genetically tuned to measure either whole populations or stage-specific subpopulations of parasites by altering the promoter driving the expression of FCU-GFP. The separate pools of labeled (transcribed) and unlabeled (stabilized) mRNAs can be isolated from developing Plasmodium parasites and comprehensively measured by DNA microarray analysis (Figs. 1A, 4B,C) or RNA-seq.
In addition to demonstrating the feasibility of this approach in Plasmodium parasites, we applied biosynthetic mRNA capture to examine the dynamic regulation imparted upon mRNA transcripts at the developmental transition between the asexual blood stage and the sexual gametocyte stage. Although previous studies aimed at elucidating the gametocyte transcriptional regulatory program using genome-wide transcriptome analyses revealed subsets of genes that are differentially abundant during gametocytogenesis (Gissot et al. 2004; Silvestrini et al. 2005; Young et al. 2005; López-Barragán et al. 2011; Pelle et al. 2015), these studies have not measured transcriptional activity or mRNA stability. Rather, they serve to identify genes that have higher transcript abundance during gametocyte development. This study is also the first to measure transcription and stabilization of genes necessary at the point of commitment without the need for physical separation of this small subpopulation from the total cell culture. Our results indicate that the mRNA dynamics of commitment to gametocytogenesis consist of a balance of transcription and stabilization in the gametocyte–producing line 3D7 (Supplemental Table S3). Interestingly, we also found that F12 initiates a limited gametocyte-associated transcriptional program (Fig. 5B) that is aborted in each asexual development cycle. Given the weak, but detectable, activity of the pfs16 promoter in F12pfs16 (Fig. 3C; Supplemental S2F), these parasites are likely attempting to follow a gametocyte commitment transcriptional program, but due to the lack of a functional AP2-G (Kafsack et al. 2014), they cannot produce mature gametocytes (Kafsack et al. 2014). Therefore both transcription and stabilization of gametocyte-specific genes is essential to gametocyte commitment. Interestingly, biosynthetic capture in F12pfs16 reveals transcription of genes essential to both asexual and sexual development raising the possibility that the mRNA dynamics are more complex than a single factor regulating sexual commitment. Our results support recent work suggesting a role for commitment events preceding the activity of AP2-G (Tiburcio et al. 2015).
We also evaluated the transcriptional dynamics of previously reported gametocyte-specific gene markers and found significant differences between the transcription and stabilization of gametocyte associated genes (Fig. 5A; Supplemental Table S2). These dynamics allowed us to identify essential metabolic processes and potential DNA regulatory motifs in transcripts associated with gametocyte commitment. Although our understanding of gametocyte-specific transcriptional regulation is in its infancy, our data demonstrate that the AP2-G DNA binding motif is associated with several mRNAs transcribed in sexually committed parasites. Although PfAP2-G is thought to transcriptionally activate gametocyte-specific genes (Kafsack et al. 2014), this has not been demonstrated on a genome-wide scale. In this study, the pfs16 promoter drives gametocyte-specific expression, although it does not contain an AP2-G motif (Eksi et al. 2008) and F12 lacks a functional PfAP2-G (Carter et al. 2013). This implies that there must be additional factors involved in the regulation of gametocyte-specific genes, which is supported by our finding that a majority of gametocyte mRNAs contain an enriched AGACA DNA motif for which an associated trans-acting factor remains to be identified. Recently, in the murine malaria model, a second ApiAP2 protein PbAP2-G2 was reported to transcriptionally repress asexual development genes (Yuda et al. 2015). The AP2-G2 motif is distinctly absent from gametocyte-associated genes (Fig. 5; Supplemental Fig. S6; Campbell et al. 2010; Yuda et al. 2015), suggesting that this repressive role may similarly be conserved in P. falciparum. Additionally, our data demonstrate that many markers of gametocytogenesis are stabilized upon commitment, supporting a significant role for translational repression of mRNAs during sexual development. Only a few RNA-binding proteins have been demonstrated to play a role in gametocyte maturation (Cui et al. 2015); however, a specific factor(s) involved in stabilization of transcripts upon commitment to gametocytogenesis remains to be identified.
Although this study has focused on the sexual stage of Plasmodium, stage-specific biosynthetic mRNA capture is poised to advance our understanding of parasite transcriptional and post-transcriptional processes at key developmental transitions throughout the entire parasite life cycle. Here, we used the well-characterized pfs16 gametocyte-specific promoter to capture mRNA dynamics of early gametocytogenesis; however, there are a number of other established gametocyte-specific genes (Pradel 2007; Liu et al. 2011; Josling and Llinás 2015) whose promoters may prove useful for biosynthetic labeling throughout the 10–12 d process of gametocytogenesis. Most recently, pfgexp05 was demonstrated to be expressed earlier than pfs16 during gametocyte commitment (Tiburcio et al. 2015). This finding highlights the need for analogous studies to be carried out using alternative promoters. Additionally, by altering the promoter driving FCU-GFP expression, this method can be adapted to examine mRNA dynamics at any other point of development using stage-specific promoters from genes known to be up-regulated in the ookinete, oocyst, sporozoite, or liver stages. These transcriptional programs can also be determined in other host cells or animals, since RNA-tagging from insects (Miller et al. 2009), human cell lines (Cleary et al. 2005), and mouse systems (Cleary 2008) have already been established. Therefore, extensions of this work can be used to explore the relationship between the Plasmodium parasite and its diverse host cell types, the mosquito midgut, hepatocytes, and red blood cells. Biosynthetic mRNA capture can also be combined with bioinformatic models to calculate real-time in vivo rates of transcription and decay on a whole-genome scale (Dolken et al. 2008; Miller et al. 2011; Sun et al. 2012; Neymotin et al. 2014) and may also be used to identify specific responses to environmental perturbations (such as drug-treatment) on short timescales.
We also envision that thiol-modified labeling will enhance efforts to define RNA-protein interactions in the malaria parasite. A recent study identified a large number of RNA-interacting proteins during asexual development; however, the methodology used was not capable of identifying the target RNAs for these proteins (Bunnik et al. 2016). Incorporation of 4-TU into parasite mRNAs enables photoactivatable UV cross-linking to RBPs that are bound to the thiolated transcripts. This allows for genome-wide RNA-protein interactions to be determined using Photoactivatable-Ribonucleoside-Enhanced Crosslinking (PAR-CL) (Baltz et al. 2012; Castello et al. 2012) or to query the interaction of specific proteins by coupling to an immunoprecipitation step (PAR-CLIP) followed by RNA sequencing that captures the exact footprint of a specific protein on each transcript (Hafner et al. 2010a,b; Ascano et al. 2012; Li et al. 2014). Ultimately, the use of this methodology in the malaria field will advance our understanding of the transcriptional dynamics required for all stages of parasite development, enable the functional characterization of RBPs, and accelerate the identification of new molecular drug targets.
Methods
Transgene construction
The open reading frame of the yeast FCU gene was PCR amplified from Plasmodium falciparum vector pCC1 (Maier et al. 2006) and cloned as a translational fusion into the unique BglII and XbaI sites of the pCBM-BSD-684 5′Pfs16-gfp vector (Eksi et al. 2008), placing the transcription of the fused gene under the control of a P. falciparum gametocyte-specific pfs16 (pf3d7_0406200) promoter, yielding the plasmid pCBM-5′pfs16-FCU–GFP. Likewise, FCU was PCR amplified and cloned into the AvrII and BsiWI sites of the pLN-ENR-GFP (Nkrumah et al. 2006) vector, resulting in the pLN-5′cam-FCU-GFP plasmid and placing transcriptional control under the P. falciparum calmodulin promotor (pf3d7_1434200). These plasmids were transformed into, replicated in, and isolated from DH5α E. coli for transfection into P. falciparum.
Strains and culture maintenance
P. falciparum strains 3D7 (Rovira-Graells et al. 2012) and F12 (Alano et al. 1995) have been described in previous studies, and parasite cultures were maintained under standard conditions (Trager and Jensen 1976) at 5% hematocrit of O+ human erythrocytes in RPMI 1640 containing hypoxanthine, NaH2CO3, HEPES, glutamine, and 5 g/L AlbuMAX II (Life Technologies). A description of gametocyte induction is included in the Supplemental Materials and Methods.
Generation of genetically modified parasite lines
Transfection of P. falciparum strains was performed as previously described (Fidock and Wellems 1997). Briefly, 5%–7% ring-stage parasite cultures were washed three times with 10 times the pellet volume of cytomix. The parasitized red blood cell (RBC) pellet was resuspended to 50% hematocrit in cytomix. In preparation for transfection, 100 µg of either pCBM-5′pfs16-FCU-GFP or pLN-5′cam-FCU-GFP plasmid was precipitated and resuspended in 100 µL cytomix. The plasmid and 250 µL of the 50% parasitized RBC suspension were combined and transferred to a 0.2-cm electroporation cuvette on ice. Electroporation was carried out using a BioRad GenePulser set at 0.31 kV, 960 uF. The electroporated cells were immediately transferred to a T-25 flask containing 0.2 mL uninfected 50% RBCs and 7 mL medium. To select for parasites containing plasmid, medium containing 1.5 µg/µL Blasticidin S (Sigma-Aldrich) was added at 48 h post-transfection. Cultures were maintained under constant 1.5 µg/µL Blasticidin S pressure, splitting weekly, until viable parasites were observed. Viable transgenic parasites were then cloned by limiting dilution, and presence of the episome encoding for either cam-fcu-gfp or pfs16-fcu-gfp was PCR verified (data not shown).
Verification of transgene expression
Western blot analysis of FCU-GFP protein expression was carried out on mixed stage transgenic P. falciparum (10% parasitemia, 5% hematocrit), isolated by saponin (0.01%) lysis. Equal volumes of protein extracts were run on a SDS-10% polyacrylamide gel and transferred to a nitrocellulose membrane. The membrane was blocked with 5% milk. To detect protein, the membrane was exposed to Sheep IgG anti-Cytosine Deaminase primary antibody (Thermo Fisher Scientific) diluted 1:500 in 3% bovine serum albumin and incubated overnight at 4°C followed by a 1-h incubation with goat-anti sheep-HRP secondary antibody (Thermo Fisher Scientific) diluted 1:1000 in 3% BSA. The membrane was incubated for 1 min in Pierce ECL-reagent (Thermo Fisher Scientific) and protein detected by exposure to film. Assessment of transgene expression by immunofluorescent imaging is described in the Supplemental Materials and Methods.
Assessment of FCU-GFP transgene ability to salvage 4-thiouracil via Northern blot
Function and stage-specific expression of the FCU-GFP transgene was verified in the 3D7cam compared to wild-type 3D7 by the addition of various concentrations (0, 10, 20, 40 µM) of 4-thiouracil (Sigma Aldrich) to the culture medium from a 200 mM stock concentration prepared in DMSO. After parasites were grown in the presence of 4-TU for 12 h, total RNA was prepared from parasites in 5 mL of TRIzol and resuspended in DEPC-treated water to a final concentration of 0.5 µg/µL. Total RNA was biotinylated using EZ-link Biotin-HPDP (Thermo Fisher Scientific) for 3 h at room temperature under RNase-free conditions to reduce the amount of RNA degradation that can occur. For 30 min, 2.5 µg of biotinylated RNA was run on an ethidium bromide 1% agarose gel at 200 V and was transferred to Hybond-N+ nitrocellulose (Amersham) membrane using traditional Northern blotting techniques. The RNA was UV-crosslinked to the membrane and probed with streptavidin-HRP (1:1000) (Thermo Fisher Scientific). Biotinylated RNA was detected via incubation with ECL-reagent and exposure to film. Additional methods using LC-MS detection of pyrimidine salvage and SYBR-green assessment of parasite survival in the presence of de novo pyrimidine synthesis inhibitors are included in the Supplemental Materials and Methods.
Thiol-labeling time course
P. falciparum transgenic parasites were cultured in human erythrocytes at 5% hematocrit in RPMI 1640 complete medium as described above. All transgenic parasites were synchronized with three successive (48 h apart) L-Alanine treatments to remove late stage parasites (Haynes and Moch 2002). To ensure that the resulting population consisted of exclusively asexual parasites, synchronized ring stages were subjected to a 60% Percoll (GE Healthcare) density gradient, and any remaining late stages were removed by centrifugation (Wahlgren et al. 1983). These highly synchronous cultures were placed back into culture and allowed to recover for at least 24 h. At 36 h post-invasion, 4-TU was added to the medium at a final concentration of 40 µM, and parasites were incubated for 12 h followed by total RNA isolation with TRIzol. This 4-TU addition, incubation, and RNA extraction was repeated every 12 h in four successive time points throughout intraerythrocytic development.
Biosynthetic modification and isolation of labeled mRNA throughout development
A detailed protocol is available in the Supplemental Materials. Briefly, transgenic P. falciparum was incubated with 4-TU, and total RNA was extracted as described above. All RNA was subjected to the biosynthetic modification and magnetic separation as previously published with a few adjustments (Cleary et al. 2005; Cleary 2008; Zeiner et al. 2008). Specifically, 80 µg total RNA (at a concentration of 0.4 µg/µL) was incubated at room temperature protected from light for 3 h in the presence of 160 µL 1 mg/mL solution of EZ-link Biotin-HPDP (Thermo Fisher Scientific). Biotinylated total RNA was precipitated and resuspended in DEPC-treated water to a final concentration of 0.5 µg/mL. Incorporation of 4-TU and biotinylation was determined by NanoDrop analysis and Northern blot probed with streptavidin-HRP. 4-TU labeled-biotinylated RNA was purified using Dynabeads MyOne Streptavidin C1 magnetic beads (Life Technologies) at a concentration of 2 µL/µg of RNA. Beads were prewashed according to the manufacturer's protocol to remove any RNases and blocked with 16 µg of yeast tRNA (Life Technologies). 4-TU-labeled biotinylated RNA was added to the bead slurry and incubated at room temperature for 20 min with rotation. The RNA-bead slurry was placed on a magnetic stand for 1 min, and liquid was carefully removed and saved for RNA precipitation. This sample contained RNA that was not thiolated or biotinylated. Then the RNA-bound beads underwent five rounds of stringent washes with buffer consisting of 1M NaCl, 5 mM Tris-HCL (pH 7.5), and 500 µM EDTA in DEPC-treated water. 4-TU-labeled biotinylated RNA was eluted from the beads with 5% 2-mercaptoethanol incubated for 10 min, placed back on the magnetic stand, and liquid was removed and saved for RNA precipitation. The RNA in this fraction contained mRNA transcribed in the presence of 4-TU from FCU-GFP-expressing cells. Reverse transcription of RNA and DNA microarray analysis are described in the Supplementary Materials and Methods.
Data access
The DNA microarray data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE72695.
Acknowledgments
We thank A.B. Vaidya, PhD, Drexel University College of Medicine, Philadelphia, for providing atovaquone, helpful discussions in establishing the experimental methodology, and review of the manuscript. We thank G. Josling, S. Lindner, T. Russell, J. Santos, and M. Szpara for critical reading of this manuscript. We thank K. Williamson (pCBM-BSD-684 5′Pfs16-gfp), D. Fidock (pLN-5′CAM-FCU-GFP), and P. Alano (P. falciparum strain F12), for providing reagents for this study. This work was funded through support from the Burroughs Welcome Fund, a National Institutes of Health Director's New Innovators award (1DP2OD001315-01), the Center for Quantitative Biology (P50 GM071508), and startup funding from the Pennsylvania State University.
Author contributions: H.J.P. and M.L. conceived and designed this study. H.J.P. generated transgenic parasite lines, conducted the experiments, analyzed the data, and generated all figures. M.C. provided technical assistance with culture maintenance and strain validation. H.J.P. and M.L. wrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.217356.116.
- Received November 11, 2016.
- Accepted March 22, 2017.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








