
Deep sequencing of natural and experimental populations of Drosophila melanogaster reveals biases in the spectrum of new mutations
Abstract
Mutations provide the raw material of evolution, and thus our ability to study evolution depends fundamentally on having precise measurements of mutational rates and patterns. We generate a data set for this purpose using (1) de novo mutations from mutation accumulation experiments and (2) extremely rare polymorphisms from natural populations. The first, mutation accumulation (MA) lines are the product of maintaining flies in tiny populations for many generations, therefore rendering natural selection ineffective and allowing new mutations to accrue in the genome. The second, rare genetic variation from natural populations allows the study of mutation because extremely rare polymorphisms are relatively unaffected by the filter of natural selection. We use both methods in Drosophila melanogaster, first generating our own novel data set of sequenced MA lines and performing a meta-analysis of all published MA mutations (∼2000 events) and then identifying a high quality set of ∼70,000 extremely rare (≤0.1%) polymorphisms that are fully validated with resequencing. We use these data sets to precisely measure mutational rates and patterns. Highlights of our results include: a high rate of multinucleotide mutation events at both short (∼5 bp) and long (∼1 kb) genomic distances, showing that mutation drives GC content lower in already GC-poor regions, and using our precise context-dependent mutation rates to predict long-term evolutionary patterns at synonymous sites. We also show that de novo mutations from independent MA experiments display similar patterns of single nucleotide mutation and well match the patterns of mutation found in natural populations.
Mutation is the ultimate driver of genetic diversity. Every genetic difference within or between species originated in the mutational process and then survived the stochastic and selective forces that act on its frequency dynamics. Any study of natural selection using genetic data thus depends fundamentally on whether we can correct for the confounding factors of mutational biases.
Our ability to study mutation, however, is severely limited. This is because mutation rates are extremely low and because a substantial fraction of new mutations are deleterious and so purged from populations by purifying selection. These two problems can, at first glance, be overcome with divergence-based measurements in which rates of substitution within nonfunctional genomic regions are calculated across taxa (Kimura 1983; Nachman and Crowell 2000; Kumar and Subramanian 2002). Here, the use of vast phylogenetic timescales permits the observation of large numbers of even rare mutational events, while the use of neutral sequences eliminates the confounding effects of natural selection. Unfortunately, these divergence-based methods can be compromised by the assumption that a genomic region is truly neutral and by the fact that even neutral regions can be subject to other selective forces (e.g., biased gene conversion, selection on genome GC content, and genome size) (Galtier et al. 2001; Vinogradov 2004; Hershberg and Petrov 2010; Neher and Shraiman 2011; Lartillot 2013; Lawrie et al. 2013; Li et al. 2013). Furthermore, divergence-based methods produce long-term averages for the mutational spectrum, and therefore may be sensitive over evolutionary timescales to changes in life history traits (e.g., generation time) and changes to mutation rates (Scally and Durbin 2012; Harris 2015).
Thus, the ideal approach for the study of mutational processes is to identify truly new mutations. The optimal study would capture de novo mutations via sequencing sets of parents and offspring and implement this strategy in a large sample to overcome both inter-individual mutation rate variation and the small number of events per individual. The advent of next-generation sequencing has made this possible; however, it is still quite expensive and only recently beginning to be realized, primarily in humans (Goldmann et al. 2016). In order to measure mutational rates and biases in organisms that do not receive the same level of funding support as humans, or in order to survey human mutational rates and patterns across diverse populations on a reasonable budget and timescale, we must use alternative approaches.
Two such alternative approaches include mutation accumulation (MA) experiments in model organisms (Halligan and Keightley 2009) and a more recently proposed method in which very rare polymorphisms in natural populations are used as a proxy for new mutations (Messer 2009; Aggarwala and Voight 2016; Zhu et al. 2017). These approaches have complementary strengths and weaknesses.
The MA approach is implemented by maintaining an organism in a population so small (N ∼ 1–2) that selection is ineffective (Ns << 1) for even strongly deleterious mutations. This allows nonlethal mutations to accumulate in the genome through many generations (Muller 1928; Halligan and Keightley 2009), thus turning the infrequent event of mutation into an observable process. This method has been applied to numerous organisms in order to precisely measure mutational rates and patterns (Farlow et al. 2015; Uchimura et al. 2015; Lovell et al. 2017); however, the method also suffers from drawbacks including that (1) mutations in the lab environment may not represent nature, (2) many mutations in one genome may change the mutational process itself, and (3) MA experiments can be laborious.
The second approach is to use rare polymorphisms as a proxy for new mutations. The rationale is that very rare polymorphisms are younger on average and have frequency dynamics dominated by stochastic noise rather than selective forces (Kimura and Ohta 1973; Messer 2009). An idealized example of this is a new germline mutation, present on a single chromosome in the population (termed a “singleton,” which in practice also refers to a single copy of an allele in a population sample). As long as it is rare, the fate of this mutation is driven mainly by chance (i.e., genetic drift). Consequently, as we look at polymorphisms at lower population frequencies, it is expected that the probability of observing different types of genetic variants should primarily be determined by mutational biases. This is an exciting approach because a large data set of genetic variation from the wild can be obtained; however, using this as a proxy for new mutations suffers from the drawbacks that (1) it is not possible to directly measure mutation rates, (2) it may be a challenge to sequence enough individuals to identify very rare variation, and (3) true genetic variation may be difficult to distinguish from sequencing and alignment errors. This last problem is particularly daunting. As Achaz (2008) noted, increasing the number of individuals in the sample does not help—the number of errors and the number of true variants both scale linearly with sequence length, but an increase in sample size causes the number of true variants to scale only logarithmically while the number of errors still scales linearly (Achaz 2008). Thus, in the pursuit of a deep catalog of genetic variation from natural populations, it is quite possible that, as more individuals are sequenced, we will be adding disproportionately more errors than real polymorphisms.
The fruit fly, Drosophila melanogaster, as both a model organism and a species with many sequenced natural isolates, provides an excellent opportunity to integrate these two approaches and thus benefit from each method's strengths while avoiding the drawbacks of either method in isolation. We combine sequencing data from MA experiments and natural populations and thus generate a large data set with which to precisely characterize the mutational spectra across the genome.
Results
In the first half of this study, we combine results from five MA experiments, including our own novel data, to arrive at a set of 2141 de novo mutations generated in the laboratory. Then, in the second half of this study, we use three publicly available data sets of sequenced natural populations in order to extract a large number (∼70,000) of high-quality, rare (<0.1%) polymorphisms which, unlike in other studies, have been fully validated via resequencing.
De novo mutations identified in MA experiments
There are two primary strategies for mutation accumulation in D. melanogaster: homozygous or heterozygous approaches (Fig. 1A). The homozygous MA strategy is essentially inbreeding in a small population (N ∼ 2) which forces new mutations to eventually be homozygosed. In contrast, the heterozygous MA experiment uses a crossing scheme to pass the chromosome through a single heterozygous male in every generation. These different approaches result in different levels of selection against recessive, strongly deleterious mutations, a class of mutational events critical to fitness and common in many natural populations (Charlesworth and Willis 2009). The heterozygous strategy has often been implemented using inversion-rich balancer chromosomes, which are prone to distortions in homology-directed repair processes. We chose to use the method that most closely mimics nature—heterozygous accumulation with noninverted chromosomes that carry recessive markers (see Methods). At the outset of this study, there were two prior studies which sequenced homozygous MA lines (Keightley et al. 2009; Schrider et al. 2013), and since we began our experiments there have been two more recent data sets published (Huang et al. 2016; Sharp and Agrawal 2016) which used a heterozygous and hybrid approach. In this work, we present our own novel data set from a heterozygous MA experiment and then combine our data with all other published experiments to-date, providing the first meta-data set created for fruit fly MA experiments, which we make publicly available.
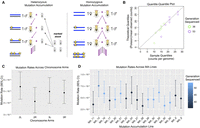
A summary of the experimental design and results for the single base pair mutation rate in this study. (A) Diagram depicting the general crossing schemes used in heterozygous (left) and homozygous (right) mutation accumulation, where this study used the heterozygous design. (B) QQ plot of the quantiles of the mutation counts on each chromosome arm of each strain, plotted against the quantiles of a Poisson distribution with mean taken from the mean counts in the MA experiment, where color indicates the generation sequenced (green = generation 36, purple = generation 53). (C) Mutation rates estimated for each chromosomal arm (Pearson's χ2 test of independence, χ2 = 2.55, df = 3, P-value = 0.47). (D) Mutation rates estimated for each strain, where color indicates the generation sequenced (Pearson's χ2 test of independence, χ2 = 7.99, df = 14, P-value = 0.89).
A new data set of 325 point mutations
Briefly, our new heterozygous MA data set was generated via the following: 17 lines of D. melanogaster were allowed to accumulate mutations for 36–53 generations in a heterozygous state (Fig. 1A, left). Each line was sequenced to ∼20–25× (Supplemental Fig. S4), reads processed (trimmed, mapped to release 5.57 of the FlyBase reference, filtered for duplicates, and realigned around indels), and variants called with a combination of GATK and VarScan. A variant was considered a de novo mutation if it was called with high confidence in one strain and simultaneously never present on more than a single sequencing read in either the ancestral strains or any other MA line. In total, 325 new mutations were identified, of which 30 were randomly chosen for visual confirmation in a pileup file, and an additional 30 were randomly chosen for PCR/Sanger sequencing. We successfully validated 29 of the 30 that were Sanger-sequenced, giving a ∼3% error rate, although we note that, upon visual inspection of the single unconfirmed mutation, we verified that both the original genotype call and the resequence data were of very high quality, and consequently, we suspect the PCR primers may have inadvertently been haplotype-specific and thus amplified the non-MA chromosome. See Methods for additional details of pipeline, and for details of the 17 MA strains, 325 de novo mutations identified, and 30 mutations Sanger-sequenced, see Supplemental Tables S1–S3 and Supplemental Data File 1.
The 325 de novo mutations identified in this study reveal a notably consistent mutation rate across strains, chromosomes, and time (Fig. 1). Plotting the quantiles of mutation counts per genome against the quantiles of a Poisson distribution (with a mean equal to the sample mean) (Fig. 1B) shows that, as expected, mutation counts appear Poisson-distributed. Consistent with previous findings, we find no significant difference in mutation counts across the major chromosomal arms (X2 test, P-value = 0.47) (Fig. 1C; Keightley et al. 2009; Schrider et al. 2013). Additionally, we find no significant difference in mutation rates between generations 36 and 53 (Poisson exact P = 0.76) (Supplemental Fig. S5), although it is likely we were underpowered to test for small variations in rates across generations. Given the presence of mutator lines in other published MA experiments, we tested for variation in the total mutation rate across strains and found no significant variation (X2 test, P-value = 0.89) (Fig. 1D; Schrider et al. 2013; Huang et al. 2016). Finally, in our experiment, we found a single base pair mutation rate of 4.9 × 10−9 per generation (95% CI 4.4–5.5 × 10−9) (Supplemental Table S7).
Five MA experiments, different single base pair mutation rates
We next compared and combined our data set with data from the four previously published MA experiments in D. melanogaster (Keightley et al. 2009; Schrider et al. 2013; Huang et al. 2016; Sharp and Agrawal 2016). In total, there are 163 lines, 36–262 generations per line, and five lines with elevated mutation rates (“mutators,” four from Schrider et al. [2013] and one from Huang et al. [2016]). To allow a fair comparison across experiments, we filtered the data to include only mutations within the 158 nonmutator strains, major autosomes 2 and 3, and nonrepetitive regions (see Methods for additional details and Supplemental Data File 2 for repeat regions masked). This procedure reduced the total number of single base pair mutations from 3220 to 2141 (the majority removed are from mutator strains). We work with these 2141 mutations for our analyses; however, we also make the entire data set available for download (Supplemental Data File 1).
A comparison of experiments and single base pair mutation rates can be found in Table 1. Our mutation rate is significantly higher than that reported by the homozygous MA studies of both Keightley et al. (2009) (Poisson exact P = 2 × 10−4) and Schrider et al. (2013) (Poisson exact P = 3 × 10−6), significantly lower than that reported by the heterozygous MA of Sharp and Agrawal (2016) (Poisson exact P = 1.5 × 10−3), and not significantly different from Huang et al. (2016) (Poisson exact P = 0.35) (see Methods and Supplemental Table S4 for additional details). These differences could be driven by differences in genetic background or experimental design, although we note that studies which used heterozygous accumulations, fewer generations, and newer sequencing technologies tended to have higher mutation rate estimates.
Summary of the five mutation accumulation experiments
The neutral expectation is reached in all five experiments
We next use functional regions of the genome to test whether the spectrum of MA mutations is truly unbiased by natural selection. In natural populations, functionally important mutations typically are at low frequencies due to purifying selection; however, we expect de novo mutations in coding regions to consist of 75% nonsynonymous mutations and 4% nonsense mutations (this expectation is not changed by codon usage bias) (see Supplemental Materials). As can be seen in Figure 2, A and B, the five mutation accumulation experiments do indeed exhibit the expected fractions of 4% nonsense (no significant difference between experiments, Fisher's exact P = 0.82) and 75% nonsynonymous mutations (χ2 test, P = 0.034). While the χ2 test for nonsynonymous mutations satisfies our P > 0.01 threshold for insignificance, note that any difference between experiments is primarily driven by the Schrider et al. (2013) study, in which 90% (CI 76%–97%) of coding mutations caused a nonsynonymous change.
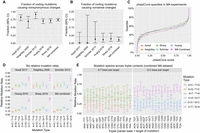
A summary of comparisons conducted between the five different MA experiments, including (A) the fraction of coding mutations which cause nonsynonymous changes, where the dotted line indicates the neutral expectation of 75%, (B) the fraction of coding mutations which cause nonsense changes, where the dotted line indicates the neutral expectation of 4%, (C) the empirical cumulative distribution for phastCons scores within each MA experiment, (D) the six relative mutation rates (i.e., sum to 1) within all nonrepetitive regions, and (E) the six relative mutation rates calculated across different triplet base contexts, within all nonrepetitive regions.
Another method to test if the MA mutations were generated in the absence of selection is to classify all sites in the genome by a conservation score and then ask whether the distribution of scores for MA mutations matches the expectation from the reference genome's distribution. To do this, we employed the publicly available phastCons scores for D. melanogaster, which is a measure of evolutionary conservation across twelve Drosophila species, mosquito, honeybee, and the red flour beetle (Siepel et al. 2005). Indeed, as we can see in Figure 2C, the distribution of phastCons scores are similar across MA experiments and not significantly different from the neutral expectation as given by the distribution of scores in the reference genome (bootstrap Kolmogorov-Smirnov [KS] test, P = 0.31).
Mutational spectra are comparable across all five experiments
We collapsed all MA mutations into their six basic mutation types and calculated the relative rate of each mutation type in each experiment (after first scaling for the D. melanogaster genome GC content of 43%) (Fig. 2D; Supplemental Table S5). We find that these six relative rates are significantly different across experiments (χ2 test, P = 0.003); however, the P-value is not very low. We find neither a specific mutation type nor a specific experiment to be driving the variation (tests of single mutation types or single experiments against the sum of the others gave χ2 test-corrected P-values > 0.01). The C → T/G → A mutation is by far the most common, and its relative frequency does not differ significantly across MA experiments (χ2 test, P = 0.15). It occurs at a relative rate of 0.4 in the combined data set (95% CI 0.38–0.42) (bottom right, Fig. 2D), which is ∼7× the rate of the least common A → C/T → G mutation. Note that this elevated rate occurs despite the paucity of cytosine methylation in D. melanogaster (known to elevate the C → T/G → A rate even higher in other organisms) (Takayama et al. 2014; Goldmann et al. 2016).
We can now also look at transition:transversion ratios and GC equilibrium of the mutational process. When considering the number of transition mutations (two possible types) and transversion mutations (four possible types), we find transition:transversion ratios that are not significantly different across experiments (G test of independence, P = 0.21), and the combined data set has a ratio of 2:1 (95% CI 1.9–2.2) (Table 1). Next, by considering mutations which change the GC content of the mutated base pair, we ask if mutation drives the genome more toward A:T pairs or more toward G:C pairs. We use the GC equilibrium metric to do this, which has only been reported by one other MA experiment (Keightley et al. 2009), and, as can be seen in Table 1, we find the GC equilibrium to be significantly different between studies (G test, P = 0.01). Interestingly, while the oldest MA paper reported a GC equilibrium of 30% (with a large CI of 24%–40%) (Keightley et al. 2009)), we find that newer studies consistently have lower values. For the combined data set, the GC equilibrium reaches ∼23% (95% CI 0.21–0.25). In contrast, the D. melanogaster genome has an actual GC content of 43%, emphasizing the importance of nonneutral processes in driving the genome GC content higher (Galtier et al. 2001; Hershberg and Petrov 2010; Lartillot 2013).
Lastly, we test for neighbor-dependent variation in the mutation spectrum. There is evidence in some organisms that single base pair mutation rates can vary depending on the neighboring base pair context (Zhu et al. 2014; Aggarwala and Voight 2016; Sharp and Agrawal 2016). To test this in D. melanogaster using our combined MA data set, we considered triplet contexts in which the center base is mutated. All possible triplets were collapsed into their forward/reverse sequence, and then we quantified the relative rates for the three mutation types that can occur within each triplet (e.g., CAG triplet can get A → T, A → C, or A → G mutations). In contrast to quantifying the total mutation rate, we use relative rates because it provides an internal control for the triplet content of the reference genome. This triplet content will vary across MA publications depending on which base pairs were masked during their analysis pipelines (information that is not consistently documented across publications). Using the combined set of 2141 de novo mutations from the five MA experiments, we do, in fact, find heterogeneity in the mutation spectrum across triplet contexts (G test of independence, P = 0.008 and P = 0.007 for GC and AT base pairs, respectively) (Fig. 2E). However, 2141 mutational events is not a large enough data set to detect whether any particular triplet is driving the heterogeneity (G test-corrected P-values > 0.01) (Supplemental Table S6). Thus, despite compiling here the largest Drosophila data set of de novo mutations yet available, an even greater number of mutational events is needed in order to perform more fine-scale analyses.
Rare polymorphisms identified in natural populations
Identification and validation of rare polymorphisms
As a proxy for new mutations, we seek to identify a class of ultra-low-frequency polymorphisms. To this purpose, we used three publicly available data sets and employed the method depicted in Figure 3 and briefly described here: (1) We identified rare genetic variants outside of repetitive regions using 621 individually sequenced monoallelic genomes (i.e., haploid or inbred) provided by the DGN (Drosophila Genome Nexus) (Lack et al. 2015), such that we had singletons at frequency ∼1/621, doubletons at ∼2/621, etc. Then (2), we filtered singletons down to a set of extremely rare polymorphisms by removing those which appeared in Nescent data, a sequencing project which pooled wild-caught flies from North America to obtain >4000 pooled genomes (SRA accession SRP075757) (Bergland et al. 2014; Kapun and Fabian 2017). This gave us a set of high-quality singletons at an order-of-magnitude lower frequency (∼1/5000). Lastly (3), we used resequence data available for 29 of the individual genomes to validate a subset of the data set (data publicly available from the DGRP (Drosophila Genetic Reference Panel) (Mackay et al. 2012) and DPGP1 (Drosophila Population Genomics Project Release 1) (http://www.dpgp.org/1K_50genomes.html#Reference_Release_1.0; SRA accession number PRJNA3009). This procedure reduces the number of polymorphisms down to only those that appeared in the 29 resequenced strains; however, this data set is of extremely high quality because each genetic variant has been observed independently at least twice—protecting our data set against the problem of confounding rare variation with sequencing and mapping errors. Note that confirmed genetic variants are annotated as ∼1c/5000, ∼1c/621, ∼2c/621, etc.

Pipeline for identification and validation of rare polymorphisms. Step 1 data set is from the Drosophila Genome Nexus (DGN) (Lack et al. 2015) which represent predominantly monoallelic genomes (i.e., either haploid or inbred) from 35 populations across three continents that were sequenced to high depth and underwent the same iterative mapping pipeline before variant calling. Step 2 data set consists of pooled sequencing data generated by our and collaborating labs which collectively represent >4000 genomes from the eastern US and Europe. Step 3 data set is resequence data made available by the Drosophila Genetic Reference Panel (DGRP) (Mackay et al. 2012) and DPGP1 (http://www.dpgp.org/1K_50genomes.html#Reference_Release_1.0; SRA accession number PRJNA3009) projects, which used Roche454 and Illumina technology (respectively) to independently resequence 29 of the strains present in the DGN.
By looking closer at the different steps of our pipeline, we found that indeed the proportion of artifactual variant calls increases as their frequency decreases. While common variation is confirmed in the resequence data at a rate of ∼100%, rare variation is only confirmed at a rate of ∼70% (Supplemental Fig. S1B–D). The observed low confirmation rates for rare polymorphisms appear to be mainly driven by low complexity and indel-rich regions (Supplemental Figs. S1C,D, S2). Furthermore, we find that applying filters to the genotype calls (using QUAL, DP, QD, etc.) only brings the confirmation rate to ∼70% for standard filters or ∼90% for severe filters (Supplemental Fig. S1E; Supplemental Materials). This means that our filters could not ameliorate the problem of artifactual calls in a data set of rare genetic variation. As a consequence, it was absolutely critical to validate rare variation using resequence data. The final count of rare polymorphisms that we will use in this study, all confirmed via resequencing, can be seen in Table 2 and variants found in Supplemental Data File 3.
Count of polymorphisms for each frequency class when looking across the entire data set, the resequenced data set, and the data set of variants resequenced and confirmed
Rare polymorphisms approach the neutral expectation within coding regions
We next sought to confirm that, in contrast to common polymorphisms, the rarest class of polymorphisms approaches the neutral expectation for new mutations. Recall that coding mutations should cause a nonsynonymous change 75% of the time, and in the MA data, 73.2% of mutations are nonsynonymous (CI 68.9%–77.1%). In our polymorphism data set, we find that common variation consists of only 17.9% nonsynonymous changes (CI 17.7%–18.2%); however, our rarest polymorphism set consists of 67.2% nonsynonymous changes (CI 64.1%–70.3%), which is significantly higher and approaches the neutral expectation (G test of independence, P-value < 2.2 × 10−16) (Fig. 4A). We can also look at the fraction of mutations in coding regions which cause nonsense changes, noting that the neutral expectation is ∼4% (MA data consist of 1.94% nonsense changes with a large CI [0.97%–3.70%] due to low counts). We find that 1.03% of rare polymorphisms are nonsense changes (CI 0.51%–1.97%, not significantly different from MA data set). This is significantly higher compared to common polymorphisms which consist of only 0.06% nonsense changes (CI 0.05%–0.09%; G test of independence, P-value = 1.9 × 10−8) (Fig. 4B).
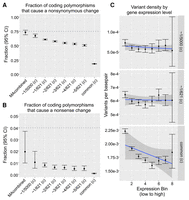
Rare polymorphisms approach the neutral expectation in terms of (A) the fraction of events causing nonsynoymous changes, (B) the fraction of events causing nonsense changes, and in (C) where, unlike common polymorphisms, rare polymorphisms occur within transcribed regions at a rate insensitive to levels of germline expression.
Another signature of natural selection we can check for is the density of polymorphisms within genes that are expressed in the germline. We would suspect that, for common polymorphisms, there would be a negative correlation between their density within a gene and expression level, reflecting natural selection purging deleterious mutations from important genes. If our rare polymorphisms indeed capture the neutral expectation, then we should find that this negative correlation would disappear (and in the case of transcription being mutagenic [Polak et al. 2010; Jinks-Robertson and Bhagwat 2014], we should find this correlation to turn positive). To test this, we downloaded the publicly available expression data generated from the D. melanogaster germline by the modENCODE project (Graveley et al. 2011) and measured the density of polymorphisms within genes that are binned by expression level (bin levels 1–8 for low-to-high expression) (see Methods). We find that common polymorphisms indeed display a negative correlation between their density and expression level within genes, and this correlation disappears for the rare frequency classes of polymorphisms (Fig. 4C). This result confirms again that these data approach the neutral expectation and suggests that transcription may have no mutagenic effect in D. melanogaster.
We have shown that rare polymorphisms indeed approach the neutral expectation; however, there remains a small “missing” fraction of deleterious events, presumably because natural selection is efficient enough to remove them even at rare frequencies. Noting that the rarest frequency class has ∼67.2% nonsynonymous mutations, this is ∼8/75 = 11% of nonsynonymous mutations that are likely strongly deleterious, as they were unable to reach a frequency of ∼1/5000 = 0.0002. Similarly, the rarest frequency class consists of only ∼1% nonsense changes where we expect 4% from neutrality, and thus approximately three-quarters of the nonsense mutations are missing. We performed additional analyses to probe the identity of this missing fraction, including looking at their phastCons score distributions and performing a GO analysis; however, we did not find any compelling results (Supplemental Table S8; Supplemental Fig. S6). We also checked whether balanced recessive lethals may account for missing deleterious mutations by looking at heterozygous sites (which are not in the primary DGN data set) and found no evidence supporting this hypothesis (Supplemental Fig. S7). This, however, does not preclude the possibility that the small missing fraction may be a result of an inbreeding process that allowed strongly deleterious recessive alleles to be purged from the genome (Charlesworth and Willis 2009).
Six relative mutation rates and how they are context-dependent among rare polymorphisms and fourfold synonymous site substitutions
We have calculated the relative rates of the six mutation types across frequency classes (coding regions only) (Supplemental Fig. S9A). We find that, while common polymorphisms have a spectra significantly different from the mutations that occurred during MA experiments (χ2-test, P-value < 2.2 × 10−16) (Supplemental Fig. S9), the rarest polymorphisms have relative mutation rates which approach the MA spectra (χ2-test comparison with MA gives P-values = 0.0003 and 0.06 for rare polymorphisms at frequencies ∼1/5000 and ∼1/621, respectively) (Supplemental Fig. S9A). Differences between the rare polymorphisms at frequency ∼1/5000 and MA data are driven by the C → T/G → A mutation type (χ2-tests without this mutation class give corrected P-values > 0.01).
Recall that, using the MA data set of 2141 mutations, we were able to detect significant heterogeneity in the mutation spectrum across triplet contexts; however, we were unable to detect whether particular triplets were driving the variation (Fig. 2E). Now, with our data set of ∼70,000 rare polymorphisms, we can again ask whether the mutational spectrum is dependent on neighbor context (Fig. 5B). To this end, we again collapsed all possible triplets into their forward/reverse sequence and then quantified within each triplet the relative rates of the three mutation types that can occur at the center base pair of the triplet. We then tested for heterogeneity in the mutation spectrum and indeed found a significant effect of triplet context (G test, P-value < 2.2 × 10−16 for both GC and AT base pairs) (Fig. 5B). Additionally, we find 6/16 triplets centered at G:C base pairs to have significant effects and 14/16 triplets centered at A:T base pairs to have significant effects (G tests-corrected P-values < 0.01) (Supplemental Table S9).
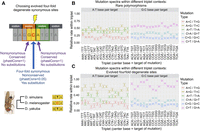
Six relative rates. (A) Schematic of how fourfold synonymous sites were chosen: The center base of the triplet acquired a substitution on the D. melanogaster branch and is conserved in the rest of the Drosophila tree, and the outer bases of the triplet are conserved across the entire Drosophila tree. (B) Six relative rates within singletons (∼1(c)/621) calculated across different triplet contexts in nonrepetitive regions, and (C) six relative rates within substitutions at fourfold synonymous sites, calculated across different triplet contexts. Note that the six relative rates within C are significantly closer to the six relative rates within B than is expected by chance (P < 0.001), indicating that mutational patterns within rare polymorphisms have predictive power for evolution at synonymous sites.
Lastly, we wished to test whether the measured heterogeneity in the mutation spectrum across triplet contexts would have any predictive power during the course of Drosophila evolution. In particular, we were curious whether our results might be applicable to codon-usage bias at 4D (fourfold degenerate) synonymous sites, in which the relative contribution of mutation has yet to be fully understood (Plotkin and Kudla 2011; Gilchrist et al. 2015). This was an intriguing question due to the fact that the nonsynonymous sites on either side of a synonymous site can provide a triplet context that is fixed over evolutionary timescales (see schematic in Fig. 5A). It is then possible that base identity, and thus codon usage, at a synonymous site may be influenced by neighboring bases that cause mutational biases.
In order to test this, we first measured triplet context-dependent patterns of 4D site substitutions (note we excluded sixfold degenerate codons L, R, and S). We identified nonconserved fourfold synonymous sites (phastCons ≤ 0.05) which have fixed and highly conserved neighbors (phastCons = 1) (Lawrie et al. 2013). As depicted in Figure 5A, we required that the conserved neighbors have a fixed identity across the Drosophila tree and required the fourfold synonymous site to have a substitution occur in only the D. melanogaster branch. Using these data, we then measured the context-dependent effects as before, where we quantified the relative substitution rates at the center base pair of each triplet (Fig. 5C). We found significant heterogeneity across triplet contexts in the spectra of substitution types in D. melanogaster (G test, P-value < 2.2 × 10−16 for both A:T and G:C base pairs) and significant effects of 10/16 and 12/16 triplets centered at A:T and G:C base pairs, respectively (G tests-corrected P-values < 0.01) (Supplemental Table S10).
We were then able to compare the measured triplet context-dependent patterns of 4D site substitutions with rare polymorphisms. We conducted a permutation test as follows: (1) The total G-value was found by summing G-values for each triplet (where rare polymorphisms give the expectation and substitutions are the observed), and then (2), the triplet labels of the substitutions were permuted and the total G-value was recalculated, and (3) this permuted G-value was obtained for 1000 different randomizations. We found that the total G-value of the original observed substitution data fell below the zero percentile of the distribution of G-values for the permuted data. This result shows that neighbor-dependent mutational patterns, as predicted by the spectrum of rare polymorphisms, indeed have a significant impact (P < 0.001) on the evolution of codon usage at fourfold synonymous sites. Thus, neutral evolution is likely contributing to codon-usage rates via the mutational biases caused by synonymous sites held within long-term triplet contexts.
Equilibrium GC content was impacted by neighbor context but not by recombination
It has been observed before that GC-rich regions tend to favor nucleotide changes toward G:C base pairs, specifically for common polymorphisms (Haddrill and Charlesworth 2008); however, it is unclear whether this pattern is driven by selective or mutational forces. To address this question, we tested whether mutational GC equilibrium in our data is dependent on the GC content of neighboring bases. To this end, we collapse triplet contexts to both strand-indifferent (i.e., an A:T neighbor base pair is the same as a T:A neighbor base pair) and site-indifferent (i.e., the center base can be A, T, C, or G) contexts, such that there are only three contexts total (see legend of Fig. 6A). Note that the only characteristics thus distinguishing these three neighbor contexts is the GC content. We can now calculate the GC equilibrium at the center site using data from the MA experiments and the rare and common polymorphisms. Interestingly, we find a positive correlation between GC equilibrium and the GC content of the neighboring base pairs (Fig. 6A; Supplemental Fig. S8). These correlations are not significant for the MA combined data set (P = 0.11), although a trend is clear in Fig. 6A, but do meet the significance threshold for the rare polymorphism data set (P = 0.01). This result suggests two equally interesting possibilities—either selection is driving GC-bias in GC-rich regions even among the rarest polymorphism class or, perhaps more likely, mutational forces are contributing to GC-biased nucleotide changes within GC-rich regions.
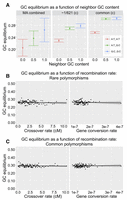
GC equilibrium. (A) The GC equilibrium in nonrepetitive regions as a function of the GC content of neighboring bases, within MA, singletons, and common polymorphisms. (B) GC equilibrium (using singletons ∼1(c)/621) in nonrepetitive regions as a function of the recombination rate, and (C) GC equilibrium (using common(c) polymorphisms) in nonrepetitive regions as a function of the recombination rate.
In some organisms, it has been found that recombination promotes mutation (Arbeithuber et al. 2015). It can be difficult to test for whether recombination is mutagenic due to the confounding effect of selection. Natural selection is more efficient in regions of higher recombination and consequently can cause a positive correlation between diversity levels and rates of recombination (Charlesworth and Campos 2014)—the same signature we would expect to find if recombination is mutagenic. However, we can employ the GC equilibrium metric to test for whether recombination affects the spectrum of mutation types. If, for example, as has been found in humans (Arbeithuber et al. 2015), recombination inflates the rate of C → T transitions relative to nonrecombining regions, we would then expect GC equilibrium to decrease with increasing recombination rate. To measure the relationship between recombination and GC equilibrium, we downloaded publicly available genome-wide estimates of both crossover and gene conversion rates (Comeron et al. 2012) and estimated GC equilibrium as a function of these recombination rates using different frequency classes of polymorphisms. We find no correlation of GC equilibrium with either crossover or gene conversion rates (Fig. 6B,C), thus suggesting that recombination does not alter the spectrum of new mutations.
Multinucleotide mutations comprise ∼4%–10% of rare polymorphisms, significantly more than is expected by chance
We last tested whether singletons cluster together significantly more often than would be expected if all events occurred independently (which, if true, suggests that single mutational events may cause multinucleotide mutations). The first measure we used was the relative proportions of the different types of multinucleotide mutations, which can be seen in Figure 7A and Table 3. The nearest neighbor distance was calculated for every singleton (i.e., distance to the closest neighboring singleton within the same strain), and the expectation was calculated by permuting the strain IDs across all singletons and recalculating the nearest-neighbor distances for each sample's singletons, and then taking the average of 500 permutations. As can be seen in Figure 7A and Table 3, 4% of singletons occur in clusters of 2–5 bp (corresponding to distances of 1–4 bp), where the expectation is only 0.02%. Note that this dramatic enrichment of multinucleotide mutations is robust to a number of strategies for calculating the expected distribution (Supplemental Fig. S3). Among the singletons occurring at distances of 1–4 bp from each other, about a quarter of them are “duples,” a pair of singletons directly next to each other, and the rest are singletons which occur up to 4 bp away from another singleton in the same strain (Table 3). Interestingly, there are even significantly more singletons clustering in the 0.3-kb- to 1.0-kb-range than is expected by chance, suggesting regional increases in mutation rate may occur as well.
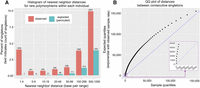
Multinucleotide mutations occur more often than is expected by chance. (A) Histogram of nearest neighbor distance, where every singleton (freq ∼ 1/621) was assigned the distance which was the shorter of the two distances on either side (within a given individual). The expectation is taken from the average of 500 permutations of sample IDs. Note that a 1–4 bp distance corresponds to a cluster of size 2–5 bp. (B) Quantile-quantile plot of distances between consecutive singletons (on both sides of singletons, within an individual), using 1% quantiles (beginning at 0.5%). The expectation is taken from an exponential distribution with a rate equal to the rate within the observed data. The purple inset shows a magnified view of the 0.5%–8.5% quantiles, such that the enrichment of multinucleotide mutations can be seen in the vertically plotted points at the start of the distribution.
Count of multinucleotide events within singletons (freq ∼ 1/621)
This skew toward shorter distances can also be seen by considering that, if mutations occurred independently, then we expect the distances between consecutive singletons (within a given individual) to be exponentially distributed. To test this, the quantiles of the distances within the sample data were plotted against the quantiles of an exponential distribution with the rate equal to the average singleton rate across all strains. As can be seen in Figure 7B, the observed sample data have a distribution that is skewed toward smaller distances.
Discussion
In order to make precise measurements of mutational rates and patterns, we have united multiple data sets and approaches, both experimental and computational, to generate the largest and highest quality data set of de novo mutations and rare polymorphisms yet available in Drosophila.
In our meta-analysis of MA data, we find that the spectrum of mutation types is remarkably similar across experiments, while the single base pair mutation rate is significantly different. At first glance, this seems a surprising result; however, upon closer consideration, these observations may not be so incongruous. The published studies which reported lower mutation rates tended to use homozygous accumulation, older technologies, and higher generation numbers. Any or all of these might cause a difference in mutational rates but not necessarily in mutational patterns. For example, if the varying detection rates of different technologies (i.e., old vs. new) do not vary by mutation type, then the mutation spectrum should not vary either. Or, alternatively, if the amount of selection against recessive deleterious mutations matters (i.e., homozygous vs. heterozygous MA), there is little reason to suspect that recessive mutations would have a different spectra of mutation types (MacArthur et al. 2012; Narasimhan et al. 2016). There is also recent evidence from Sharp and Agrawal (2016) that suggests the single base pair mutation rate and spectra are robust to experimental design. In their work, the MA experiment was done in different genetic backgrounds (wild type and deleterious), and they found a difference in the fitness decline over time that was mediated by a difference in the indel mutation rate, not the single base pair mutation rate or spectra. Interestingly, it has also been found in yeast that the single base pair mutation rate is consistent across MA strains and experiments, but the indel mutation rate is not (Behringer and Hall 2016b). Overall, we think the significantly different total mutation rates across Drosophila MA strains do not reflect a fundamentally different mutational spectrum across strains but rather a difference in experimental methods.
Overall, these observations validate the MA approaches for characterizing the spectra of new single base pair mutations and allow combining the data across the five experiments and 158 lines into one large data set. We have made the entire meta-data set available.
In our rare polymorphism analysis, we have united disparate genomic resources in the D. melanogaster community to generate the first massive (∼70,000) set of high-quality, fully resequenced, rare polymorphisms in D. melanogaster, with which we precisely measure mutational patterns across the genome. Our finding that rare variants are conflated with artifactual genotype calls at a high rate, even when called in high-coverage genomes and with severe filtering on quality scores, is of broad interest to the genomics community because the majority of genetic variants segregating in natural populations are rare. Additionally, many widely used statistical tests that rely on the site frequency spectrum are sensitive to erroneous rare variant calls (Johnson and Slatkin 2008). Our work rearms that artifactual variant calls disproportionately affect rare variants and that it would be best to incorporate resequencing into any study which analyzes them.
Our data set of rare polymorphisms consists of ∼68% nonsynonymous changes, close to the neutral expectation reached in MA experiments (∼73%). This corroborates recent work in yeast, which demonstrated that ∼1400 rare polymorphisms display minimal signatures of selection in clinical strains (Zhu et al. 2017). Using our data set, we were able to detect significant fine-scale heterogeneity in the mutation spectrum across different sequence contexts (“triplets”). Note that context-dependency of mutation has been detected in other organisms, including humans (Aggarwala and Voight 2016; Sharp and Agrawal 2016; Zhu et al. 2017). Our novel contribution here is, in addition to the highest precision estimate of context-dependency yet available in Drosophila, a demonstration that our detected mutational patterns are relevant to the course of evolution within coding regions. The context-dependent rates of mutations, as measured from rare polymorphisms, predict the spectra of substitutions which occurred at fourfold synonymous sites in the D. melanogaster phylogenetic branch. This shows that, in addition to forces like selection for translational efficiency (Plotkin and Kudla 2011; Poh et al. 2012) or biased gene conversion (Clément and Arndt 2013; Figuet et al. 2014), the mutation process itself is contributing to biased codon usage patterns at synonymous sites.
Additionally, we established in both MA and rare polymorphism data that mutational processes by themselves are expected to drive the genome GC content to ∼25%, despite the fact that genome-wide GC content in Drosophila is ∼43%. This low GC equilibrium is largely due to an elevated C → T/G → A mutation rate (∼7× the least common), occurring despite the paucity of cytosine methylation in D. melanogaster (which, for example, in humans drives the rate to ∼11× higher than the least common mutation type [Takayama et al. 2014; Goldmann et al. 2016]). Our finding is consistent with previous work in Drosophila and yeast in which elevated C → T rates were found despite minimal methylation in the genome (Petrov and Hartl 1999; Zhu et al. 2014; Behringer and Hall 2016a), suggesting that the sensitivity of cytosines to mutation may be a general feature of cytosines in a cellular context.
Many species have an actual genome GC content significantly higher than expected from mutation alone (Hershberg and Petrov 2010), which presents the question of what forces are driving the genome GC content to such high values in general, and in Drosophila in particular. Although weak selection and/or biased gene conversion have been implicated in the evolution of high GC content in many species, it is unlikely in Drosophila. This is because the common polymorphisms do not display a substantial bias toward higher GC values (at only ∼28%), and neither does this bias increase with recombination rates. Both of these patterns would be expected under the models of weak selection or biased gene conversion. Instead, the high GC content of the Drosophila genome likely reflects its high functional density and the elevated GC content of those functional sequences (coding at ∼54% GC). Consistent with that model, the parts of the genome that are expected to have lower functional density do have substantially lower GC content. For example, the average GC content of short introns is ∼32% (Clemente and Vogl 2012), although given our finding of ∼25% GC equilibrium, this means even the gold standard for neutrality within the Drosophila genome (i.e., short-introns) may possibly still be under constraint.
We observe another interesting relationship between genome GC content and GC equilibrium—a correlation between the mutational GC equilibrium and the local genome GC content such that mutational processes drive the GC content up in GC-rich neighborhoods (or, since GC equilibrium is only ∼25%, we can say GC is driven lower in already GC-poor regions). It has been observed before that common polymorphisms in intergenic regions display this same pattern (Haddrill and Charlesworth 2008; Clemente and Vogl 2012), and it has been thought that such a pattern is largely driven by selective forces. However, our data set of de novo MA mutations and also rare polymorphisms is large enough to show that the pattern persists even among genetic variants that have little to no filtering from both natural selection and biased gene conversion. Thus, while mutational processes drive genome GC content down and selective forces drive genome GC content up, we find that mutation is most effective at driving GC content down in regions that are already GC-poor.
Lastly, our finding that multinucleotide mutations occur significantly more than is expected by chance both confirms and extends previous findings in the literature. In Drosophila MA studies which used a set of ∼1000 de novo mutations (Schrider et al. 2013; Sharp and Agrawal 2016), it was found that ∼3%–4% of single base pair events occur in clusters of size ≤50 bp, coinciding closely with our finding of ∼6% (in which ∼4% are ≤5 bp clusters and ∼2% are 6–50 bp clusters). Similar rates of small multinucleotide clusters have been found in other organisms (Schrider et al. 2011; Harris and Nielsen 2014; Besenbacher et al. 2016). With our larger data set, we can take the analysis a step further and find that as many as ∼10% of single base pair mutations occur in clusters of size 0.3–1 kb (where the expectation is only ∼4%). This result is consistent with some recent work done in humans (Besenbacher et al. 2016; Goldmann et al. 2016) which also suggests that such regional increases in mutation rate may be a common occurrence in the genome.
In combination, the MA approach and the rare polymorphism approach have provided complementary methods for studying the spectrum of new mutations, enabling a precise estimate of both total mutation rates and subtle mutational biases. We hope, with an ever growing catalog of deep sequence data from natural populations being made available to the scientific community, that researchers will take advantage of the opportunity to apply the methods described here to studying mutational patterns in other organisms.
Methods
Mutation accumulation
The strains used were DGRP RAL-765 (ancestor) and an hshid strain 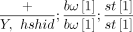 (marked stock). A single male RAL-765 fly (note that males have little-to-no recombination) was crossed to six virgin hshid
females and a single red-eyed male progeny then crossed to six hshid virgins. From this, 50 red-eyed male progeny were used
to found 50 MA strains. In every generation, a single red-eyed male was crossed to three hshid virgin females. Seventeen lines
out of the original 50 were sequenced (it is common for MA lines to die off). We used 5–15 red-eyed flies in generations 36,
37, 49, and 53 to extract DNA (Huang et al. 2009). Paired-end barcoded DNA sequencing libraries were prepared with an Illumina Nextera DNA Library Preparation kit (#FC-121-1031)
and Index kit (#FC-121-1012) and a KAPA Biosystems Library Amplification kit (#KK2611). The DGRP, hshid, and MA libraries
were sequenced on a HiSeq 2000 to a depth of 20–25×, and genetic variants unique to a MA strain were labeled de novo mutations.
Repetitive regions filtered included RepeatMasker (http://www.repeatmasker.org), a run of TRF (Benson 1999) on the Drosophila reference, and a list of annotated transposable elements (Fiston-Lavier et al. 2011). After masking repetitive regions, the total genome length for Chromosomes 2 and 3 was 87,130,614 base pairs. The total
number of MA generations (762) was multiplied by the total number of post-filtered base pairs to get 66,393,527,868, the denominator
of the mutation rate calculation.
(marked stock). A single male RAL-765 fly (note that males have little-to-no recombination) was crossed to six virgin hshid
females and a single red-eyed male progeny then crossed to six hshid virgins. From this, 50 red-eyed male progeny were used
to found 50 MA strains. In every generation, a single red-eyed male was crossed to three hshid virgin females. Seventeen lines
out of the original 50 were sequenced (it is common for MA lines to die off). We used 5–15 red-eyed flies in generations 36,
37, 49, and 53 to extract DNA (Huang et al. 2009). Paired-end barcoded DNA sequencing libraries were prepared with an Illumina Nextera DNA Library Preparation kit (#FC-121-1031)
and Index kit (#FC-121-1012) and a KAPA Biosystems Library Amplification kit (#KK2611). The DGRP, hshid, and MA libraries
were sequenced on a HiSeq 2000 to a depth of 20–25×, and genetic variants unique to a MA strain were labeled de novo mutations.
Repetitive regions filtered included RepeatMasker (http://www.repeatmasker.org), a run of TRF (Benson 1999) on the Drosophila reference, and a list of annotated transposable elements (Fiston-Lavier et al. 2011). After masking repetitive regions, the total genome length for Chromosomes 2 and 3 was 87,130,614 base pairs. The total
number of MA generations (762) was multiplied by the total number of post-filtered base pairs to get 66,393,527,868, the denominator
of the mutation rate calculation.
MA data from references
MA data were drawn from the following references: Keightley et al. (2009); Schrider et al. (2013); Huang et al. (2016); Sharp and Agrawal (2016). Lists of mutations were downloaded from each publication and combined to generate a VCF of all mutations. For the “MA combined” data set, we filtered out repetitive regions, removed mutator lines (line 19 from Huang et al., and 33-27, 33-45, 33-5, and 33-55 from Schrider et al.), and subsetted to major autosomes 2 and 3. For comparisons of the mutation rates with a Poisson exact test, we required information on genome size, which was incomplete across publications. Given the mutation rate µ = m/(n × t × l) (where m = mutation count, n = strain count, t = generation count, and l = base pair count), we back-calculated l, which is in the Supplemental Materials.
DGN rare variant calling
Sequences of the 623 genomes provided by the Drosophila Genome Nexus (Lack et al. 2015) were downloaded and and repeat regions masked. Additionally, the DGN indel VCFs were downloaded and indel locations masked (±5 bp). Resequence data used to confirm a subset of the variants was obtained from the DPGP1 project's Solexa (now Illumina) sequencing (http://www.dpgp.org/solexa_release_1/dpgp_solexa_r1.0.tar) and from the DGRP (Mackay et al. 2012) project's Roche 454 sequencing (ftp://ftp.hgsc.bcm.edu/DGRP/freeze1_July_2010/snp_calls/454/). Pooled Nescent data were from Bergland et al. (2014), Kapun and Fabian (2017), and SRA accession SRP075757.
Variant annotation and analysis
VCFs for MA and DGN data were generated with in-house Perl scripts and final data loaded into R/Bioconductor (Huber et al. 2015; R Core Team 2015). Downstream analyses were performed with Bioconductor tools, including BSgenome.Dmelanogaster.UCSC.dm3 and TxDb.Dmelanogaster.UCSC.dm3.ensGene. Functional impacts of variants were annotated using predictCoding and locateVariants tools.
Data access
Sequence data from the MA experiment from this study have been submitted to the Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) under accession number SRP116884. Additional data available include: the mutations of all five MA experiments (Supplemental Data File 1), the coordinates of repetitive regions masked prior to analysis (Supplemental Data File 2), and the confirmed rare and common polymorphisms (after polarization) (Supplemental Data File 3).
Acknowledgments
We thank the members of the Petrov lab, particularly Alan Bergland, Ryan Taylor, Heather Machado, David Lawrie, and interns Leslie Chan and Katelyn Haduong. We also thank the Associate Editor and the three reviewers for helpful comments. We also thank the Nescent consortium which generated the pooled sequence data used here. This work was supported by the 6 Dimensions (Drosophila) National Institutes of Health Grant R01GM100366.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.219956.116.
- Received December 19, 2016.
- Accepted October 20, 2017.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








