
An atlas of alternative splicing profiles and functional associations reveals new regulatory programs and genes that simultaneously express multiple major isoforms
- Javier Tapial1,2,
- Kevin C.H. Ha3,4,6,
- Timothy Sterne-Weiler3,6,
- André Gohr1,2,6,
- Ulrich Braunschweig3,
- Antonio Hermoso-Pulido1,2,
- Mathieu Quesnel-Vallières3,4,
- Jon Permanyer1,2,
- Reza Sodaei1,2,
- Yamile Marquez1,2,
- Luca Cozzuto1,2,
- Xinchen Wang3,7,
- Melisa Gómez-Velázquez5,
- Teresa Rayon5,8,
- Miguel Manzanares5,
- Julia Ponomarenko1,2,
- Benjamin J. Blencowe3 and
- Manuel Irimia1,2
- 1EMBL/CRG Systems Biology Research Unit, Centre for Genomic Regulation (CRG), The Barcelona Institute for Science and Technology, 08003 Barcelona, Spain;
- 2Universitat Pompeu Fabra (UPF), 08003 Barcelona, Spain;
- 3Donnelly Centre, University of Toronto, Toronto, Ontario M5S 3E1, Canada;
- 4Department of Molecular Genetics, University of Toronto, Toronto, Ontario M5S 3E1, Canada;
- 5Centro Nacional de Investigaciones Cardiovasculares Carlos III (CNIC), 28029 Madrid, Spain
-
↵6 These authors contributed equally to this work.
Abstract
Alternative splicing (AS) generates remarkable regulatory and proteomic complexity in metazoans. However, the functions of most AS events are not known, and programs of regulated splicing remain to be identified. To address these challenges, we describe the Vertebrate Alternative Splicing and Transcription Database (VastDB), the largest resource of genome-wide, quantitative profiles of AS events assembled to date. VastDB provides readily accessible quantitative information on the inclusion levels and functional associations of AS events detected in RNA-seq data from diverse vertebrate cell and tissue types, as well as developmental stages. The VastDB profiles reveal extensive new intergenic and intragenic regulatory relationships among different classes of AS and previously unknown and conserved landscapes of tissue-regulated exons. Contrary to recent reports concluding that nearly all human genes express a single major isoform, VastDB provides evidence that at least 48% of multiexonic protein-coding genes express multiple splice variants that are highly regulated in a cell/tissue-specific manner, and that >18% of genes simultaneously express multiple major isoforms across diverse cell and tissue types. Isoforms encoded by the latter set of genes are generally coexpressed in the same cells and are often engaged by translating ribosomes. Moreover, they are encoded by genes that are significantly enriched in functions associated with transcriptional control, implying they may have an important and wide-ranging role in controlling cellular activities. VastDB thus provides an unprecedented resource for investigations of AS function and regulation.
A major goal of functional genomics is to identify and characterize the full repertoires of RNA and protein products that govern the development and maintenance of diverse cell and tissue types. Definition of such repertoires is further critical for understanding disease mechanisms. Alternative splicing (AS) is a widespread process by which splice sites are differentially selected in pre-mRNA to generate distinct RNA and protein isoforms. It provides a critical layer of gene regulation that impacts most if not all biological pathways in vertebrate species. Consequently, AS often causes or contributes to human diseases when misregulated (Licatalosi and Darnell 2006; Scotti and Swanson 2016).
Genome-wide profiling of AS using microarrays and RNA-seq revealed networks of functionally coordinated cassette exons that are significantly enriched in genes that operate in specific functional processes and pathways (Licatalosi and Darnell 2010; Kalsotra and Cooper 2011; Irimia and Blencowe 2012). Identification of these networks provided a valuable entry point for elucidating the functions of individual AS events. For example, characterization of splice variants controlled by the neural RNA binding regulators Nova1, Nova2, and Srrm4 (also known as nSR100) linked specific AS events to phenotypes seen in Nova1-, Nova2-, and Srrm4-deficient mice (Ule et al. 2005; Ruggiu et al. 2009; Allen et al. 2010; Quesnel-Vallières et al. 2015). Similarly, definition of a program of cardiomyocyte-specific AS revealed individual exons that regulate vesicular trafficking genes during postnatal heart development and adulthood (Giudice et al. 2014, 2016). These examples illustrate a few of the many cases in which genome-wide surveys of AS led to the discovery and characterization of important new gene functions at an exon-level resolution.
These studies emphasize the importance of compiling a comprehensive resource of quantitative AS profiles that can be mined for valuable information on exon and intron regulation and function. There thus exists a major need in the research community for readily accessible AS profiling data across the most widely studied cell and tissue types, as well as developmental stages. Moreover, when such information is overlaid with protein features, such as domains and motifs, it is possible to infer new and interesting testable hypotheses for the functional impact of specific isoforms (e.g., disruption of a protein–protein interaction). A resource compiling such information would provide immediate insight into which exon and intron sequences in a gene are differentially spliced in specific biological contexts as well as their possible functional roles.
In the present study, we have addressed these goals in the form of a new resource, the Vertebrate Alternative Splicing and Transcription Database (VastDB; http://vastdb.crg.eu/), that provides easily accessible, genome-wide information on the regulation and protein-level associations of tens of thousands of AS events across dozens of diverse tissues, cell types, and developmental stages in human, mouse, and chicken.
Results
New landscapes of AS in vertebrate species
We used vast-tools (Fig. 1; Supplemental Fig. S1; https://github.com/vastgroup/vast-tools) to comprehensively detect and quantify all major types of AS events from raw RNA-seq reads, including individual and complex combinations of cassette exons and microexons (AltEx), Alternative 5′ and 3′ splice site choices (Alt5 and Alt3, respectively), and intron retention (IR). This tool has been extensively used to detect and quantify tissue-specific and splicing factor–regulated AS programs in multiple organisms and experimental systems, yielding measurements of inclusion levels that have been validated at a high rate by RT-PCR assays (Han et al. 2013; Braunschweig et al. 2014; Irimia et al. 2014; Raj et al. 2014; Giampietro et al. 2015; Gueroussov et al. 2015; Quesnel-Vallières et al. 2015, 2016; Solana et al. 2016) (and see below, “Neural and muscle AS programs dominate tissue-differential transcriptomic profiles”). Moreover, vast-tools compares similarly or favorably with commonly used software for AS quantification in terms of computing time and memory usage (Supplemental Fig. S2A) and AS event discovery rate, particularly for microexons in the 3–15 nt range (Supplemental Fig. S2B,C). We benchmarked AS quantifications provided by vast-tools against MAJIQ (Vaquero-Garcia et al. 2016), MISO (Katz et al. 2010), and rMATS (Shen et al. 2014). For all types of AS events, vast-tools showed a comparable or higher accuracy for both Percent Splice In (PSI) and ΔPSI estimates using simulated (Supplemental Fig. S3; Supplemental Table S2) as well as RT-PCR data (Supplemental Fig. S4).
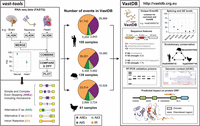
VastDB: An atlas of alternative splicing profiles and functional associations in vertebrate cell and tissue types. (Left) vast-tools was used to profile 1478 independent RNA-seq data sets comprising 108 human, 139 mouse, and 61 chicken sets of tissues, cell types, and developmental stages, which produced thousands of AS events of all major types (center). Each AS event possesses a unique and constant event identifier (EventID) across vast-tools and VastDB, allowing profiling of new RNA-seq samples with vast-tools and direct contextualization of interesting events in VastDB. (Right) For each EventID, VastDB provides functional information, including a graphical display of splicing and gene expression levels across samples, sequence features, suggested primers for RT-PCR validation, genomic context through the UCSC Genome Browser, and multiple protein features (e.g., domains, disorder regions, 3D structure). Moreover, EventIDs have a homology annotation interconnecting the AS events across the different VastDB species.
VastDB was assembled using vast-tools AS profiles from 1478 independent RNA-seq data sets comprising 108 human, 139 mouse, and 61 chicken sets of tissues, cell types, and developmental stages (Supplemental Table S1). We identified thousands of AS events representing all major AS event types (Fig. 1; Supplemental Fig. S5A). Comparison with Ensembl annotations revealed that substantial fractions of these AS events were not previously annotated: 6133 (13.9%) AS events in human, 6646 (20.2%) in mouse, and 11,704 (48.3%) in chicken (Supplemental Fig. S5B). Extensive RT-PCR validation data from the present and previous studies supported the robustness of our PSI measurements, despite using independent RNA samples for RT-PCR and RNA-seq (R2 = 0.81 for all exons, and R2 = 0.88 for exons shorter than 100 nt, which are less prone to PCR amplification bias) (Supplemental Fig. S5C,D). All raw PSI values are provided in Supplemental Table S3.
Interestingly, investigation of intragenic relationships between VastDB AS events reveals that pairs of cassette exons (especially in the case of neighboring exons) and pairs of retained introns within the same gene show highly significant correlation of inclusion or retention levels across samples from each species (P < 10−5, Wilcoxon rank-sum test) (Supplemental Fig. S6A,B). Specifically, most pairs of IR events within the same gene had strong positive coregulation, whereas pairs of exons from multiexonic AltEx events displayed a pronounced bimodal distribution, in which most pairs of exons are strongly positively correlated. For example, the five exons encoding the plectin domains in MACF1 in mammals are included or skipped as a unit (Supplemental Fig. S7), but a small fraction is strongly negatively correlated (involving mutually exclusive exons) (Supplemental Fig. S6C,D, blue arrows). In the case of IR, the coordinated retention of multiple introns may serve to increase the efficiency of this mechanism to down-regulate transcript levels to dynamically regulate or functionally tune gene expression (Braunschweig et al. 2014) (e.g., in the human HES1 gene) (Supplemental Fig. S8A,B). Remarkably, genes that contained at least five highly retained introns (but that generally contain additional introns that are not retained) (Supplemental Methods) were strongly enriched for Adenyl nucleotide binding and related terms in both human and mouse (Supplemental Fig. S8C). Furthermore, sets of genes with multiple retained introns significantly overlapped between these species (P = 1.89 × 10−53, one-sided Fisher's exact test), suggesting evolutionary selection to maintain this regulatory mechanism.
A subset of AS events is alternatively spliced across all tissues and cell types
When considered individually, most alternative exons are skipped or included in only a small fraction of the samples and together predominantly display a bimodal PSI distribution, with the majority of events displaying either high or low inclusion (Fig. 2A). However, when alternative exons are binned according to the percentage of samples in which they are alternatively spliced, this bimodal distribution progressively converts into a unimodal distribution centering on a median PSI of approximately 50 for exons that are alternative in most (>80%) of the samples (3516 exons in 2627 genes in human) (Fig. 2A). Remarkably, these AltEx events (hereafter “PanAS” events) are significantly enriched in genes that encode DNA binding proteins and that function in transcriptional and chromatin regulation (Fig. 2B, top). In contrast, switchlike AltEx AS events (hereafter “SwitchAS” events; events with a PSI > 90 or PSI < 10 in >80% of the samples, but with a range of PSIs ≥ 80 across samples) and tissue-specific AS events are enriched in gene functions such as cytoskeleton, cell adhesion, or GTPase regulator activity, consistent with previous studies (Fig. 2B, bottom; Ule et al. 2005; Fagnani et al. 2007; Warzecha et al. 2009; Han et al. 2013; Giudice et al. 2014; Irimia et al. 2014). Genes harboring PanAS exons include transcription factors with key developmental roles, e.g., TCF7L2, MEIS2, PBX1, MEF2A, MBD1 (Supplemental Fig. S9A), histone modifiers (particularly related to deacetylation and lysine modifications; e.g., HDAC7, NCOR1, KMT2A, and SETD2), and numerous genes linked to human disease (e.g., MECP2 and EIF4E) (Supplemental Table S4). RT-PCR assays confirmed the midrange PSI levels across tissues for all 12 tested AS events (Supplemental Fig. S9A,B), and PSI measurements obtained from 400 independent samples from 16 tissue types from the GTEx consortium (Melé et al. 2015) provided similar intermediate inclusion levels (Supplemental Fig. S10A). Likewise, Alt3 and Alt5 (but not IR) events that are alternatively spliced in most human samples displayed comparable Gene Ontology (GO) enrichment (Supplemental Fig. S10B–F), and similar results were obtained from analyzing mouse data (Supplemental Fig. S10G).
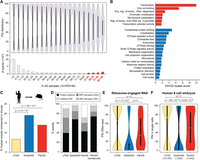
A subset of exons in genes encoding DNA binding proteins is alternatively spliced across most cell and tissue types. (A) Distribution of PSIs across all samples with sufficient read coverage (violin plots, top) and total number of exons (histograms, bottom) for bins of alternative exons that are alternatively spliced (10 < PSI < 90) in an increasing fraction of samples. PanAS exons correspond to those exons that are alternatively spliced in >80% of the samples (i.e., red histograms). (Red dots) median PSI of the bin. (B) Enriched Gene Ontology categories using DAVID scores for genes harboring exons that are alternatively spliced in >80% of the samples (PanAS events; red, four last bins on A) or show switchlike inclusion patterns (SwitchAS; blue). (C) Percentage of human low-frequency AS (LFAS; yellow), SwitchAS (blue), or PanAS (red) AltEx events that have the same class of regulation in mouse orthologous exons. (D) Percentage of human low-frequency AS, SwitchAS, PanAS, and conserved PanAS exons that are predicted to generate alternative ORF-preserving isoforms (black), disrupt the ORF when included/excluded (light/dark gray), or overlap noncoding sequences (white). (E) For each AS group, PSI distributions obtained from ribosome-engaged RNA-seq data from multiple cell types. (F) For each AS group, PSI distributions in individual human eight-cell stage embryo blastomeres. The number of tested exons for each category is provided in parentheses. All P-values correspond to two-sided Fisher's exact tests; for E and F, the numbers of events with PSIs corresponding to alternative (10 < PSI < 90) versus nonalternative events is compared for each AS group.
PanAS exons are significantly more often evolutionarily conserved between human and mouse than are exons that are alternatively spliced at a low frequency (hereafter “low-frequency AS” or “LFAS” events; events that are alternatively spliced [10 < PSI < 90] in 10%–25% of the samples; P = 1.9 × 10−26, two-sided Fisher's exact tests) (Fig. 2C). Moreover, a higher proportion of PanAS exons were predicted to preserve reading frame both when included and skipped (particularly for conserved PanAS events) or to reside in untranslated regions (UTRs) compared to low-frequency AS events, which are mostly frame-disruptive (Fig. 2D), suggesting that they have the potential to create coexisting alternative protein isoforms or perform regulatory functions in UTRs. In fact, exon–exon junction-based PSI quantifications using ribosome-engaged RNA-seq (Ribo-seq) data for different cell lines from various independent sources (Weatheritt et al. 2016) show that both transcript isoforms generated by PanAS exons are generally engaged by ribosomes and are thus likely translated (65.2% of PanAS events had PSI values between 10 and 90 in Ribo-seq data compared to 4.6% and 15.6% of SwitchAS and low-frequency AS exons, respectively; P < 2.2 × 10−16, Fisher's exact tests) (Fig. 2E). Moreover, similar to switchlike and tissue-regulated AS events (Buljan et al. 2012; Ellis et al. 2012), PanAS exons are significantly enriched in disordered protein regions (Supplemental Fig. S10H) and underrepresented in structured domains (Supplemental Fig. S10I) compared to low-frequency AS events.
Importantly, single-cell RNA-seq data indicate that PanAS patterns are, in most cases, due to coexpression of the isoforms in individual cells rather than averages of expression across populations of cells that express primarily one or the other isoform. For instance, PanAS exons are alternatively spliced in the vast majority of individual blastomeres from eight-cell human embryos, showing a broad PSI distribution centered around 50, in stark contrast to low-frequency AS and SwitchAS events (P < 10−38 for all pairwise comparisons, two-sided Fisher's exact test) (Fig. 2F). Other cell types from multiple human and mouse biological sources (Supplemental Table S1), including other early embryonic stages, embryonic stem cells (ESCs), neurons, and immune and cancer cells, showed qualitatively similar results (Supplemental Fig. S11). Altogether, these observations suggest that evolutionary selection pressure has acted to ensure relatively balanced levels of splice isoforms that function in transcription and chromatin regulation across diverse cell and tissue types. Collectively, these observations contrast with recent reports suggesting that the vast majority of mammalian genes generally only express one dominant splice isoform (Discussion).
Neural and muscle AS programs dominate tissue-differential transcriptomic profiles
Principal Component Analyses (PCA) of PSIs revealed that AS profiles formed four main distinct cell/tissue-specific groups in the three species: neural, muscle, pluripotent (ESCs, induced pluripotent stem cells [iPSCs], and early embryo stages) and the rest of cell and tissue types (Supplemental Fig. S12A–C), with precursor and differentiating cells lying at intermediate positions between pluripotent samples and the corresponding differentiated tissues. In contrast, PCA using gene expression values for the same samples showed less clear separation of neural, muscle, and pluripotent cells into independent clusters based on the first two principal components in the three species (Supplemental Fig. S12D–F), indicating lower dominance of these samples in gene expression compared to AS tissue-specific variance. These observations were further supported by unsupervised clustering of PSI measurements: samples from cells and tissues with similar ontogenies formed distinct clusters in human (Fig. 3A), mouse (Supplemental Fig. S13A), and chicken (Supplemental Fig. S13B), particularly for neural and muscle samples.
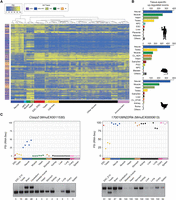
Tissue regulation of AS is dominated by neural and muscle. (A) Heatmap and hierarchical clustering of highly regulated AS events (standard deviation of PSIs across samples higher than 20) in widely expressed human genes (events with sufficient read coverage in at least 40 samples). Samples with >80% missing values (i.e., events with read coverage below VLOW) were discarded. Immune-hem: immune-hematopoietic samples. (B) Bar plots showing the number of exons with increased PSI in a specific tissue compared to all other tissue types. (Early Dev) early embryonic stages in mouse (from oocyte to eight-cell stage) and chicken (from Stage X to HH6). (C) RT-PCR validations and corresponding VastDB RNA-seq PSI estimates for exons in Clasp2 (neural, increased PSI) and 1700106N22Rik (testis, decreased PSI).
We next investigated the relative extent to which different AS types are specific to a cell or tissue type (Supplemental Methods). Tissues of neural origin more often displayed specific increased exon inclusion levels compared to all other cell and tissue types, followed by muscle/heart and testis in the three species (Fig. 3B). These tissues together comprise between 88.1% and 92.6% of all identified tissue-specific exons. Tissues with specifically decreased exon inclusion, as well as other types of AS, were also strongly dominated by these tissues, together with early embryonic and pluripotent cell samples, although the relative tissue dominance was more variable among species compared to exons with increased inclusion (Supplemental Fig. S14). RT-PCR assays validated all nine tested tissue-regulated AltEx events probed, comprising exons with specific increase and decrease in PSI in a wide variety of tissues (Fig. 3C; Supplemental Fig. S5, S15). Moreover, comparison of sets of pronounced tissue-dependent differences in PSIs (|ΔPSI| > 25) between pairs of human tissue types represented in VastDB and the equivalent GTEx tissue samples revealed a high concordance between the two independent sources, both within and between tissue types (Supplemental Fig. S16).
Coregulation network analysis identifies multiple layers of tissue-specific regulation
In order to capture more complex regulatory patterns across tissues, we next used Graphical Model Selection methods to construct coregulated splicing networks (Papasaikas et al. 2015). Furthermore, to allow direct evolutionary comparisons, we used as input 1033 pairs of homologous alternative exons that are highly alternatively spliced (with 20 < PSI < 80 in at least 10% of the samples or a PSI range ≥50 across samples) in both human and mouse, and have sufficient read coverage across a wide range of comparable cell and tissue types in both species (see Methods). The final data set included 23 matched pairs of diverse cell/tissue types with two or more replicates in each species (Supplemental Fig. S17A; Supplemental Table S1). For each species, we then built a network of exons (nodes) based on their coregulatory relationships (edges) across these samples. The distribution of edges across the networks was highly unequal: most exons had very few edges (e.g., 56.3% of the exons had a degree lower than 5 in human, whereas 20.0% of the exons showed more than 50 regulatory interconnections) (Supplemental Fig. S17B). Detection of coregulated exon modules (communities) produced eight and nine communities with more than 10 exons in human and mouse, respectively (Fig. 4A; Supplemental Fig. S17C). Consistent with the preceding results, the largest communities for this subset of conserved AltEx events also corresponded to neural- and muscle-specific exons, as indicated by their increased mean absolute Z-scores compared to all other samples (Fig. 4A; Supplemental Fig. S17C). Next, since each node had by definition a one-to-one correspondence in the other species’ network, we evaluated the conservation of each community at the level of node (exon) and edge (coregulation). Remarkably, neural and muscle communities showed a high level of node conservation, reaching up to 84.4% of node overlap between neurally enriched communities in both species (P ≤ 0.001 for all comparisons, Bonferroni-corrected Fisher's exact test) (Fig. 4C). Similarly, we found a highly significant conservation of the edges within these communities compared to randomized networks (P < 0.001 permutation tests) (Fig. 4D).
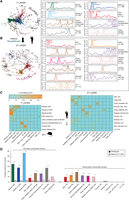
Coregulated splicing network reveals two layers of tissue AS regulation. (A,B) Left: Graphical representation of the human splicing networks, highlighting the different exon communities for the first (A) and second (B) layer networks. Right: Plot of the mean absolute Z-score across tissues for each community of the first (A) and second (B) layer human networks. Dominant tissues are highlighted. Communities were named based on dominant tissues, when possible. For simplicity, only communities with 10 or more AS events are displayed. (C) Heatmap showing the percentage of node (exon) overlap between human (y-axes) and mouse (x-axes) communities for the first (left) and second (right) layer networks. P-values correspond to Bonferroni-corrected one-sided Fisher's exact tests. (D) Percentage of edge conservation for each main human community in the mouse network, for the first (left) and second (right) layer networks, and expected percentage of conservation based on randomized networks of the same size of the tested community. Error bars in controls represent first and third quartiles of the distribution. P-values correspond to permutation tests with 1000 random networks: (***) P-value ≤ 10−3; (**) 10−3 < P-value < 0.01; (*) 0.01 ≤ P-value < 0.05.
Despite the strong dominance of neural and muscle samples driving the largest coregulated and most conserved splicing modules, the plots of mean absolute Z-scores also revealed PSI variation across other cell and tissues types, even if less pronounced (Fig. 4A; Supplemental Fig. S17C), particularly for immune/hematopoietic tissues and ESCs, which also formed conserved exon communities. Therefore, we reasoned that an additional “layer” of coregulation may exist among the other cell and tissue types that is partially masked by the strong, distinct patterns of neural and muscle samples. To address this possibility, we repeated the same network analysis for the exact same set of conserved AltEx events, but excluding neural, muscle, and heart samples. Although the resulting networks were more sparse (Supplemental Fig. S17B), this analysis revealed 11 and 12 communities with more than 10 exons in human and mouse, respectively (referred to as “second layer” communities) (Fig. 4B; Supplemental Fig. S17D). Remarkably, these included exon communities dominated, individually or in combinations, by ESCs, testis, immune/hematopoietic, adipose, colon, and other specific subsets of tissues in both species. Importantly, many of these exon communities were also significantly conserved both at the level of node overlap (Fig. 4C) and edge preservation (Fig. 4D). Remarkably, similar results were found when using a subset of 434 exons that are also conserved and highly alternatively spliced in chicken across 14 matched samples (Supplemental Fig. S18). Moreover, unsupervised clustering of scaled PSIs of these orthologous exons for human, mouse, and chicken showed a strong tissue dominance (Supplemental Fig. S19), suggesting that the evolutionary conservation of tissue-dependent AS regulation among vertebrates is higher than previously appreciated (Barbosa-Morais et al. 2012; Merkin et al. 2012). Overall, these data, together with the presence of various subsets of tissue-specific exons identified above (Fig. 3B; Supplemental Fig. S14), indicate that AS is tightly regulated in a conserved and coordinated manner across a broad range of vertebrate cell and tissue types.
VastDB: a web resource for investigation of AS regulation and function
To facilitate the access to VastDB by the research community, we built a public web interface that allows interactive visualization of AS events (http://vastdb.crg.eu; see Supplemental Methods for details on the web page architecture) (Fig. 1). AS events can be searched directly in VastDB by their vast-tools event IDs, gene or coordinate, or through the UCSC Genome Browser with a built-in VastDB custom hub. The web resource has two major visual interfaces: Gene views and Event views. Each Gene view page (Supplemental Fig. S20) displays several gene features, a link to the UCSC Genome Browser with all AS events, an interactive plot with the expression levels across all samples, and a list of AS events harbored by the gene, including an interactive plot to display their PSIs individually or in combinations. Each Event view page (Supplemental Fig. S21) contains comprehensive information about sequence and genomic features, impact on protein coding sequence and 3D structures (where available) as well as on predicted domains and disordered regions (Supplemental Fig. S21), links to event-level orthologous in other species, suggested primers for RT-PCR validation, and interactive plots displaying PSI values across all 108, 139, and 61 developmental stages and cell and tissue types for human, mouse, and chicken, respectively. Moreover, several additional data sets of special interest are displayed individually under the “Special Data sets” sections in the Gene and Event view. For human, these include (1) 8378 samples from 49 tissues and 543 individuals from GTEx version 6; (2) three groups of BA41 and BA9 brain regions from control subjects and patients with idiopathic or Dup15q autism spectrum disorder (Parikshak et al. 2016); and (3) four cell-type–specific pancreas samples from young and old individuals. For mouse, the special interest data sets include (1) time course of glutamatergic neuronal differentiation in vitro (Hubbard et al. 2013); (2) time course of myoblast differentiation in vitro (Trapnell et al. 2010); (3) spermatogenesis (Soumillon et al. 2013); and (4) a comprehensive catalog of isolated cells from the hematopoietic lineage obtained from multiple sources (Supplemental Table S1).
Discussion
The advent of RNA-seq has transformed our understanding of the complexity, regulation, and function of AS by revealing programs of AS that function in normal physiology and that are misregulated in human diseases and disorders (Lee and Cooper 2009; Parikshak et al. 2016; Quesnel-Vallieres et al. 2016; Scotti and Swanson 2016; Sebestyen et al. 2016). However, despite the steady rise in the frequency of transcriptome-wide studies, a comprehensive and central resource that compiles valuable and readily extractable information on the regulation and functional associations of AS events to facilitate further investigation has been lacking in the field. In this study, we have described such a resource.
Other databases of AS events have been reported. In particular, FasterDB (Mallinjoud et al. 2014) provides exon-level information for human and mouse from microarray data, including expression quantification in various tissues and cell lines, associated protein features, and CLIP-seq data for splicing regulators. Moreover, an associated resource has been recently published (Exon Ontology) (Tranchevent et al. 2017), which affords the identification of enrichment of functional protein features among custom sets of human alternative exons. Other databases are focused on specific characteristics relating to AS. For example, AS-ALPS (Shionyu et al. 2009) predicts the impact of full-length transcript isoforms on protein structures and interactions, and APPRIS (http://appris.bioinfo.cnio.es) attempts to annotate “principal” and minor protein isoforms. However, unlike VastDB, these databases do not integrate extensive and quantitative RNA-seq-based measurements, nor do they provide data on all classes of AS. Furthermore, they generally do not annotate individual AS events in terms of their predicted functional impact using as many sequence and protein feature characteristics as does VastDB (Fig. 1). A particularly valuable feature of VastDB to end users is that it graphically and interactively displays a comprehensive set of splicing and mRNA expression levels across diverse precursor and differentiated cells, tissue types, and developmental stages that have been derived from RNA-seq data. Importantly, this feature of VastDB is further enhanced by its coupling with vast-tools, which provides AS quantifications from new RNA-seq samples that are directly comparable to those in VastDB through the use of a unique event identifier, thus providing immediate contextual information to any user's custom transcriptomic analysis. Furthermore, the event-level homology annotation is another unique feature of VastDB that facilitates evolutionary studies of AS, both at the level of individual events and on a genome-wide level.
Illustrating the power of the VastDB resource, in the present study we discovered new principles of AS coordination and regulation on a global scale, observing a much greater degree of differential regulation of AS across diverse cell and tissue types than previously documented. Based on the analysis of approximately 57 billion reads from 1478 individual RNA-seq data sets represented in the current version of VastDB, we identify 23,113 AS events of all major types in 9131 (47.8%) human multiexonic genes that undergo pronounced switchlike regulation (ΔPSI ≥ 50) between at least one pair of cell and/or tissue types (for details, see Supplemental Methods). These data also indicate that IR is the most prevalent type of AS, expanding previous results from analyzing fewer data sets (Braunschweig et al. 2014) and supporting recent reports on the roles of IR as a major contributor to multiple biological processes in mammals, including spermatogenesis (Naro et al. 2017), granulocyte differentiation (Wong et al. 2013), erythropoiesis (Pimentel et al. 2016), neuronal function and differentiation (Yap et al. 2012; Braunschweig et al. 2014; Mauger et al. 2016), and response to stress (Shalgi et al. 2014; Boutz et al. 2015). Moreover, our analyses show that neural and muscle samples have the strongest and most conserved AS signal, followed by testis and ESCs, extending previous observations (Yeo et al. 2004; Barbosa-Morais et al. 2012; Merkin et al. 2012; Melé et al. 2015). Interestingly, however, our coregulated splicing network analysis further revealed a robust and conserved “second layer” of tissue regulation beyond neural and muscle tissues, indicating that the same tissue-regulated exons may also play important roles in diverse cell and tissue types.
In addition to the widespread differential cell/tissue regulation, we identified hundreds of genes (representing 18.5% of all multiexonic genes with comparable expression across our sample set) that have cassette exons that are alternatively spliced to produce coexisting isoforms in nearly all profiled cell and tissue types. Strikingly, the genes containing these “PanAS” exons are enriched in DNA binding proteins and transcriptional regulators, thus likely expanding their regulatory capabilities. For example, in a transcription factor with key developmental roles, MEIS2, a deeply conserved PanAS exon identified in the VastDB data generates distinct C termini, which are known to confer different transcriptional activation capacities (Huang et al. 2005; Irimia et al. 2011). Similarly, the Wnt pathway effector TCF7L2 harbors three PanAS exons, whose combinations have been reported to impact promoter-binding and transcriptional activation properties on Wnt targets (Weise et al. 2010) and to have different effects on β-cell turnover and function (Le Bacquer et al. 2011). Importantly, single-cell analyses demonstrate that the isoforms resulting from these AS events are generally coexpressed within the same cells. Therefore, our results from analyzing the comprehensive transcriptomic data represented in VastDB contrast with the conclusions of previous transcriptomic and proteomic analyses suggesting that the vast majority of genes produce a single major isoform (Tress et al. 2016).
In summary, VastDB provides the research community with the most comprehensive annotation of AS events in human, mouse, and chicken, together with measurements for their inclusion levels across a wide range of developmental stages and cell and tissue types, as well as data on multiple sequence- and protein-related features that inform possible functions. We anticipate that this rich resource will allow further in-depth investigations of AS both at the individual event and transcriptomic-wide levels, by both wet laboratories and computational biologists. Furthermore, addition of new vertebrate and invertebrate species to VastDB in the near future will allow further unprecedented comparative studies that will deepen our understanding of gene regulation at an exon-level resolution.
Methods
RNA-seq data sets and genome assemblies
All RNA-seq samples used in the current study are listed in Supplemental Table S1. For VastDB, we collected 108, 139, and 61 RNA-seq sets for human, mouse, and chicken, respectively. Many of these sets consist of pools of multiple individual RNA-seq experiments from comparable tissues or cell types to increase the read depth and thus PSI estimation accuracy. VastDB is currently available for the following assemblies: hg19 and hg38 (human), mm9 and mm10 (mouse), and galGal3 (chicken).
Profiling of inclusion levels of AS events across multiple RNA-seq samples
All AS profiles in VastDB have been generated using vast-tools. This software (https://github.com/vastgroup/vast-tools) consists of multiple utilities to align and process raw RNA-seq reads to derive PSIs for all types of AS in different species (Supplemental Fig. S1).
Identification of PanAS events
To define groups of events based on their degree of AS potential, we performed the following steps. First, to ensure a broad expression level of the gene across tissues, we required AS events to have sufficient read coverage in at least 20 samples using a strict criteria (“LOW” coverage score or higher in vast-tools). From the events that passed this filtering step, we then defined three sets of AS events: (1) PanAS, which were alternatively spliced (i.e., 10 < PSI < 90) in >80% of the samples with sufficient read coverage; (2) low-frequency AS, corresponding to those that were alternatively spliced in 10%–25% of the samples; and (3) SwitchAS, which were not alternatively spliced (i.e., had a PSI > 90 or PSI < 10) in >80% of the samples but had a range of PSIs across samples of at least 80.
Coregulated splicing network analyses
We first selected a subset of 23 pairs of cell/tissue types in human and mouse that could be directly matched in a one-to-one manner between the two species and that had at least two replicate samples for each of them (Supplemental Table S1). Next, to allow direct comparisons between the resulting networks for human and mouse, we used only the pairs of orthologous alternative exons that satisfied the following criteria in both species:
-
Sufficient read coverage (“VLOW” coverage score or higher) in at least two replicates of at least 80% of the 23 selected cell/tissue types.
-
Highly alternatively spliced: 20 ≤ PSI ≤ 80 in at least 10% of all VastDB samples with sufficient coverage and/or a range of PSI ≥ 50.
-
A range of average PSI values of at least 25 in the 23 selected cell/tissue types, to filter out AS events with constant inclusion patterns in the selected samples.
For these events, PSI values for each cell/tissue type not satisfying the coverage threshold were set to NA, and then imputed from the 10 nearest events using knn.impute from the impute Bioconductor package, with default parameters. All PSI values in the matrix were then scaled and centered, first by event and then by sample. The resulting matrix was used to compute the covariance between all pairs of AS events, and these were used to derive a correlation matrix between events, which was subjected to graphical lasso L1 regularization, as described in Papasaikas et al. (2015), setting the regularization parameter value (rho) to 0.80 for both species. Community detection in the networks was performed using the greedy modularity optimization algorithm, as implemented in the igraph R package (Csárdi and Nepusz 2006).
To build the chicken networks, we first selected 14 cell/tissue types that could be matched between chicken and mammals (Supplemental Table S1). Next, chicken exons that were conserved with mammals and that also fulfilled the same coverage and AS conditions were used to build a network using the same parameters described above. Node and edge comparisons between human and chicken were done using chicken as reference.
Data access
RNA-based sequencing data from stage X chicken embryos used in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE90957.
Acknowledgments
The authors thank Panagiotis Papasaikas for assistance in splicing network analyses, Nuno Barbosa-Morais for advice on statistical analyses, Baldomero Oliva for advice on protein structural model validation, Lawrence Kelley and Michael Sternberg for privileged access to the Phyre2 server, and the Donnelly Sequencing Centre for generating RNA-seq data for this study. This work has been supported by grants from the European Research Council (ERC-StG-LS2-637591 to M.I.), the Spanish Ministry of Economy and Competitiveness (Centro de Excelencia Severo Ochoa 2013–2017, SEV-2012-0208 to the CRG, SEV-2015-0505 to the CNIC, and BFU2014-55076-P to M.I.), McLaughlin Foundation to B.J.B., and Canadian Institutes of Health Research to B.J.B. J.T. holds a FPI Severo Ochoa fellowship and was supported by Grant SVP-2014-068315. K.C.H. was supported by an Ontario Graduate Scholarship and a CIHR Frederick Banting and Charles Best Canada Graduate Doctoral Award. Y.M. holds an EMBO LTF fellowship.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.220962.117.
- Received January 23, 2017.
- Accepted August 9, 2017.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








