
Specific down-regulation of spermatogenesis genes targeted by 22G RNAs in hybrid sterile males associated with an X-Chromosome introgression
- Runsheng Li1,7,
- Xiaoliang Ren1,7,
- Yu Bi1,
- Vincy Wing Sze Ho1,
- Chia-Ling Hsieh2,
- Amanda Young2,
- Zhihong Zhang2,
- Tingting Lin3,
- Yanmei Zhao3,
- Long Miao3,
- Peter Sarkies4,5 and
- Zhongying Zhao1,6
- 1Department of Biology, Hong Kong Baptist University, Hong Kong, China;
- 2Illumina Incorporated, San Diego, California 92122, USA;
- 3Institute of Biophysics, Chinese Academy of Sciences, Beijing 100190, China;
- 4MRC Clinical Sciences Centre, London W12 0NN, United Kingdom;
- 5Institute of Clinical Sciences, Imperial College London, London SW7 2AZ, United Kingdom;
- 6State Key Laboratory of Environmental and Biological Analysis, Hong Kong Baptist University, Hong Kong, China
- Corresponding authors: p.sarkies{at}csc.mrc.ac.uk, zyzhao{at}hkbu.edu.hk
-
↵7 These authors contributed equally to this work.
Abstract
Hybrid incompatibility (HI) prevents gene flow between species, thus lying at the heart of speciation genetics. One of the most common HIs is male sterility. Two superficially contradictory observations exist for hybrid male sterility. First, an introgression on the X Chromosome is more likely to produce male sterility than on autosome (so-called large-X theory); second, spermatogenesis genes are enriched on the autosomes but depleted on the X Chromosome (demasculinization of X Chromosome). Analysis of gene expression in Drosophila hybrids suggests a genetic interaction between the X Chromosome and autosomes that is essential for male fertility. However, the prevalence of such an interaction and its underlying mechanism remain largely unknown. Here we examine the interaction in nematode species by contrasting the expression of both coding genes and transposable elements (TEs) between hybrid sterile males and its parental nematode males. We use two lines of hybrid sterile males, each carrying an independent introgression fragment from Caenorhabditis briggsae X Chromosome in an otherwise Caenorhabditis nigoni background, which demonstrate similar defects in spermatogenesis. We observe a similar pattern of down-regulated genes that are specific for spermatogenesis between the two hybrids. Importantly, the down-regulated genes caused by the X Chromosome introgressions show a significant enrichment on the autosomes, supporting an epistatic interaction between the X Chromosome and autosomes. We investigate the underlying mechanism of the interaction by measuring small RNAs and find that a subset of 22G RNAs specifically targeting the down-regulated spermatogenesis genes is significantly up-regulated in hybrids, suggesting that perturbation of small RNA-mediated regulation may contribute to the X-autosome interaction.
Hybrid incompatibility (HI) refers to any measurable reduction in fitness commonly seen in interspecific hybrids. One of the most extreme types of HI is hybrid male sterility that may block gene flow between species. However, genetic and molecular mechanisms underlying an observed HI vary dramatically between species, making it necessary to dissect HI across taxa to achieve a global view of genetic or genomic conflict in the species hybrid that leads to HI. It is widely observed that heterogametic hybrid progeny are more likely to suffer HI than their homogametic siblings, which is dubbed Haldane's rule (Turelli and Orr 1995; Schilthuizen et al. 2011). One explanation for this is the dominance theory, which proposes that if alleles causing HI are recessive and sex-linked, the heterogametic hybrid progeny will manifest full effects because of hemizygosity, whereas homogametic hybrid progeny will not, owing to compensation by a second copy of a wild-type allele (Turelli and Orr 2000). Dominance theory has gained wide support in genetic studies of hybrid sterility in both animal and plant species (Masly et al. 2006; Yang et al. 2012).
However, expression and genetic analyses have revealed seemingly opposite roles of the X Chromosome in hybrid male sterility. For example, genes expressed in the germline are enriched on the autosomes but depleted on the X Chromosome during spermatogenesis in both the Drosophila and Caenorhabditis species (Reinke et al. 2004; Sturgill et al. 2007; Ortiz et al. 2014; Vicoso and Bachtrog 2015). Moreover, histone markers indicative of active gene transcription are enriched on autosomes but depleted on the X Chromosome in the Caenorhabditis elegans male germline, whereas an opposite pattern is observed for repressive histone markers (Kelly et al. 2002; Schaner and Kelly 2006). These observations support the hypothesis of X demasculinization or sexual antagonism and X inactivation (SAXI) (Wu and Xu 2003). Nevertheless, the X Chromosome is found to play a disproportionately larger role than autosomes in the development of hybrid male sterility (Masly and Presgraves 2007; Bi et al. 2015). One possible explanation for this discrepancy is the presence of a genetic interaction between the X Chromosome and autosomes to maintain the correct expression ratios between the X Chromosome and the autosomes (hereafter termed as X:A imbalance), which is essential for spermatogenesis (Wu and Xu 2003). The clearest way to test for such an interaction would be to use hybrid sterile males carrying a defined introgression from another species in an otherwise isogenic genetic background. Indeed, expression analyses of testis genes between hybrid Drosophila simulans males that carry a fertile or sterile introgression revealed that autosomal genes were more likely to be misexpressed than those on the X Chromosome (Lu et al. 2010). These hybrid males differ only in a small region of the X Chromosome containing the Ods-site homeobox (OdsH) locus of hybrid sterility (Ting et al. 1998; Sun et al. 2004), minimizing the complications associated with heterogeneous genetic background as is the case in F1 hybrids. The study supports a genetic interaction between X and autosomes, but how the interaction is maintained and whether a similar interaction is present in other taxa are largely unknown.
The control of transposable elements (TEs) is another important factor contributing to HIs (Maheshwari and Barbash 2011). TEs may become aberrantly activated when introduced into a new host that lacks a specific control mechanism, leading to a widespread invasion of the host genome and hybrid sterility or lethality. For example, TE mobilizations have been observed in hybrids between different marsupial species (Metcalfe et al. 2007) and between Drosophila species (Shpiz et al. 2014; Erwin et al. 2015), though whether the TE activation is the only cause of HI remains to be determined. Two HI genes, Hmr and Lhr, genetically interact to cause hybrid lethality between D. melanogaster and D. simulans (Brideau et al. 2006). RNA-seq analyses revealed that Hmr and Lhr are required to repress transcription from satellite DNAs and many families of TEs in its native host (Satyaki et al. 2014). One possible cause of aberrant TE expression in hybrids is altered expression of Piwi-interacting small RNAs (piRNAs), a class of small RNAs that interacts with the Piwi family of Argonaute proteins to control the expression of TEs in the germline (Di Giacomo et al. 2013). This is because the piRNA population in a host rapidly adapts to the TE content through generation of new piRNA clusters, allowing de novo production of piRNA and other types of small RNAs for silencing of the invading TE (Shpiz et al. 2014; Senti et al. 2015). This rapid divergence can be seen clearly through the phenomenon of hybrid dysgenesis, in which intraspecific crosses between different Drosophila lines with and without a particular TE produce sterile progeny due to a requirement for maternally deposited piRNAs for silencing a paternally derived TE (Brennecke et al. 2008). Consistent with this, Drosophila interspecific hybrids phenocopy piRNA pathway mutants (Kelleher et al. 2012). Whether desilencing of TEs is directly involved in interspecific hybrid sterility remains largely unexplored in other species. It is worth noting that in most of the studies on TE-mediated hybrid sterility or lethality, total RNAs were extracted from hybrid F1 animals with a largely heterozygous genetic background. Consequently, both sets of piRNA loci on autosomes are present, making it difficult to demonstrate directly that misexpression of piRNAs is actually causative for TE dysfunction and, indeed, whether TE dysfunction itself is responsible for sterility. This also means that it is unclear whether an imbalance between TEs and piRNAs would occur to the same extent in species without maternal deposition of piRNAs.
Isolation of a Caenorhabditis briggsae sister species called Caenorhabditis nigoni, previously known as Caenorhabditis sp.9 (Felix et al. 2014), opens the possibility of using nematode species to study interspecific HI. Results from the preliminary crossing between the two support Haldane's rule (Woodruff et al. 2010; Kozlowska et al. 2012). To facilitate the species pair as a model for isolation of HI loci, we have recently developed random labeling of C. briggsae chromosomes with GFP markers and mapped them into defined genomic regions (Bi et al. 2015). Multiple independent introgressions from a C. briggsae X Chromosome produce sterile males in a C. nigoni background (Bi et al. 2015), but the molecular mechanism underlying the observed sterilities remains unclear. Here we examined the molecular mechanisms of hybrid male sterility between C. briggsae and C. nigoni mainly through genomic approaches.
Results
A C. nigoni draft genome was generated with approximately 20× coverage of Illumina synthetic long reads
A current draft assembly of C. nigoni genome was produced using paired end next-generation sequencing (NGS) data with an average read length of ∼100 bp, making it difficult to generate sequence contigs with sufficient length and accuracy for the downstream analysis (Kumar et al. 2012). It is worth noting that the draft genome is further complicated by sequence contamination from Caenorhabditis afra (previously known as Caenorhabditis sp.7) to an unknown extent (Felix et al. 2014). In particular, the short reads are problematic for assembling contigs that are rich in repetitive sequences, which will prevent accurate annotation of these sequences, including TEs and some small RNAs that are to be addressed in this study. We previously demonstrated that Illumina synthetic long reads not only are able to recover nonrepetitive sequences but also are capable of recovering most types of repetitive sequence except for those arranged in a long stretch of tandem repeats (Li et al. 2015). To facilitate comparative analysis of expression of small RNAs, TEs, and protein-coding genes between wild-type and hybrid strains, we produced approximately 20× coverage of Illumina synthetic long reads for C. nigoni as described previously for a C. elegans genome assembly (Li et al. 2015). Most of the reads are ∼10 kbp in length with a minimum size of 1.5 kbp.
A total of 6882 contigs were assembled using the long reads with an N50 size of 57 kbp. We next aligned the contigs against C. briggsae (AF16) reference genome “cb4” (Ross et al. 2011) using LAST (Kielbasa et al. 2011) to locate the syntenic regions between the two, the output of which was used to construct C. nigoni pseudochromosomes. Sequences equaling a total of 119 Mbp were anchored into the six pseudochromosomes, while the remaining 22 Mbp were retained as so-called unassigned sequences. Taking into account residual heterozygosity, we estimated the total size of the C. nigoni genome to be ∼130–140 Mbp. We used the draft genome that is called “cn1” hereafter for the subsequent analysis. We also performed preliminary annotation for the “cn1” genome, including prediction of structural genes, which were refined with our RNA-seq data along with their homolog in C. briggsae (Fig. 1; Supplemental Fig. 1; Supplemental Table 1). To facilitate the use of the draft genome by the research community, we established an online genome browser that allows sequence retrieval, visualization of C. nigoni gene models, and gene expression, as well as synteny between C. briggsae and C. nigoni genome (see “Data access”). At present, the pseudochromosomal assembly strategy used means that this genome will have a limited use in determining complex structural rearrangements between C. nigoni and C. briggsae. Future work incorporating long-read, single molecule, real-time (SMRT) sequencing technology from Pacific Biosciences (PacBio) or sequencing of mate pair genomic libraries may help to resolve structural variations through generating an unbiased contiguity (Huddleston et al. 2014). Nevertheless, the “cn1” genome assembly is expected to cover the majority of DNA elements, including small RNAs, TEs, and protein-coding genes that will be analyzed in this study.
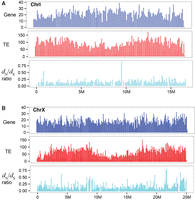
Overview of C. nigoni genome. Shown are densities of coding genes (blue), transposable elements (TEs; red), and dN/dS ratio (cyan) of orthologous regions between C. nigoni and C. briggsae over Chromosome I (A) and Chromosome X (B) in a window size of 100 kbp.
Replacement of different fragments of the C. nigoni X Chromosome with homologous regions from C. briggsae results in defective spermatogenesis and sterility
Our previous efforts in systematic introgression of GFP-linked C. briggsae genomic fragments into the C. nigoni background produced multiple independent introgression lines that demonstrate complete male sterility (Bi et al. 2015). Here we focused on two male sterile lines, each of which carries an independent, nonoverlapping fragment from the C. briggsae X Chromosome (Fig. 2). The females carrying either of the introgressions as a heterozygote are fertile. One of the lines, ZZY10330, carries a fragment from the right arm of the C. briggsae X Chromosome that is ∼5.1 Mb in size, while the other line, ZZY10307, contains a fragment of ∼7.6 Mb in size from the middle of the C. briggsae X Chromosome (Fig. 2A–C). The introgression was achieved by repeated backcrossing of the GFP-linked C. briggsae genomic fragment into C. nigoni for at least 15 generations (Fig. 2D; Bi et al. 2015), meaning the two male sterile lines essentially carry pure C. nigoni background except for the introgression region. To verify the introgression boundaries defined by single-worm PCR (Yan et al. 2012), we performed NGS to determine the introgression boundaries as previously described (Bi et al. 2015). As expected, the introgression boundaries defined by NGS agree well with those by the PCR-based genotyping in both strains (Fig. 2B). No extra C. briggsae fragments were found in the introgression lines (Supplemental Fig. 2), indicating the genetic background of the two lines is essentially C. nigoni JU1421 except for a C. briggsae genomic fragment that is tightly linked to a chromosomal insertion of cbr-myo-2::GFP.
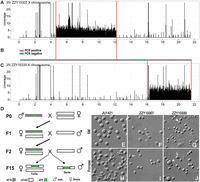
Characteristics of two hybrid male sterile lines. (A–C) Confirmation of introgression boundaries by NGS for ZZY10307 (A) and ZZY10330 (C), each carrying an introgression derived from C. briggsae X Chromosome in an otherwise C. nigoni background. Read coverage (y-axis) is shown across C. briggsae X Chromosome coordinates (x-axis). Genotyping results by single-worm PCR are shown in the middle (B), with PCR positive and negative corresponding to the presence and absence of C. briggsae–specific amplification, respectively. (D) Schematic diagram showing steps of generating two hybrid sterile lines. (E–J) Sperm morphology and activation in C. nigoni (JU1421) or hybrid males. (E–G) DIC micrographs showing sperm morphologies for JU1421, ZZY10307, and ZZY10330, respectively. (H–J) DIC micrographs showing sperm activations for JU1421, ZZY10307, and ZZY10330, respectively. Residual bodies and sign of activation in hybrids are indicated with an arrowhead and arrow, respectively. (SM) Sperm media.
To investigate whether the observed sterilities are caused by defective spermatogenesis, we first examined germline morphology in the hybrid males. We found that instead of the typical “U” shape as seen for C. nigoni male germline, the ZZY10307 and ZZY10330 male germlines are disorganized and appear to lack an obvious turn (Supplemental Fig. 3A,B). In addition, sperm cells were displaced anteriorly comparable to those observed in the F1 hybrids between the two species (Ting et al. 2014), suggesting a defect in spermatogenesis. We next performed mating tests to examine whether the sterility is produced by incapability of sperm transfer using MitoTracker-stained sterile males that were mated with C. nigoni virgin female. We found sperm transfer was successful for both ZZY10307 and ZZY10330 (Supplemental Fig. 4A,B), indicating the sterility is not caused by the failure in sperm transfer during copulation. We finally evaluated the morphology of the sperm cells and their competences for activation in both hybrid and C. nigoni males. We found that sperm cells in the two lines showed similar phenotypes. For example, overall sperm cell shapes tend to be irregular, and their sizes are smaller in both hybrids than in C. nigoni (Fig. 2E–J). Residual bodies were frequently found in the sperm cells from ZZY10330, suggesting a defect in spermatogenesis (Liu et al. 2013). We next evaluated the activation potential of the sperm in both JU1421 and the sterile males. Unlike C. elegans sperm, which can be activated by either Pronase, zinc, monensin, or TEA (Liu et al. 2013), C. nigoni sperm cells can only be activated by Pronase (Fig. 2H; data not shown). Despite the defective size and shape, the sperm cells from both sterile males also showed a sign of activation (Fig. 2I,J), but they may not be competent enough for fertilizing C. nigoni oocytes. Taken together, the data demonstrate that the sterilities in both ZZY10307 and ZZY10300 are mainly caused by defective spermatogenesis.
Independent introgressions on X Chromosome produce a similar pattern of down-regulation in genes that are significantly enriched on autosomes
Previous expression studies using microarray, RNA-seq, or antibody staining demonstrate a biased expression of spermatogenesis genes between the X Chromosome and autosomes; namely, most genes expressed in male germline are located on autosomes but depleted from the X Chromosome (Albritton et al. 2014; Ortiz et al. 2014). We asked whether replacement of part of the C. nigoni X Chromosome with its homologous sequence from C. briggsae would disrupt the “imbalanced expression” between the X Chromosome and autosomes, which could be associated with the observed sterilities. To test this, we performed RNA-seq analysis to quantify the mRNA transcripts in the two sterile male lines and its parental males from C. nigoni and C. briggsae (Supplemental Table 2). We produced around 8 million reads for each sample with three replicates each. All RNA reads were mapped against annotated coding sequences in C. briggsae (AF16) (Harris et al. 2014). The reads were also de novo assembled into transcripts using Trinity (Haas et al. 2013) for the purpose of small RNA mapping as detailed below.
We identified a total of 574 and 922 genes that are significantly up-regulated in the males of ZZY10307 and ZZY10330, respectively, and 1242 and 1317 genes that are significantly down-regulated, respectively, in the two compared with C. nigoni males (Supplemental Table 3). A total of 358 and 860 genes are significantly up- and down-regulated, respectively, in both hybrid lines compared with C. nigoni males (Fig. 3A). Interestingly, the two independent introgressions lead to down-regulation of a highly overlapping set of genes (P < 1 × 10−5 by random sampling), whereas the up-regulated genes do not show the similar overlapping pattern (Fig. 3B). Overall similarity of gene expression between the two sterile lines is much higher than that between either of the two and JU1421 (Fig. 3C,F–H). Intriguingly, we observed that the ratio of down-regulated genes is disproportionately higher on autosomes than on the X Chromosome where the introgression fragments are located (P < 1 × 10−17, Fisher's exact test). For example, 96% of the shared down-regulated genes are located on the autosomes and only 4% on the X Chromosome (Fig. 3D,E), though the X Chromosome carries ∼17% of C. nigoni protein-coding genes. Notably, such enrichment could not be observed for up-regulated genes in the hybrids (Fig. 3E).
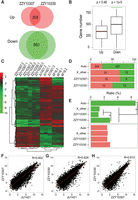
Expression profiling of coding genes in male hybrids and their parental males. (A) Venn diagrams showing shared numbers of up- or down-regulated genes between both hybrid males and C. nigoni. (B) The bootstrap test for the intersection of misregulated genes between two hybrids. The observed number of overlapping down-regulated genes (860, green line) is significantly higher than random, while the observed number of overlapping up-regulated genes (358, red line) is not higher than random. The bootstrap sampling was performed 100,000 times with the one-tailed P value shown on top. (Green) P < 0.05; (red) P > 0.05. (C) A heat map showing hierarchical clustering of normalized expression for the shared up-regulated (red) and down-regulated (green) genes in A for each RNA-seq sample of C. briggsae (AF16), C. nigoni (JU1421), ZZY10330, and ZZY10307. Expression of each gene is normalized against the average of 12 samples. (D) Chromosomal distribution of up-regulated (red) and down-regulated (green) genes as defined in C. (E) Percentages of down-regulated (green) or up-regulated (red) genes on autosomes or the X Chromosome. Note the percentage of down-regulated genes is significantly higher on the autosomes than on the X Chromosome regardless of whether the genes are located within or outside the introgressions, whereas no significant enrichment is found for up-regulated genes. (*) P < 0.01, Fisher's exact test. (F,G) A pairwise comparison of overall expression in males between ZZY10307 (F) or ZZY10330 (G) and C. nigoni, respectively. Correlation coefficient (R) is indicated. (H) A pairwise comparison of overall expression in males between ZZY10307 and ZZY10330.
To evaluate whether genes located within and outside of the introgressions demonstrated differential patterns in misregulation, we divided the C. briggsae X Chromosome into three distinct regions, that is, introgression from ZZY10330 and ZZY10307 and the remaining region, which are about 5.1, 7.7, and 8.7 Mbp in size, respectively. We counted the number of C. briggsae genes within each introgression as well as the number of C. nigoni genes orthologous to the C. briggsae genes within the remaining 8.7 Mbp (X_other in Fig. 3D,E) that showed misregulation in the hybrid background. We found no significant enrichment of up-regulated genes located within and outside of both introgressions on the X Chromosome (Fig. 3D,E). However, genes within the introgression region of ZZY10330 do show a significant enrichment compared with those outside of both introgressions on the X Chromosome (P = 1 × 10−4, Fisher's exact test) (Fig. 3E). A pairwise comparison of genes located within the introgression regions between a hybrid and its native parental strain reveals that the genes within the ZZY10307 introgression seem to be expressed in a more similar way as those in C. briggsae than in C. nigoni, while those located within the ZZY10330 introgression show an expression pattern that is comparable to both parents (Supplemental Fig. 5). In addition, correlations of expression between genes within introgression and those in both parental species are higher in ZZY10307 (with a correlation coefficient of 0.817 and 0.903, respectively) than in ZZY10330 (with a correlation coefficient of 0.744 and 0.717, respectively), indicating there is a higher level of gene misregulation in the introgression of ZZY10330 than in the introgression of ZZY10307. In summary, independent replacements of the X Chromosome fragment lead to preferential down-regulation of autosome-linked genes associated with male sterilities, supporting an interaction between the X Chromosome and autosomes, which is consistent with the hypothesis of demasculinization of X Chromosome.
Only spermatogenesis genes are significantly enriched in the down-regulated gene set in both hybrid sterile males
Given the primary phenotype associated with the two introgressions is the male sterility that seems to be a product of defective spermatogenesis (Fig. 2E–J), we investigated the relationship between the sterility and the misregulated genes shared in both sterile strains. We took advantage of the classification of C. elegans male- and female-specific germline genes, as well as C. briggsae sex-biased genes defined in recent studies using RNA-seq (Thomas et al. 2012; Albritton et al. 2014; Ortiz et al. 2014). We examined whether the categories of spermatogenesis or male-specific genes were enriched in our misregulated gene list (see Methods) (Supplemental Table 4). We performed enrichment analysis separately for the down- and up-regulated genes shared between the two hybrids against spermatogenesis or male-specific genes as stated above. Only spermatogenesis genes were significantly enriched in our down-regulated gene list (FDR < 0.01), whereas oogenesis and gender-neutral ones show no enrichment in the list (Fig. 4A; Supplemental Table 4). However, no significant enrichment was found in up-regulated genes. Similar enrichment analysis was performed using sex-biased genes defined previously (Thomas et al. 2012; Albritton et al. 2014). Again, male-specific genes were significantly enriched in the down-regulated gene list (Fig. 4B; Supplemental Table 4). In addition, a category of “low male” also showed a significant enrichment in the up-regulated list, albeit at a much lower scale compared with that in the down-regulated list. Gene ontology (GO) analysis demonstrates that genes involved in regulation of cell shape or protein phosphorylation are significantly enriched (Fig. 4C), suggesting that these genes may control spermatogenesis through these pathways. Notably, genes up-regulated in both hybrids do not show enrichment for either spermatogenesis (Fig. 4A) or autosomes (Fig. 3E), suggesting that these genes are involved in pathways other than spermatogenesis, such as general developmental pathway or physiology. Consistent with this, GO analysis reveals that these genes are primarily involved in response to nutrient levels or extracellular stimuli (Fig. 4C). Similar enrichment patterns were observed when the misregulated genes were analyzed separately for the two hybrid strains (Supplemental Fig. 6). Intriguingly, a significant enrichment on the autosomes versus the X Chromosome was observed for the down-regulated but not the up-regulated genes (Supplemental Fig. 7). Taken together, nonoverlapping replacements of X Chromosome fragments produce male sterilities through preferential down-regulation of spermatogenesis genes that are mainly located on autosomes.
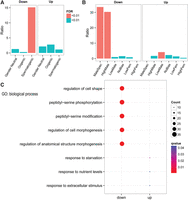
Enrichment analysis of down- or up-regulated genes against sex-biased genes. Analysis was performed separately for down-regulated and up-regulated genes found in both hybrids against the gene categories defined by two previous studies (see text). Note a significant enrichment of spermatogenesis genes (A) or male-specific genes (B) in the down-regulated gene list. “Low male” category also shows a significant enrichment for up-regulated genes, but its ratio is not comparable to those in down-regulated list. (C) Gene ontology analysis of down- or up-regulated genes shared by the two sterile lines.
Few TEs show aberrant expression in the hybrid sterile males compared with C. nigoni males
Overexpression of TEs is frequently found in F1 hybrids and is associated with male sterility (Rozhkov et al. 2013; Erwin et al. 2015). We wondered whether misregulation of TEs might be associated with the observed male sterility in the introgression lines despite the fact that they were subjected to backcrossing for at least 15 generations (Bi et al. 2015). To analyze TE expression, we compiled a TE list de novo from both C. briggsae and C. nigoni genomes (Supplemental Fig. 8, see Supplemental Methods). We defined a total of 247 and 319 families of TE in C. briggsae and C. nigoni, respectively (Supplemental Table 5). Interestingly, the C. nigoni genome seems to carry all the TE families found in the C. briggsae genome but also contains unique TE families. Read mapping against TEs was performed in a similar way as that for protein-coding genes but with modified parameters (see Methods and Supplemental Methods). Overall expression of TEs in both hybrids was similar to those in C. nigoni (Fig. 5). Out of all the measured TE families, we detected a significant elevation of expression for only one family in ZZY10307 males (cb_rnd-3_family-789 [DNA/PiggyBac]) and two families in ZZY10330 males (cb_rnd-3_family-381 [DNA/Merlin] and c_sp9_rnd-4_family-428 [LINE/CR1]) compared with C. nigoni males; a single TE family (c_sp9_rnd-5_family-1528 [LINE]) showed significant down-regulation in ZZY10307 (fold change > 2 and P < 0.01) (Fig. 5C,D; Supplemental Table 5). Since a single TE family can contain multiple members, we further investigated whether the member count contributed to the significant increase in TE expression. The average numbers of normalized reads between three replicates in the hybrids that mapped to the three families were 38, 34, and 25, respectively, with an average of fewer than two copies per member, arguing against a significant role of overexpression of the three families in the observed male sterilities. This is based on empirical data on TE misregulation in other species. For example, despite the significantly elevated expression of TEs relative to C. nigoni, the expression of these TEs in our hybrids is much lower than the misregulated TEs caused by two HI proteins, Hmr and Lhr in sterile hybrids between D. melanogaster and D. simulans, where over 50 families demonstrated overexpression in F1 hybrids with a much higher read count (up to 1 million reads) relative to both parents (Satyaki et al. 2014). It should be noted that due to difficulty in isolating germlines from hybrid sterile males, the whole animals were used for RNA extraction. Therefore, all expression assays do not distinguish between germline and somatic tissues.
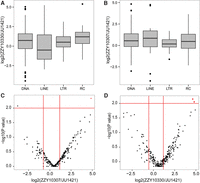
Comparison of TE expression between hybrid and C. nigoni (JU1421) males. (A,B) Boxplots showing the comparison of overall expression of TEs in the males at TE class level between ZZY10330 or ZZY10307 and JU1421, respectively. (C,D) Volcano plots showing the comparison of overall expression of TEs in the males at TE family level between ZZY10307 or ZZY10330 and JU1421, respectively. Note there are only one and two TE families showing a significantly higher expression (highlighted in red) in ZZY10307 (C) or ZZY10330 (D) than in JU1421, respectively. (LINE) Long interspersed nucleotide elements; (LTR) long terminal repeats; (RC) rolling circle.
Misregulation of 22G RNAs but not piRNAs in the hybrid sterile males
piRNAs are key regulators of genome stability via regulation of TEs. piRNA mutants in both D. melanogaster and Mus musculus show defects in spermatogenesis, leading to sterility, which is associated with altered TE expression (Cox et al. 1998; Brennecke et al. 2007; Carmell et al. 2007). Moreover, due to their critical role in TE regulation, differences in piRNA content cause hybrid dysgenesis between different Drosophila species (Kelleher et al. 2012). In C. elegans, piRNAs, also known as 21U-RNAs, associate with the Piwi protein PRG-1 and target TEs for silencing via the induction of 22G RNAs (Batista et al. 2008; Das et al. 2008). piRNAs also target both transgenes and endogenous loci for heritable silencing. Loss of the piRNA pathway leads to progressive sterility associated with increased repetitive element expression (Batista et al. 2008; Das et al. 2008; Simon et al. 2014). We therefore tested whether differences in piRNA expression between sterile hybrids and the parental lines might underlie defective spermatogenesis and male sterility in these lines.
Given the highly similar phenotypes in sperm as well as in the mRNA and TE expression patterns between the two sterile lines, we focused on small RNAs in one of the two sterile lines, ZZY10330. In C. briggsae, as in C. elegans, piRNAs are produced from individual loci located in clusters, with each individual piRNA associated with a specific upstream promoter identified by a highly conserved motif (the Ruby motif) (Ruby et al. 2006; de Wit et al. 2009; Shi et al. 2013). The C. briggsae piRNA clusters have previously been shown to reside on Chromosomes I and IV (de Wit et al. 2009; Shi et al. 2013). We therefore aligned small RNAs from both C. nigoni parent males (hitherto wild type) and sterile hybrid males to the C. nigoni scaffolds “cn1” as described above. In both cases, we found strong enrichment of piRNA alignments within the regions of the C. nigoni genome corresponding to the C. briggsae piRNA loci, with little difference in the alignment positions, as would be expected given that the region of the C. briggsae genome inserted in the hybrid strain is outside of these loci. Although there was a reduction in the total number of overall reads from piRNA loci in the hybrid (P < 2 × 10−16, Mann-Whitney U test), this difference was rather small (median, eight reads per million for C. nigoni versus seven reads per million in ZZY10330) (Fig. 6A,B).
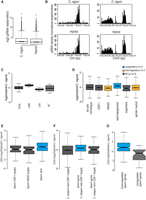
Comparison of expression of piRNAs and 22G RNAs between the males of ZZY10330 and C. nigoni (JU1421). (A) Comparison of read counts for piRNAs between C. nigoni males and the hybrid males (ZZY10330). (B) Distribution of piRNAs along the piRNA clusters on Chromosome I and Chromosome IV for C. nigoni (top) and the hybrid (bottom). The y-axis shows the number of unique piRNA sequences per million in each genomic window of 100 kbp. The x-axis shows position along the chromosome in base pairs. (C) Boxplot showing differences in 22G RNAs mapping antisense to different classes of TEs between hybrid males and C. nigoni males. Box shows interquartile range (IQ) with a line at the median, and the whiskers show the furthest point within 1.5 times the IQ range. (D) Boxplot showing differences between hybrid and C. nigoni in 22G RNAs mapping antisense to C. nigoni genes categorized by annotations from C. briggsae (CSR-1) or C. elegans (other categories). (E) Breakdown of spermatogenesis genes from D into CSR-1 and WAGO targets. (F) Differences between hybrid and C. nigoni either in all male-specific CSR-1 targets as defined in C. elegans or in the male-specific CSR-1 targets that overlap with spermatogenesis genes from D. (herm) Hermaphrodite. Boxplot parameters as in C. (G) Boxplot showing differences between hybrid and C. nigoni 22G RNAs mapping antisense to spermatogenesis genes found either up-regulated or down-regulated in hybrid males by mRNA-seq analysis (Fig. 3). Boxplot parameters as in C.
Although the reduction in piRNA expression was very small, it is conceivable that this might lead to differences in silencing of TEs downstream. To test this, we examined whether 22G RNAs mapping to the TEs compiled above were different between the hybrid and wild-type males. Overall, there was excellent correlation between the wild type and the sterile hybrids in terms of the reads mapped to consensus TE sequences (Supplemental Fig. 9), indicating that 22G RNAs mapping to TEs were mostly unchanged in the hybrids. There were five TEs showing greater than fourfold difference in read count, but none of these corresponded to the TEs showing differential expression by RNA-seq (Supplemental Table 5). Moreover, there were no significant shifts in the levels of 22G RNAs mapping to different classes of TEs between the sterile hybrid and wild type (P > 0.1 for each, Wilcoxon paired test) (Fig. 6C). Taken together with the absence of differences in the expression of TEs, this suggests that altered silencing of TEs by small RNAs is unlikely to explain the male sterility phenotype in the hybrid.
In addition to TEs, 22G RNAs also map to genic loci and can be generated from many different primary small RNA triggers, both piRNA dependent and piRNA independent (Gu et al. 2009; de Albuquerque et al. 2015; Phillips et al. 2015). We therefore considered whether 22G RNAs mapping to coding genes might show differences between the hybrid and the wild type. Given the sterility phenotype of the hybrids, we focused on germline genes. In C. elegans, there are two major classes of 22G RNAs that are found in the germline, CSR-1, which binds to the Argonaute CSR-1 (Claycomb et al. 2009), or WAGO, which binds to WAGO-family Argonautes (Yigit et al. 2006; Gu et al. 2009). CSR-1–dependent small RNAs activate target gene expression whereas WAGO-dependent small RNAs generally reduce target gene expression (Seth et al. 2013; Wedeles et al. 2013). In order to classify genes in C. nigoni, we used previously published C. briggsae annotations of CSR-1 RNAs based on a direct immunoprecipitation approach for CSR-1 (Tu et al. 2015) and homology mapping from C. elegans for WAGO (Gu et al. 2009). We then examined levels of 22G RNAs mapping antisense to these genes in the hybrid and wild type (Supplemental Table 6).
Interestingly, putative spermatogenesis genes showed a clear increase in median levels of 22G RNAs in the hybrid relative to wild-type C. nigoni (P = 1.5 × 10−10, Wilcoxon signed rank test) (Fig. 6D). This increase did not correspond to increased 22G RNAs either for CSR-1 target genes, which showed no significant change (P > 1 × 10−3, Wilcoxon signed rank test), or for WAGO-dependent genes, which showed a decrease in 22G RNA levels (P = 2 × 10−4, Wilcoxon signed rank test), suggesting that neither CSR-1 nor WAGO-1 target genes could explain the increased levels of 22G RNAs mapping to spermatogenesis genes. Consistent with this interpretation, neither CSR-1–targeted nor WAGO-1–targeted spermatogenesis genes showed altered levels of 22G RNAs, with the changes predominantly affecting genes that were not targeted by either Argonaute (Fig. 6E). However, importantly, these annotations refer specifically to hermaphrodites. In C. elegans males, CSR-1 was shown to target spermatogenesis genes (Conine et al. 2013). We therefore examined 22G RNAs mapping to male-specific CSR-1 targets. Intriguingly, orthologs of spermatogenesis genes that were targets of CSR-1 in C. elegans males were significantly up-regulated in hybrids relative to C. nigoni (P = 1 × 10−10, Wilcoxon signed rank test). This did not affect the C. nigoni orthologs of CSR-1 targets in C. elegans males that were not sperm genes, which were not significantly different between hybrids and C. nigoni (Fig. 6F).
We wondered whether the down-regulation of spermatogenesis gene expression we observed (Fig. 4A) could be associated with the up-regulation of 22G RNAs. We therefore subdivided spermatogenesis genes into up-regulated and down-regulated genes. This analysis showed that only down-regulated spermatogenesis genes demonstrated increased 22G RNAs mapping to them, while up-regulated spermatogenesis genes showed no significant change in 22G RNAs (Fig. 6G). Importantly, the misregulation of 22G RNAs that we observe is not directly due to cis-acting differences in the X Chromosome. None of the spermatogenesis genes with 22G RNAs that increased by at least twofold and that showed significantly altered gene expression by RNA-seq are found within the introgressed region (Supplemental Table 6), and these genes indeed were more likely to be found on autosomes than expected even given the biased distribution of spermatogenesis genes with altered expression by mRNA-seq (odds ratio >15 P = 0.02 Fisher's exact test) (Supplemental Table 6). Thus both increased 22G RNAs and the decreased expression in spermatogenesis genes are likely to be an indirect response to the introgression region. We speculate that misregulation of 22G RNAs caused by disruption of X Chromosome integrity might be a general response to the disrupted X:A imbalance that occurs in hybrids.
Differential cis-acting regulation of miR-237 between C. briggsae and C. nigoni is associated with sterility in hybrid males
In order to further examine possible molecular differences between sterile hybrids and the parent C. nigoni strain, we compared expression of miRNAs between hybrid males and those of C. nigoni. Although some miRNAs are remarkably highly conserved across species, the short sequence lengths and relatively straightforward requirements for processing give them the potential to evolve rapidly (Marco et al. 2013). Moreover, their important roles in development make differences in miRNAs attractive potential causes of species-specific differences (Niwa and Slack 2007; Tang et al. 2010), although examples of clear roles for miRNAs in speciation have yet to be described.
In order to identify possible miRNAs that are different between C. briggsae and C. nigoni, we first identified homologs to previously identified C. briggsae miRNAs (de Wit et al. 2009; Shi et al. 2013; Kozomara and Griffiths-Jones 2014) in the wild-type C. nigoni. The majority of miRNAs had identical sequences between C. briggsae and C. nigoni, and of the six for which we found single-nucleotide polymorphisms in the mature miRNA sequence, none showed any change in the critical “seed region” between nucleotides 2–7 (Bartel 2004) responsible for targeting (Supplemental Table 7). Thus miRNA sequences are highly conserved between the two species. We next analyzed the expression of these miRNAs in wild-type males and ZZY10330 sterile males. The majority of miRNAs showed highly consistent expression between the two strains; however, one miRNA, miR-237, showed greater than fivefold reduced reads in the hybrid ZZY10330 relative to wild-type C. nigoni. (Fig. 7A; Supplemental Table 8). Intriguingly, when we examined the expression of this miRNA in C. briggsae, we found that C. briggsae males also showed strongly reduced reads relative to its counterpart in C. nigoni (Fig. 7B). Notably, this miRNA is found within the introgression region (Supplemental Table 8). We therefore speculated that the difference in expression of miR-237 might reflect sequence changes in the locus. The mature miRNA shows only a 1-nucleotide (nt) difference, outside of the seed region, which is unlikely to directly affect expression. Moreover, using RNAfold, we predicted that miRNA-precursors would form with highly comparable free energy (−39 kJ/mol for C. nigoni vs. −41 kJ/mol for C. briggsae) (Fig. 7C,D). Thus this difference in expression is unlikely to come from differences in the processing of the precursor miRNA by Drosha or Dicer. However, the region immediately 5′ to the miR-237 sequence showed appreciable sequence divergence (Fig. 7E). Thus it is possible that a sequence difference in the promoter of mir-237 might drive the differential expression of this miRNA in both C. briggsae and C. nigoni background. The differential expression we observed may contribute to the sterility of hybrid lines. In this regard, it is interesting that miR-237 is expressed in the somatic gonads of C. elegans (Harris et al. 2014), so it might contribute to reproductive capacity in C. nigoni; testing the consequences of reduced miR-237 expression in C. nigoni males will be an interesting area for further research. However, because the two introgression regions on the X Chromosome that give rise to sterility are nonoverlapping, the cis-acting differences in the mir-237 promoter will only be found in one of the sterile lines arguing that this cannot be the sole cause of sterility.

miR-237 is differentially expressed between males of ZY10330 and C. nigoni. (A,B) Scatterplots showing miRNA read counts in C. nigoni (x-axis) against either hybrid males (A) or C. briggsae (B). miR-237 mature (5p) and star (3p) are highlighted. (C,D) Secondary structure predictions of the miRNA precursors in C. briggsae and C. nigoni. Predictions were made by RNAfold. (E) Alignment of mir-237 genomic locus in C. briggsae and C. nigoni showing some sequence divergence in the putative promoter region.
Discussion
Despite intensive studies of HI by expression analyses, its regulatory mechanism across species remains poorly understood. Most of these studies concentrate on F1 hybrid incompatibilities, including hybrid male sterility. However, the mechanisms underlying F1 HI can be different from that in animals carrying an introgression genomic segment in an otherwise pure genetic background, which provides an opportunity for identifying region-specific interaction that could be responsible for a given HI. Consistent with this, a recent study demonstrated that the lethality of hybrid F1 males between C. nigoni and C. briggsae could be suppressed by cbr-him-8 (Ragavapuram et al. 2015). However, the gene did not show misregulation in either of the two hybrids used in this study (Supplemental Table 2). In addition, most of the F1 hybrid males between the two species are atypically small, and 37% of them have no gonad (Woodruff et al. 2010; Kozlowska et al. 2012); whereas nearly all of the hybrid males used in this study have gonads with obvious sperm cells (Bi et al. 2015). Here we investigated the molecular mechanism of hybrid male sterilities between the nematodes C. briggsae and C. nigoni through detailed analysis of expression changes of coding genes, TEs, and small RNAs. The sterilities are caused by independent introgressions of an X-Chromosome linked fragment from C. briggsae in an otherwise C. nigoni background. Our results support a genetic interaction between the X Chromosome and autosomes and suggest a role of the endogenous RNAi pathway in mediating the interaction.
It is widely held that misregulation of piRNAs and their associated TEs is one of the major contributors to HI, especially in F1 hybrids (Erwin et al. 2015). However, this is apparently not the case for the hybrid that carries a homogenous genetic background except for an X-linked introgression fragment because we detected few changes in expression for both piRNAs and TEs in the two hybrid lines (Figs. 5, 6). It is possible that misregulation of piRNAs and their associated TEs could nevertheless occur in the F1 hybrid males between C. briggsae and C. nigoni, but the hybrids might develop immunity against these TEs during the subsequent introgression steps through generation of 22G RNAs downstream from piRNA targeting (Ashe et al. 2012; Shirayama et al. 2012). It would be informative to investigate these possibilities by assessment of transcriptomes of both TEs and piRNAs in the F1 hybrid males between the two species.
An alternative explanation for the observed hybrid male sterilities is the disruption of a genetic interaction between the X Chromosome and the autosomes (X:A imbalance) as demonstrated previously in the Drosophila species (Lu et al. 2010). It has long been observed that genes expressed in the male germline tend to be enriched on the autosomes but depleted on the X Chromosome (Albritton et al. 2014; Ortiz et al. 2014), suggesting an interaction between X Chromosome and autosomes, which is presumed to be essential for proper spermatogenesis. So far, direct evidence in favor of this hypothesis is limited to Drosophila species (Lu et al. 2010). Our transcriptome data of the hybrid sterile males and its parental C. nigoni males demonstrate the interaction in Caenorhabditis species. The two sterile lines each carry an independent introgression fragment from the X Chromosome of another nematode species C. briggsae but in an otherwise C. nigoni background achieved through multiple generations of backcrossing (Fig. 2D), which minimizes the complications of the mixed genomes as is the case of F1 hybrids. However, in contrast to the Drosophila study, in which the sterility was attributed to one specific locus on the X Chromosome (Lu et al. 2010), the two C. briggsae introgressions in the C. nigoni background are quite large nonoverlapping fragments (Fig. 2A–C), yet the two sterile lines demonstrate very similar defects in germline and sperm, suggesting they suffer from similar defects in spermatogenesis. In addition, spermatogenesis genes are down-regulated in a similar way between two sterile lines, and these expression changes are significantly enriched in autosomal genes (Figs. 3, 4). Altogether this suggests that a correct expression balance between the X-linked and autosomal genes is important to ensure proper spermatogenesis. The fact that distinct nonoverlapping regions can produce a similar effect suggests that maintaining the correct balance requires broad regions of the X Chromosome and is not limited to one or two “master regulators.” Instead, a perturbed X:A imbalance caused by disrupted X Chromosome integrity involving multiple loci is a plausible explanation for the sterilities as a result of introgression of the C. briggsae X into the C. nigoni background. Evidence of an interaction between the X Chromosome and autosomes also comes from studies of dosage compensation (Meyer 2005). Whether the disrupted X Chromosome perturbs dosage compensation in cis remains an open question.
How the interaction between autosomes and the X Chromosome is maintained is a longstanding enigma. Our analysis of small RNAs provides insights into the interaction. In sterile hybrids, 22G RNAs targeted to spermatogenesis genes were specifically up-regulated. Importantly, up-regulated small RNAs corresponded well to down-regulated spermatogenesis genes in the hybrids (Fig. 6D). Thus up-regulation of 22G RNAs mapping to spermatogenesis genes may cause failure of spermatogenesis by reducing the expression of spermatogenesis genes, possibly through inducing epigenetic changes in chromatin. In C. elegans, CSR-1–type and WAGO-type 22G RNAs are the two major classes of small RNAs enriched in germline. CSR-1–dependent small RNAs are enriched for spermatogenesis genes (Conine et al. 2013); thus we might expect that the spermatogenesis genes showing increased 22G RNAs are CSR-1 dependent. Indeed, the spermatogenesis genes we identified with increased 22G RNAs are enriched for genes homologous to CSR-1 targets previously annotated as enriched in C. elegans males (Fig. 6F; Conine et al. 2013). However, CSR-1 functions to protect gene expression, in contrast to WAGO-dependent 22G RNAs, which act to silence their targets (Seth et al. 2013; Wedeles et al. 2013). Thus we speculate that 22G RNAs targeted to spermatogenesis genes that bind to CSR-1 in wild-type C. nigoni males and thus normally support gene expression become rerouted in sterile hybrids into the WAGO pathway and result in silencing. As “nonself” DNA triggers stable 22G RNA-mediated silencing of the “nonself” region in C. elegans (Ashe et al. 2012; Lee et al. 2012; Shirayama et al. 2012), it is interesting to speculate that the 22G RNA up-regulation that occurs as a result of disrupted X:A imbalance might be a more widespread response to foreign sequences that leads to global misregulation of spermatogenesis genes. Definitive tests of such a possibility will require further experiments, for example, pull-down assays using various types of Argonantes followed by RNA-seq for small RNAs, which are beyond the scope of this study.
Our improved C. nigoni draft genome “cn1” will become an invaluable resource for the community, which will not only facilitate the studies of HIs between the two nematode species but also provide a foundation for comparative analysis of other fundamental biological processes. One of the fascinating biological processes is sex determination, which is known to have been subjected to fast evolution between Caenorhabditis species (Ellis and Lin 2014). Despite C. briggsae being the closest relatives of C. nigoni and their hybrid progeny being viable, the two nematode species have distinct modes of sex determination, with the former adopting a hermaphroditic mode of reproduction and the latter using a dioecious mode. Availability of an improved C. nigoni genome, as well as transcriptomes produced in this study, will facilitate the study of the sex determination pathway and its modifiers.
Methods
Worm strains and maintenance
All worms were maintained at 25°C on NGM plates seeded with OP50 as food source. Strains AF16 and JU1421 were used as wild isolate of C. briggsae and C. nigoni, respectively, throughout the paper. Hybrid male sterile strains ZZY10307 and ZZY10330 were produced previously by backcrossing with GFP-labeled C. briggsae strains with JU1421 for at least 15 generations (Bi et al. 2015). The GFP-containing introgression fragments were maintained as a female heterozygote.
Genotyping of introgression boundaries by NGS
Introgression boundaries of ZZY10307 and ZZY10330 were genotyped as previously described (Bi et al. 2015). A total of 1.5 and 1.7 million reads were generated for ZZY10307 and ZZY10330, respectively, which were mapped back to combined C. briggsae (“cb4”) and C. nigoni reference genomes (“cn1”). Read coverage was visualized across the C. briggsae genome as shown in Figure 2 and Supplemental Figure 2.
C. nigoni genome assembly
Around 20× coverage of Illumina synthetic long reads were produced for C. nigoni (JU1421), and its genome assembly was produced as previously described (Li et al. 2015). C. nigoni pseudochromosomes were produced using syntenic information with C. briggsae, and preliminary genome annotation was performed using the BAKER1 pipeline (see Supplemental Methods) (Hoff et al. 2015).
Compilation of TEs in C. nigoni and C. briggsae genome
To de novo annotate putative TEs, RepeatModeler (version 1.0.7, http://www.repeatmasker.org/RepeatModeler.html) was run with the genome sequences of C. nigoni (“cn1”; this study) and C. briggsae (“cb4”), respectively, as an input, using default parameters. The consensus sequences of TEs produced were used as an input to RepeatMasker (version 4.0.4; http://www.repeatmasker.org) to annotate all the possible TE loci over the two genomes. RMblast (version 2.28) was used in the two annotation pipelines to align genome sequence against possible TEs.
Sequencing of mRNAs by NGS
Three hundred young adult males were picked for mRNA extraction for each sample for C. briggsae (AF16), C. nigoni (JU1421), ZZY10307, and ZZY10330 with three replicates each. For collection of ZZY10307 and ZZY10330 young adult males, fertile females that carry the introgression were mated with JU1421 males. The male progeny that carry the introgression as judged by the expression of cbr-myo-2::GFP were picked under a fluorescence microscope for mRNA extraction. Initial attempts of using dissected germlines for RNA extraction were made, but dissecting of germline from hybrid sterile males turned out to be impractical, as hydrostatic pressure in the sterile males is not comparable to parental males (data not shown). Total RNAs were extracted using TRIzol (Invitrogen) following the manufacturer's instructions. mRNA purification and fragmentation, cDNA synthesis, end repairing, adapter ligation, and DNA fragment enrichment were performed using Illumina's TruSeq stranded mRNA library preparation kit based on the kit's manual. Each library was barcoded and sequenced to obtain paired-end (2 × 150 bps) reads using Illumina MiSeq. We obtained approximately 8 million reads of high-quality score (greater than 30 mean quality score) on average per sample.
Sequencing of small RNAs by NGS
RNA was extracted from C. briggsae (AF16), C. nigoni (JU1421), and hybrid (ZZY10330) males expressing GFP for small RNA sequencing. RNA was treated with 2 µL RppH (NEB) to remove both 5′ caps and 5′ triphosphate and to enable cloning of 22G RNAs. Libraries were constructed using the Illumina TruSeq small RNA library prep kit according to the manufacturer's instructions.
Data analysis of mRNA-seq reads
All the mRNA-seq reads were mapped against C. briggsae genome (“cb4”) using CLC genomic workbench 8.0. To ensure the same mappability between C. nigoni and C. briggsae reads against C. briggsae ORFs, a relaxed mismatch cutoff was used for reads derived from C. nigoni and hybrids than for those derived from C. briggsae (see Supplemental Methods). The gene with fold change > 2 and FDR < 0.01 between C. nigoni (JU1421) and a hybrid was considered as a DEG (differentially expressed gene). For quantification of TE expression, we mapped all reads from mRNA-seq against the TE consensus sequences as described above using CLC genomic workbench 8.0, with a cutoff of at least 75% similarity and 75% length. Total number of the mapped reads was counted for calculation of the differential expression between hybrids and JU1421 in a similar way as that for mRNAs.
Enrichment analysis of misregulated genes
GO terms for C. elegans and the C. elegans/C. briggsae ortholog table were fetched from WormBase (WS250). The gonad-specific gene categories were previously defined by the comparison of RNA-seq results between C. elegans male- and female-specific gonads (Ortiz et al. 2014). The C. briggsae GO term and gonad-specific gene categories were built from C. elegans data, based on the ortholog table. The sex-biased gene categories for C. briggsae were fetched from the comparison of RNA-seq results of C. briggsae male and female worms (Albritton et al. 2014). The enrichment analysis for GO terms, gonad-specific, and sex-biased gene categories were carried out using ClusterProfiler (Yu et al. 2012) by hypergeometric test in an R statistical computing environment (R Core Team 2016). The P-value and FDR were calculated for each category, and the categories with FDR < 0.05 were reported.
Data analysis on 22G RNAs
To identify piRNAs, 21U-RNAs that either did not map exactly or that matched with up to one mismatch to a C. briggsae miRNA were aligned to the C. nigoni genome assembly (“cn1”) produced in this study using Bowtie, allowing zero mismatches and reporting only one alignment per sequence. We visually examined plots of the density of piRNA loci across the C. nigoni genome using histograms made in R, verifying that piRNAs were strongly enriched at the syntenic regions to the C. briggsae piRNA clusters described previously (Shi et al. 2013). We then prepared plots of the piRNA clusters to compare the number of unique sequences per million total unique sequences. To assess the difference in the overall piRNA read counts, we used the Wilcoxon test (unpaired), which makes no assumption about the distribution involved.
To analyze 22G RNAs, we aligned 22G RNAs to either TEs or transcripts from C. nigoni (see Data access section and files at http://158.182.16.70:8080/). We then assessed the total counts for 22G RNAs mapping to individual genes or TEs, and this total read count was normalized to the total number of mapped 22G RNAs. To compare the 22G RNAs mapping to different gene classes, we first used annotations of 22G RNAs mapping to C. briggsae (Claycomb et al. 2009). This study reports CSR-1 targets in C. briggsae hermaphrodites by a direct immunoprecipitation approach and also adds information about WAGO targets by homology. We supplemented these annotations with identification of CSR-1 targets in C. elegans males (Conine et al. 2013). We then used BLAST to identify the best-matching C. nigoni transcript to the C. briggsae transcriptome, discarding genes that failed to map with an e-value of <10−4, for which we could not assign a homolog with a better than 10−4 e-value. The significance of up or down-regulation of 22G RNAs was assessed using the Wilcoxon test (paired between individual loci), which does not make any assumption of the underlying distribution. Data analysis on miRNAs and piRNAs is outlined in the Supplemental Methods.
Data access
The Illumina synthetic long reads and the “cn1” genome assembly for C. nigoni from this study have been submitted to the NCBI BioProject database (http://www.ncbi.nlm.nih.gov/bioproject/) under BioProject ID PRJNA306403 with accession number LWKT00000000 for the assembly and accession number SRR3031106 for the Illumina synthetic long reads. The mRNA and small RNA sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE76306 and GSE75763, respectively. The DNA sequencing data for introgression boundary mapping of ZZY10307 and ZZY10330 have been submitted to the NCBI Sequence Read Achieve (SRA; http://www.ncbi.nlm.nih.gov/sra/) under accession numbers SRR3081358 and SRR3081363, respectively. A genome and synteny browser for this project is available at http://158.182.16.70:8080/.
Acknowledgments
We thank Chung Wai Shing for logistic support and the members of Z. Zhao's laboratory for helpful discussion and comments. This work was supported by the Early Career Scheme Fund (HKBU263512), the General Research Fund (HKBU12103314), and the Collaborative Research Fund (HKBU5/CRF/11G) from the Hong Kong Research Grant Council, and the Strategic Development Fund of Hong Kong Baptist University to Z. Zhao; a grant from the United Kingdom Medical Research Council (MC-A652-5PZ80) to P.S.; and an Imperial College London Research Fellowship to P.S.
Author contributions: R.L., X.R., Y.B., and V.W.S.H. performed introgressions and worm preparation. X.R. conducted mRNA sequencing; R.L. and Z. Zhao performed data analysis on mRNA-seq and TEs. C.H., A.Y., and Z. Zhang provided long reads. T.L., Y.Z., and L.M. characterized sperm phenotypes. P.S. performed small RNA sequencing and data analysis and contributed to manuscript writing. Z. Zhao conceived the project, provided the reagents, and wrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.204479.116.
- Received January 18, 2016.
- Accepted May 16, 2016.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








