
A new class of temporarily phenotypic enhancers identified by CRISPR/Cas9-mediated genetic screening
- Yarui Diao1,7,
- Bin Li1,
- Zhipeng Meng2,
- Inkyung Jung1,
- Ah Young Lee1,
- Jesse Dixon1,3,4,
- Lenka Maliskova5,
- Kun-liang Guan2,
- Yin Shen5,7 and
- Bing Ren1,6
- 1Ludwig Institute for Cancer Research, University of California, San Diego, La Jolla, California 92093, USA;
- 2Department of Pharmacology and Moores Cancer Center, University of California, San Diego, La Jolla, California 92093, USA;
- 3Medical Scientist Training Program, University of California, San Diego, La Jolla, California 92093, USA;
- 4Biomedical Sciences Graduate Program, University of California, San Diego, La Jolla, California 92093, USA;
- 5Institute for Human Genetics and Department of Neurology, University of California, San Francisco, San Francisco, California 94143, USA;
- 6Department of Cellular and Molecular Medicine, Institute of Genomic Medicine and Moores Cancer Center, University of California, San Diego, La Jolla, California 92093, USA
- Corresponding authors: yin.shen{at}ucsf.edu, biren{at}ucsd.edu
-
↵7 These authors contributed equally to this work.
Abstract
With <2% of the human genome coding for proteins, a major challenge is to interpret the function of the noncoding DNA. Millions of regulatory sequences have been predicted in the human genome through analysis of DNA methylation, chromatin modification, hypersensitivity to nucleases, and transcription factor binding, but few have been shown to regulate transcription in their native contexts. We have developed a high-throughput CRISPR/Cas9-based genome-editing strategy and used it to interrogate 174 candidate regulatory sequences within the 1-Mbp POU5F1 locus in human embryonic stem cells (hESCs). We identified two classical regulatory elements, including a promoter and a proximal enhancer, that are essential for POU5F1 transcription in hESCs. Unexpectedly, we also discovered a new class of enhancers that contribute to POU5F1 transcription in an unusual way: Disruption of such sequences led to a temporary loss of POU5F1 transcription that is fully restored after a few rounds of cell division. These results demonstrate the utility of high-throughput screening for functional characterization of noncoding DNA and reveal a previously unrecognized layer of gene regulation in human cells.
A remarkable feature of multicellular organisms is that they develop distinct sets of highly specialized cells using the same genetic blueprints. The developmental program in each species is governed by complex transcriptional regulatory circuitry composed of large numbers of transcription factors and cis-regulatory elements (Lee and Young 2013; Gorkin et al. 2014; Levine et al. 2014). Large-scale studies such as ENCODE and Roadmap Epigenomics projects have annotated millions of candidate cis-regulatory elements in the mammalian genome (Gerstein et al. 2012; Shen et al. 2012; Xie et al. 2013) based on biochemical signatures such as histone modification, transcriptional factor binding, and DNase I hypersensitivity. These putative regulatory regions harbor a disproportionally large number of sequence variants associated with human traits and diseases, leading to the notion that genetic lesions in cis-regulatory sequences contribute substantially to common human diseases (Maurano et al. 2012; Roadmap Epigenomics Consortium et al. 2015). A major bottleneck in advancing this hypothesis is a lack of high-throughput means to functionally characterize the large number of predicted cis-regulatory elements with regard to their contributions to target gene expression.
Traditional molecular genetic approaches have uncovered a small number of cis-regulatory sequences that confer spatiotemporal gene expression patterns to specific target genes in human cells (Li et al. 2002), but more effective methods are needed for functional characterization of the large number of candidate enhancers annotated recently in the human genome (The ENCODE Project Consortium 2012; Roadmap Epigenomics Consortium et al. 2015). Transgenic mouse experiments used to validate enhancer activities in vivo (Visel et al. 2009) are limited to elements functioning in embryonic tissues, are of a modest throughput, and do not inform about the cognate target gene. High-throughput reporter assays in cultured cells, another strategy for validating tens of thousands of putative enhancers simultaneously (Melnikov et al. 2012; Patwardhan et al. 2012), test enhancer activities using heterologous promoters outside of their native chromatin context and do not inform on the target genes of the elements either.
In order to fully assess the contribution of a candidate enhancer to gene expression, it is necessary to delete or modify the element in its endogenous location. The bacterial clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 genome editing method has proven as a versatile tool for rapid alteration of genetic sequences in cells (Cong et al. 2013; Mali et al. 2013) with high specificity and efficiency (Jinek et al. 2012). CRISPR/Cas9 is capable of interrogating genome function for both transcribed genes and noncoding sequences. For example, deletion of a candidate super-enhancer located 130 kb downstream from the Sox2 gene resulted in a 90% reduction of Sox2 transcription in mouse embryonic stem cells (Li et al. 2014; Zhou et al. 2014a). High-throughput CRISPR/Cas9 strategies have been developed for functional screening of protein coding genes essential for several physiological traits, such as cell vibration and intoxication in human cells (Shalem et al. 2014; Wang et al. 2014; Zhou et al. 2014b). Conceivably, this high-throughput CRISPR/Cas9 strategy can be used for large-scale identification and functional characterization of cis-regulatory elements. A recent study has identified the functional cores of human-specific erythroid enhancers by utilizing pooled single guide RNA (sgRNA) libraries targeted to three candidate enhancers of the BCL11A gene (Canver et al. 2015). Similarly, several enhancers were identified for TP53 and ESR1 genes in human BJ cells with this approach (Korkmaz et al. 2016).
Here, we describe a large-scale CRISPR/Cas9-mediated functional screening of cis-regulatory elements in human genome. Application of this strategy to POU5F1 in the human embryonic stem cells (hESCs) uncovered both classical cis-regulatory elements and a class of noncanonical elements that regulate transcription in an unexpected manner.
Results
High-throughput screening for functional cis-regulatory elements in the POU5F1 locus
To identify cis-regulatory elements involved in the transcriptional control of POU5F1 we focused on 174 candidate cis-regulatory elements located within the gene's topological associated domain (TAD) (Fig. 1A; Dixon et al. 2012; Gerstein et al. 2012; Xie et al. 2013). Previous studies have shown that the human genome is partitioned into highly conserved and stable TADs (Dixon et al. 2012; Nora et al. 2012; Sexton and Cavalli 2015), within which the majority of the enhancer-promoter looping events occur (Jin et al. 2013). The TAD containing the POU5F1 gene (Chr 6: 30,520,000–31,561,000) consists of 87 RefSeq annotated genes and 174 putative cis-regulatory elements with chromatin features of enhancers, CTCF binding, and/or DNase I hypersensitivity (Supplemental Table S1). Presently, it is unclear which candidate cis-regulatory elements are involved in POU5F1 expression. Given the presence of many other genes within this TAD, we expect that only a small number of elements are involved in regulating POU5F1 transcription.
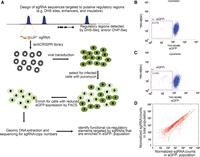
Experimental design of a high-throughput CRISPR/Cas9-mediated screen for identifying cis-regulatory elements. (A) Workflow of lentiCRISPR screening strategy to identify functional regulatory elements. sgRNA library sequences were designed to create random mutations at 174 predicted regulatory regions (blue peaks) in the POU5F1 TAD via nonhomologous end joining (NHEJ). sgRNA sequences were synthesized in an array-based oligo pool, cloned into the lentiCRISPR plasmid, and packaged into lentiviral libraries. We then used lentiviral libraries to infect H1 POU5F1-eGFP cells to generate random mutagenesis of the 174 candidate regions. hESCs with mutations at cis-regulatory elements affecting POU5F1 expression can be identified as eGFP− cells. (B) Control H1 POU5F1-eGFP without lentiviral infection were dissociated into single cells and subjected to FACS analysis to determine the eGFP− (P4) gate for eGFP− population; 0.31% indicates the ratio of P4 in parental live singlets. (C,D) The H1 POU5F1-eGFP cells were infected with lentiCRISPR library by spin infection at low multiplicity of infection (MOI). Twenty-four hours after infection, the cells were cultured for 7 d under puromycin selection; for another 10 d, without puromycin. (C) The cells were subjected to FACS analyses. (2.16%) The ratio of P4 in parental population. (D) The eGFP− P4 population was collected by FACS sorting. Genomic DNA was isolated from P4 and nonsorted control cells, followed by PCR amplification of sgRNA sequence and deep sequencing. Scatter plot for sgRNA read counts in eGFP− cells compared with the control cells after LOESS normalization. Dots underneath the green line are sgRNAs with at least twofold enrichment in the eGFP− cells compared with the control population.
We designed 1964 sgRNA sequences (Supplemental Table S2) targeting these putative cis-regulatory elements with an average of 11 sgRNA per element (Supplemental Fig. S1A). On average, each sgRNA could result in genetic disruption of the target locus with 30% probability under our experimental conditions (Li et al. 2014). Therefore, we expect >98% probability of introducing mutations to an element with 11 sgRNAs. As negative controls, we also designed 1415 ineffective sgRNA sequences that complement sequences in the 174 elements but lack a NGG/NCG protospacer adjacent motif (PAM) sequence necessary for effective targeting in vivo (Supplemental Table S2). Additionally, we also included 539 sgRNA to target regions bearing no evidence for regulatory function. We infected the H1 POU5F1-IRES-eGFP reporter line (Zwaka and Thomson 2003) with the lentiviral libraries expressing the above sgRNAs along with the Cas9 protein. The infected cells were selected in puromycin media for 7 d and expanded in regular media (Fig. 1A) before being sorted based on the eGFP signals, which reflects transcription levels of the cotranscribed POU5F1 gene.
We next isolated cells with reduced eGFP signals (eGFP−) (Fig. 1B,C), amplified the sgRNA inserts from the integrated lentiviral sequences, and determined the enriched sgRNAs by deep sequencing. We computed the relative enrichment of each sgRNA in the eGFP− cells compared with the total cell population after LOESS normalization. We performed the experiment four times and identified a list of sgRNA sequences with at least twofold enrichment in the eGFP− population in each experiment (Fig. 1D; Supplemental Fig. S1B–D). Unexpectedly, many negative control sgRNAs, those lacking the PAM sequence, were tested positive in each experiment. These false hits were most likely due to the small fraction of eGFP− population (∼0.3%) that naturally exists in the parental H1 POU5F1-eGFP line (Fig. 1B). To eliminate these false positives, we required that a positive cis-regulatory element should have at least two distinct sgRNAs enriched by twofold or more in the eGFP− population in at least three out of the four independent experiments (Supplemental Table S3; Supplemental Fig. S2A). By use of this criterion, no negative control sgRNA passed the filter, while six cis-regulatory elements were identified as positives (Fig. 2A). These elements are located with various linear genomic distances from the POU5F1 transcription start site (TSS), ranging from −1.4 to 491 kbp (Fig. 2A,B). Among these positive hits are the POU5F1 promoter and a proximal enhancer (DHS_115) located 1.4 kbp upstream of the TSS, confirming the essential role of POU5F1 promoter in controlling gene expression, and also providing additional functional evidence for the POU5F1 proximal enhancer (Chia et al. 2010; Xie et al. 2013).
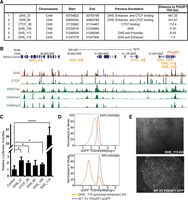
Characterization of cis-regulatory elements identified near POU5F1. (A) A list of regulatory elements regulating POU5F1 transcription identified from the lentiCRISPR screening with coordinates (hg18). Each element was identified by at least two distinct sgRNAs enriched by twofold or more in the eGFP− population in at least three out of the four independent screenings. (B) Genome browser snapshot shows the location and epigenetic environment for each element in H1 hESCs. (C) Reporter assays for the six identified cis-regulatory elements. H1 hESC cells were transfected with various luciferase reporter plasmid as indicated. Two days post transfection, cells were lysed and subjected to luciferase reporter assays. All tested elements are cloned into the control reporter plasmid containing the 360-bp POU5F1 minimal promoter sequence driving reporter gene expression. DHS_115 exhibits the highest enhancer activities ([****] P < 10−6), while the other four elements show either minimal (DHS_37, CTCF_28, and DHS_108 with [*] P < 0.05) or no enhancer activities (DHS_65 with P = 0.27) compared with the control reporter plasmid containing POU5F1 promoter only. (D) FACS analysis showed that the DHS_115 biallelic mutant hESCs (orange line) gradually lose POU5F1-eGFP expression in culture over time compared with parental H1 POU5F1-eGFP line (dash line). (E) Phase images of hESCs with biallelic mutations at DHS_115 locus (top) and wild-type hESCs (bottom). Deletions of DHS_115 sequences lead hESCs to lose pluripotency and become differentiated in the culture (top).
Next, we tested whether the identified elements could function as enhancers in the classical reporter assays (DHS_37, CTCF_38, DHS_65, DHS_108, and DHS_115). We cloned genomic fragments corresponding to each element into a reporter plasmid containing the 360-bp POU5F1 core promoter region and a luciferase reporter gene. All elements, with the exception of DHS_65, exhibited significant enhancer activities in the H1 cells compared with the control plasmid containing only the POU5F1 core promoter sequence (Fig. 2C). DHS_115, located 1.4 kb upstream of POU5F1, activated the reporter more than 26-fold, and this activity can be further boosted by DHS_37 when tested in combination (Supplemental Fig. S3A). These results suggest that most of the elements testing positive in the CRISPR/Cas9 sgRNA screening may function as enhancers.
Consistent with the results from the reporter assay, a cell clone with mutations at DHS_115 (one allele with 13-bp deletion and the other allele with 4-bp substitution of the 17-bp original sequences) exhibited reduced eGFP levels, which decreased further in additional passages (Fig. 2D; Supplemental Fig. S3B). Cells with mutations at this proximal enhancer could not be expanded as they quickly differentiated (Fig. 2E), suggesting that DHS_115 is a major regulator of POU5F1 expression in hESCs. Of note, this region is within a previously defined hESC-specific proximal enhancer region (Chr 6: 31,247,052–31,248,218) that controls POU5F1 expression in primed hESCs (Gafni et al. 2013).
Temporary phenotype after deletion of DHS_37, DHS_65, and DHS_108
Of the remaining four distal regulatory regions, DHS_37, CTCF_38, DHS_65, and DHS_108, three (DHS_37, DHS_65 and DHS_108) display strong DNase I hypersensitivity in hESCs, and we decided to further study their functionalities. We deleted these elements individually in the H1 POU5F1-eGFP cells with a pair of sgRNAs flanking each region. In contrast to cells with mutations at DHS_115, cells with biallelic deletion of DHS_37, DHS_65, or DHS_108 do not show any growth defects. In addition, all biallelic KO clones exhibit temporary loss of POU5F1 transcription in a subpopulation of cells: Reduced POU5F1 transcription during initial culturing, indicated by decreased eGFP signals, was seen in a variable fraction of each clonal population (Fig. 3A; Supplemental Fig. S4A). Interestingly, after two additional passages in culture, each clonal population reverted to wild-type POU5F1 expression (Supplemental Fig. S4B). To confirm the transient nature of the POU5F1 down-regulation in these cells, we used hESC-specific markers SSEA4 and Tra1-60 to isolate the SSEA4+/Tra1-60+/eGFP− cell population (Supplemental Fig. S5) and found that this eGFP− pluripotent hESC population rapidly regained eGFP signals after a few cell divisions in culture (Fig. 3B). Furthermore, loss of two or even all three of these elements in hESCs led to transient reduction of eGFP levels that was quickly regained upon additional passages (Fig. 3D,E; Supplemental Fig. S4A). This observation confirmed the involvement of these elements in regulating POU5F1 expression in hESCs. More intriguingly, the existence of both eGFP+ and eGFP− cells from the same genotype indicated that deletion of these elements produced heterogeneous effects (Fig. 3A).
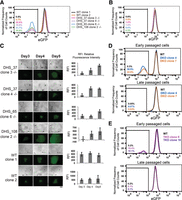
Characterization of Temp enhancers. (A–C) Biallelic clones with single deletions at DHS_37, DHS_65, and DHS_108 loci were generated by cotransfection of a pair of pX330 plasmids expressing sgRNAs flanking each locus. Each clone was cultured for 2 wk before their POU5F1 expression levels were assessed using FACS analysis of eGFP in early passages (A) and late passages (B). (A) In early passages, biallelic mutant clones showed heterogeneous eGFP expression levels. (B) eGFP− cells isolated from (A) restored eGFP level. (C) Colony image tracing experiments showed that mutant clones gradually regain eGFP expression in culture over time. Bar graph represents average relative fluorescence intensity (RFI) for each colony quantified by ImageJ. Scale bar, 200 μm. (D) DHS_37 and DHS_108 (−/−, −/−) double knockout (DKO) clones were generated by two rounds of deletion of each region by CRISPR/Cas9 and confirmed by Sanger sequencing. DKO cells exhibit transient loss of eGFP expression that can be recovered after long-term culture. (E) DHS_37, DHS_65, and DHS_108 (−/−, −/−, −/−) triple knockout (TKO) clones were generated by three rounds of deletion of regions by CRISPR/Cas9 and confirmed by Sanger sequencing. TKO cells exhibit transient loss of eGFP expression that can be recovered after long-term culture.
The transient loss of POU5F1-eGFP expression in the above clones is surprising. In order to rule out the possibility that contaminating parental POU5F1-eGFP hESCs outgrew mutant clones over time, we followed the growth and eGFP signals of single-cell clones with fluorescence microscopy over a period of 3 d after clonal isolation and expansion post transfection with CRISPR/Cas9 genome editing vectors (Fig. 3C). DHS_37, DHS_65, and DHS_108 KO clones (−/−) gradually gained eGFP expression, while the wild-type clones displayed constant eGFP signals. Therefore, the temporary, but detectable, loss of eGFP signals in the above clones could not be due to contaminant parental POU5F1-eGFP hESCs.
The eGFP− cells are positive for the hESC marks SSEA4 and Tra1-60, indicating these cells can maintain pluripotency even with the transient loss of POU5F1 expression. Additionally, despite the reduced expression level of POU5F1, these cells showed wild-type expression levels for other stem markers, including NANOG and SOX2 (Supplemental Fig. S6A). ChIP-seq analysis of H3K27ac in two distinct DHS37−/− clones revealed little change in active chromatin landscape compared with the parental cells (Supplemental Fig. S6B). Furthermore, the recovered eGFP+ cells have no obvious defects for early differentiation as the embryoid bodies (EBs) derived from these cells expressed similar levels of marker genes for three germ layers (endoderm-specific marker AFP, ectoderm-specific marker SOX1, and mesoderm-specific marker T brachyury transcription factor) (Supplemental Fig. S6C).
DHS_65 and DHS_108 act in cis to regulate POU5F1 expression
DHS_37, DHS_65, and DHS_108 sequences overlapped with protein coding genes or lncRNA, namely, PPP1R18, LINC00243, and TCF19, respectively (Supplemental Fig. S4A). Both PPP1R18 and TCF19 are transcribed in H1 cells (Djebali et al. 2012). To rule out the possibility that POU5F1 reduction may be due to disruption of their gene products, we generated cell clones with monoallelic deletion of these elements and examined if genetic disruption at these loci would affect POU5F1 transcription in cis (on the same allele) but not in trans (from the homologous chromosome). If the phenotype of transient loss of POU5F1-eGFP expression is due to indirect effects such as mutations in other gene products, we would expect to see reduced eGFP no matter on which allele the deletion of those elements occurs. On the other hand, if these elements act in cis to modulate POU5F1 transcription, we should observe the transient down-regulation of eGFP expression only in mutants bearing deletion on that allele.
To carry out this analysis, we took advantage of the recently phased H1 genome (Selvaraj et al. 2013; Dixon et al. 2015) and identified the haplotype of the eGFP allele (P1) and wild-type allele (P2) by targeted sequencing of the region that contains multiple SNPs across the POU5F1 gene (Supplemental Fig. S7A). We then inferred the allelic information for DHS_65 and DHS_108 based on the phased SNPs in each element. We were able to distinguish clones with a deletion on the eGFP-knockin allele versus the clones with a deletion in the other allele by linking SNPs near DHS_65 and DHS_108 to the SNPs on the eGFP allele. We obtained two heterozygous clones for the DHS_65 KO (clone 8−/+ with deletion on P1 allele and clone 7+/− with deletion on P2 allele) and three heterozygous clones for the DHS_108 KO (clone 1−/+ and clone 6−/+ with deletions on P1 allele and clone 4+/− with deletion on P2 allele) (Supplemental Fig. S7B,C). We followed their POU5F1-eGFP levels from initial isolation over the period of a week and observed only clones with mutations on the P1 allele exhibited a transient loss of eGFP expression. In contrast, the mutant clones (DHS_65 clone 7 and DHS_108 clone 4) with deletion on the P2 allele showed normal levels of eGFP expression over this period (Fig. 4A). Additionally, cells with the P1 deletion (−/+) regained POU5F1-eGFP expression upon additional passages in culture (Fig. 4B,C). Those results support our model that DHS_65 and DHS_108 act in cis to regulate POU5F1 transcription.
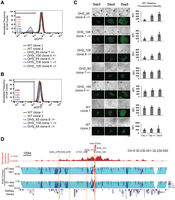
DHS_65 and DHS_108 regulate POU5F1 in cis. (A–C) Cell clones harboring monoallelic deletion of DHS_65 and DHS_108 were generated by cotransfection of H1 POU5F1-eGFP cells with a pair of pX330 plasmids expressing sgRNAs flanking each locus. P1 is the eGFP-containing allele, while P2 is the non-eGFP allele according to the SNP phasing information in the H1 genome (see Methods; Supplemental Fig. S7). Genotype information for mutant clones is indicated by + (WT) or − (KO) labeling in the order of P1/P2. FACS analysis was performed for the parental cell line and monoallelic mutant clones for DHS_65 and DHS_108 at early passages (A) and late passages (B). Note only the P1 allele (eGFP allele) deletion impaired eGFP expression in a subpopulation of cells for each clone, while the eGFP levels in the clones for P2 allele (non-eGFP allele) deletion were not affected. The GFP− population sorted from clones in A regained eGFP signals comparable with the wild-type clones in B. (C) Time-course colony image tracing experiments showed monoallelic mutant clones with P1 allele deletion gradually regain eGFP expression in culture, while mutant clones with P2 allele deletion and WT clones exhibit constant eGFP signal levels. Bar graphs represent RFI for each colony quantified by ImageJ. Scale bar, 200 μm. (D) A genome browser snapshot for chromatin interaction frequencies originating from the POU5F1 promoter. (Top) Normalized chromatin interaction counts in a published Hi-C data set (Dixon et al. 2012). (Bottom) Contact profiles at the POU5F1 locus from two replicates of 4C-seq with the POU5F1 promoter as the bait (highlighted in dark orange). Contact profiles (curves) indicate the frequencies of chromatin interactions, while the probability of interactions is quantified in a multiscale analysis as previously described (van de Werken et al. 2012). Five million H1 POU5F1-eGFP cells were used for each 4C-seq experiment. 4C-seq data were processed with 4C-seq pipeline (van de Werken et al. 2012).
Taken together, the above results indicate that DHS_65, DHS_108, and possibly DHS_37 are a novel class of cis-regulatory elements that exert detectable and temporary phenotypes on target gene expression. The main feature of these noncanonical elements is that their disruption leads to a temporary loss of target gene expression. With comparison to the classical enhancers such as DHS_115, these temporarily phenotypic (“Temp”) enhancers show weak enhancer activity in reporter assays (Fig. 2C). However, they display other common characteristics of enhancers, such as chromatin accessibility, and the requirement for gene activation in vivo. For example, DHS_108 region is enriched for RNA polymerase II ChIP-seq signals (The ENCODE Project Consortium 2012), while three other distal elements (DHS_37, DHS_65, and CTCF_38) are associated with multiple transcription factors that are highly expressed in hESCs such as TEAD4, YY1, and SIN3A (The ENCODE Project Consortium 2012).
RAD21, a subunit of the cohesin complex, is also found at DHS_37, DHS_65, and DHS_108 (Supplemental Fig. S8; The ENCODE Project Consortium 2012). Since the cohesin complex has been implicated in mediating chromatin interactions between enhancers and promoters (Kagey et al. 2010; Zuin et al. 2014), we hypothesized that chromatin loops may exist between these three elements and the POU5F1 promoter. We first analyzed previously published Hi-C data for H1 cells (Dixon et al. 2012) and examined the chromatin interactions centered at the POU5F1 promoter. We found that all Temp enhancers, namely, DHS_37, DHS_65, and DHS_108, are located near regions that show high levels of chromatin interactions with the POU5F1 promoter. To confirm this observation, we also performed 4C-seq experiments in H1 POU5F1-eGFP cells. Again, we found these elements near regions displaying high and consistent interaction frequencies with the POU5F1 promoter (Fig. 4D). Taken together, our results suggest that Temp enhancers may be involved in local higher-order chromatin structure to regulate POU5F1 expression, a hypothesis that requires additional in depth investigation in the future.
Discussion
In the current study, we describe a high-throughput CRISPR/Cas9 screening approach for functional analysis of transcriptional regulatory sequences. Applying it to 174 candidate cis-regulatory elements in the 1-Mbp POU5F1 locus, we identified not only classical regulatory elements but also a class of noncanonical enhancers, termed Temp enhancers. The Temp enhancers carry the following properties: (1) They reside in the open chromatin regions, and (2) their loss leads to temporary reduction in transcription of the target gene. Similar to shadow enhancers (Perry et al. 2010), these elements show weak enhancer activities in in vitro reporter assays and are located distal from their cognate target promoter. However, they differ from shadow enhancers in their transitory characteristics in gene regulation. The high-throughput screening described in the current study can pick up such elements because the genetic labeling of POU5F1 with the eGFP reporter allowed isolation of single cells with transient reduction in POU5F1 transcription by FACS.
Functions of known cis-regulatory elements are generally classified based on their roles in driving or influencing transcription at a population level in specific cell types. For example, promoters are required for initiation of transcription, while enhancers can drive transcription from a heterologous promoter independent of relative locations. Insulators, on the other hand, block transcriptional activation when inserted in between an enhancer and a promoter. Loss of these elements typically leads to stable and permanent effects on transcription. In contrast to these well-characterized cis-regulatory elements, the Temp enhancers reported in the current study exhibit temporal and reversible defects in transcription. While there is a requirement for the Temp enhancers in expression, evident in the early stage of sequence disruption, the cell can quickly adapt to loss of the sequence after a few cell divisions, suggesting a high degree of robustness of the transcriptional regulatory process. On the other hand, Temp enhancers are conserved in the mouse Pou5f1 locus with the same genomic distributions as in human POU5F1 locus and contain features of potential regulatory activities marked by histone marks such as H3K4me3, H3K9ac, or H3K27ac (Supplemental Fig. S9).
How do Temp enhancers contribute to transcription? We can envision at least three models for their action: First, activation of a gene by the Temp enhancers may be needed only transiently at the initiation phase of transcription. Such a “hit-and-run” model was previously proposed for the immunoglobulin heavy-chain enhancer but was subsequently disputed upon further experimentation (Wabl and Burrows 1984; Schaffner 1988; Porton et al. 1990). Second, the Temp enhancers may play a role in local chromatin organization, by facilitating or stabilizing enhancer and promoter contacts, in a similar way as CTCF binding sites at the super-enhancer domains (Dowen et al. 2014; Narendra et al. 2015). Indeed, a recent study showed that scaffold associated regions (SARs) are enriched near actively transcribed genes (Keaton et al. 2011). The elements discovered in this study could be SARs supporting POU5F1 expression by maintaining a proper chromatin structure. Upon disruption of a stable transcriptional state due to loss of an existing SAR, other SARs can replace the previous one in mutant cells to re-establish or maintain the chromatin structure. In a static state, when the transcriptional network in a cell population has been established, only a portion of such elements that are in use can be detected by our screening method. Detection of more elements will require additional studies in populations of cells at different transcriptional states. Third, Temp enhancers may facilitate the movement of POU5F1 to remote transcription factories for activation. RNA polymerases have been shown to exist in discrete loci in the nuclear space, which are termed transcription factories (Jackson et al. 1993). It has been posited that genes need to move into these nuclear space in order for RNA synthesis to take place. We speculate that Temp enhancers may play a role in the maintenance of association between the gene and the factories. Regardless of which model may explain the nature of the Temp enhancers, our results highlight the importance of analyzing cis-regulatory element function in a temporally sensitive manner. Previous studies examining dynamics of transcriptional control of integrated transgenes after disruption of a canonical enhancer have provided deep insights into mechanisms of enhancer function, suggesting that enhancers increase the likelihood of gene activation rather than the expression level (Magis et al. 1996; Walters et al. 1996; Francastel et al. 1999). The Temp enhancers, although demonstrating a temporary phenotype in gene expression instead of more long-lasting effects typical of canonical enhancers, may function similarly by increasing the probability of transcription of the targeted gene, possibly with the help of a strong proximal enhancer.
Our study may not identify all regulatory elements that could play a compensatory role in supporting POU5F1 expression. For example, cis elements that lack canonical epigenetic features in hESCs used for their annotations would not be targeted by our sgRNA design. A better strategy will be to delete all regions in the POU5F1 TAD regardless of their epigenetic status utilizing the dual sgRNA approach. From the number of positive elements that we detected, we expect that Temp enhancers to be abundant in the human genome. This approach can be applied to identify regulatory sequences for any gene provided that a protein tag could be engineered into the gene. It would be interesting to utilize this approach to identify regulatory elements necessary to fine-tune the expression levels of broadly expressed genes in multiple cell types.
Methods
Design of sgRNA in the putative regulatory regions in POU5F1 locus
We extracted sequence information of mapped DNase I hypersensitivity (DHS) sites, CTCF binding sites, and predicted enhancers in H1 cells from the 1-Mb TAD containing POU5F1 (Chr 6: 30,520,000–31,561,000) according to previous studies (Gerstein et al. 2012; Xie et al. 2013). To design sgRNAs, we first identified all potential 20-bp sequences followed by the PAM sequences (NGG) in those regions. Second, in order to maximize the Cas9 target specificity, we narrowed down the list by ensuring that the guide sequences together with the PAM sequences didn't map to other genomic loci when three mismatches are allowed. The control sgRNA sequences were designed so that they are still within the 1-Mb TAD but no overlap with putative regulatory regions. In addition, we incorporated an additional 1415 ineffective sgRNA sequences within the putative regulatory regions, which are not followed by PAM sequences. These ineffective sgRNA sequences served as negative controls. The sgRNA oligo library was synthesized by LC Sciences. The sequences of the sgRNA are described in Supplemental Table S1.
Cell culture
The POU5F1-eGFP H1 hESC line was purchased from WiCell and described previously (Zwaka and Thomson 2003). The cells were cultured on Matrigel-coated (Corning, catalog no. 354277) plates, maintained in Essential 8 medium (Life Technologies, catalog no. A1517001), and passaged by Accutase (Stemcell Technologies, catalog no. A1517001) with 10 µM ROCK inhibitor Y-27632 (Stemcell Technologies, catalog no. 72302) supplement.
Lentiviral CRISPR/Cas9 screening
Briefly, the screening was performed following previous protocol described by Shalem et al. (2014) with minor modifications. For detailed information, see the Supplemental Material.
CRISPR/Cas9 gRNA sequence data analysis
A total of eight short-sequence-reads data sets were generated from four independent experiments and analyzed together for the present study. YD36 corresponds to eGFP− population 14 d post infection. YD37 corresponds to the total, nonsorted population from the same experiment, and is used as the control for YD36. YD38 corresponds to eGFP− population 10 d post infection from a second replicate, and YD39 is the total, nonsorted population control. YD44 corresponds to the eGFP− population 10 d post infection from the third replicate, and YD45 is the total, nonsorted population. YD46 correspond to the eGFP− population 10 d post infection from the fourth replicate, and YD47 is the total, nonsorted population. The sequence read lengths of each data set are 50 bp. (See Supplemental Table S3 for details.)
The sequence reads were first mapped to sgRNA library based on exact match; then the frequencies of each sgRNA in each sample are normalized using the LOESS normalization method implemented in R version 3.0.2 (R Core Team 2013). A subsequence with length of either 20 or 21, with the prefix of the sequence matching ACACC and suffix matching GTTTT, is extracted and compared to the whole length of the sgRNA. For each pair of eGFP− population and the control, the sgRNA frequencies in control samples are normalized using the standard LOESS normalization method; the normalized control sgRNA frequencies are then used as the expected frequencies to compare with sgRNA frequencies from the eGFP− population sample. The sgRNAs with a minimum of 50 reads in either eGFP− population or control and also with a minimum of twofold enrichment are identified as positive hits. To further identify cis-regulatory elements involved in POU5F1 expression, we determined the cis-regulatory elements with two positive sgRNA hits in at least three replicates. By use of this criterion, none of the negative control sgRNAs passed the filter (Supplemental Fig. S2).
Transfection and luciferase reporter assay
Luciferase assays were conducted as previously described (Heintzman et al. 2007). For detailed information, see the Supplemental Material.
Knockout of POU5F1-eGFP cis-regulatory elements with CRISPR/Cas9 system
CRISPR/Cas9 constructs targeting DHS_37, DHS_65, and DHS_108 was made following a protocol described earlier (Cong et al. 2013). The ssDNA oligo pairs (F and R for each DHS site) are listed below and cloned into the pX330-U6-Chimeric_BB-CBh-hSpCas9 (Addgene plasmid no. 42230) vector: DHS_37_5′_F, caccGAAATCACAGGGTGGGTCGAC; DHS_37_5′_R, aaacGTCGACCCACCCTGTGATTTC; DHS_37_3′_F, caccGCCCCTCCGAGAGTTCGGTAC; DHS_37_3′_R, aaacGTACCGAACTCTCGGAGGGGC; DHS_65_5′_F, caccGCCAACTTGTACAGGCGCCCC; DHS_65_5′_R, aaacGGGGCGCCTGTACAAGTTGGC; DHS_65_3′_F, caccGTGAATCTTGATCCCCATCGC; DHS_65_3′_R, aaacGCGATGGGGATCAAGATTCAC; DHS_108_5′_F, caccGCGCGGGTAGATCCGAAACG; DHS_108_5′_R, aaacCGTTTCGGATCTACCCGCGC; DHS_108_3′_F, caccGCATAACTGGTTGAACCTCCG; and DHS_108_5′_R, aaacCGGAGGTTCAACCAGTTATGC.
After validating the sgRNA sequences by Sanger sequencing, a pair of plasmids targeting 5′- and 3′- boundary of the same element was mixed at 1:1 ratio and cotransfected with plasmid expressing mCherry into POU5F1-eGFP cells by hESCs Nucleofector Kit 2 (Lonzo, catalog no. VPH-5022) according to the manufacturer's instruction. Three days post transfection, the cells were sorted into a Matrigel-coated 96-well plate (single cell per well) in E8 media supplemented with 10 µM ROCK inhibitor. After 10 to 12 d recovering, the viable single cell in each well would form a visible single colony. These colonies were defined as P1 passage described in this study. The P1 colonies were then transferred to 24-well plates for expanding, and they were defined as P2 passage. When the P2 cells were confluent in the 24-well plate, the cells were dissociated and analyzed or sorted using BD FACSAria II. A subpopulation of P2 cells was collected and treated with QuickExtract DNA extraction solution (Epicentre, catalog no. QE0905T), followed by genotyping PCR, TOPO cloning (Life Technologies, catalog no. K2800-20), and Sanger sequencing for sequence verification. The genotyping experiment was conducted as previously described (Shalem et al. 2014) with the following primers: DHS_37_GT_F, GCATGAGCCACAGGAGGTAG; DHS_37_GT_R, CGCTTTCTCTCCCTCAACC; DHS_65_GT_F, GAGGCAGCATCTAACCTTGC; DHS_65_GT_R, TCCTTACCATGTGGCATTTG; DHS_108_GT_F, GAATTCCGAAGGAGGGGTAG; DHS_108_GT_R, CGTGCAATACGAACACATCA; DHS_115_GT_F, GATGCTAGGGAATTCGATCCCCT; and DHS_115_GT_R, ATCCGAGCTCTGCAGGATTTGCT.
FACS sorting and analysis
The wild-type and mutant POU5F1-eGFP cells were dissociated with Accutase and stained with APC-SSEA-4 (1:100, R&D Systems, catalog no. FAB1435) and PE-Tra1-60 (1:50, eBioscience, catalog no. 12-8863-82) for 30 min, followed by FACS sorting and analysis. The FACS data were analyzed using FlowJo software.
Cell imaging
Fluorescence microscopy was performed with a LEICA DM microscope, and a LEICA DFC400 digital camera captured the images. The images were taken under 10× objective lens with the following setting: exposure time 10 sec, gain 6×, saturation 3.00, and gamma 2.0. The fluorescent images were quantified with ImageJ software.
H1 POU5F1-eGFP phasing
Determination of the haplotypes containing the targeted eGFP fusion and the nontargeted native POU5F1 allele was performed as follows. As the homology arms of the targeting vector contain DNA not native to the H1 genome, only sequencing from the nontargeted allele can provide useful information in terms of the haplotype of the targeted and nontargeted alleles. Therefore, we designed primers that would span from a common locus in the 5′ region of the POU5F1 gene to the 3′ end of the POU5F1 gene, downstream to where the fusion with the eGFP transgene would occur. Therefore, these primers will only amplify the native, nontargeted allele. TOPO cloning and Sanger sequencing of the PCR product identified that five out of five possible genotypes were derived from the P2 allele in four out of four independent clones, indicating that the P2 allele contains the native locus, and by deduction, the P1 allele must contain the targeted eGFP transgene: POU5F1 5′ common primer, AACAGGGAATGGGTGAATGA; POU5F1 3′ specific primer, TTTAAGTGTGTCTATCTACTGTGTCC.
Data access
The sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE75450.
Acknowledgments
We thank Yunjiang Qiu, Zhen Ye, and Samantha Kuan for technical assistance. This work is supported by National Institutes of Health (NIH) (2P50 GM085764) and by the Ludwig Institute for Cancer Research (to B.R.). Y.D. is supported by the Human Frontier Science Program Long Term Fellowship.
Author contributions: Y.S., Y.D., and B.R. designed research; Y.S., Y.D., A.Y.L., J.D., and L.M. performed experiments; Y.S., Y.D., B.L., and I.J. analyzed data; Z.M. and K.G. packaged lentiCRISPR library; and Y.S., Y.D., and B.R. wrote the paper.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.197152.115.
- Received July 18, 2015.
- Accepted January 20, 2016.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








