
Models of human core transcriptional regulatory circuitries
- Violaine Saint-André1,5,
- Alexander J. Federation2,5,
- Charles Y. Lin2,
- Brian J. Abraham1,
- Jessica Reddy1,3,
- Tong Ihn Lee1,
- James E. Bradner2,4 and
- Richard A. Young1,3
- 1Whitehead Institute for Biomedical Research, Cambridge, Massachusetts 02142, USA;
- 2Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, Massachusetts 02215, USA;
- 3Department of Biology, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA;
- 4Department of Medicine, Harvard Medical School, Boston, Massachusetts 02115, USA
- Corresponding authors: young{at}wi.mit.edu, violaine.saintandre{at}gmail.com, james_bradner{at}dfci.harvard.edu
-
↵5 These authors contributed equally to this work.
Abstract
A small set of core transcription factors (TFs) dominates control of the gene expression program in embryonic stem cells and other well-studied cellular models. These core TFs collectively regulate their own gene expression, thus forming an interconnected auto-regulatory loop that can be considered the core transcriptional regulatory circuitry (CRC) for that cell type. There is limited knowledge of core TFs, and thus models of core regulatory circuitry, for most cell types. We recently discovered that genes encoding known core TFs forming CRCs are driven by super-enhancers, which provides an opportunity to systematically predict CRCs in poorly studied cell types through super-enhancer mapping. Here, we use super-enhancer maps to generate CRC models for 75 human cell and tissue types. These core circuitry models should prove valuable for further investigating cell-type–specific transcriptional regulation in healthy and diseased cells.
The pathways involved in complex biological processes such as metabolism have been mapped through the efforts of many laboratories over many years and have proven exceptionally valuable for basic and applied science (Krebs 1940; Kanehisa et al. 2012). Although we know much about the general mechanisms involved in control of gene transcription (Roeder 2005; Rajapakse et al. 2009; Bonasio et al. 2010; Conaway and Conaway 2011; Novershtern et al. 2011; Adelman and Lis 2012; Peter et al. 2012; Spitz and Furlong 2012; Zhou et al. 2012; de Wit et al. 2013; Gifford et al. 2013; Kumar et al. 2014; Levine et al. 2014; Ziller et al. 2014; Dixon et al. 2015; Tsankov et al. 2015), the complex pathways involved in the control of each cell's gene expression program have yet to be mapped in most cells. For some cell types, it is evident that core transcription factors (TFs) regulate their own genes and many others, forming the central core of a definable pathway. For most mammalian cell types, however, we have limited understanding of these pathways. These gene control pathways are important to decipher because they have the potential to define cell identity, enhance cellular reprogramming for regenerative medicine, and improve our understanding of transcriptional dysregulation in disease.
There is considerable evidence that the control of cell-type–specific gene expression programs in mammals is dominated by a small number of the many hundreds of TFs that are expressed in each cell type (Graf and Enver 2009; Buganim et al. 2013; Lee and Young 2013; Morris and Daley 2013). These core TFs are generally expressed in a cell-type–specific or lineage-specific manner and can reprogram cells from one cell type to another. In embryonic stem cells (ESCs), where transcriptional control has been most extensively studied, the core TFs POU5F1 (also known as OCT4), SOX2, and NANOG have been shown to be essential for establishment or maintenance of ESC identity and are among the factors capable of reprogramming cells into ESC-like induced pluripotent stem cells (iPSCs) (Young 2011). These core TFs bind to their own genes and those of the other core TFs, forming an interconnected auto-regulatory loop (Boyer et al. 2005), a property that is shared by the core TFs of other cell types (Odom et al. 2004, 2006; Sanda et al. 2012). The core TFs and the interconnected auto-regulatory loop they form have been termed “core regulatory circuitry” (CRC) (Boyer et al. 2005). Because the ESC core TFs also bind to a large portion of the genes that are expressed in an ESC-specific manner, we can posit that regulatory information flows from the CRC to this key portion of the cell's gene expression program, thus forming a map of information flow from CRC to cell-type–specific genes (Young 2011).
With limited knowledge of CRCs in most cell types, attempts to map the control of gene expression programs have thus far been dominated by efforts to integrate global information regarding gene-gene, protein-protein, gene-protein, and regulatory element interactions nested in these networks (Lefebvre et al. 2010; Gerstein et al. 2012; Neph et al. 2012; Yosef et al. 2013; Kemmeren et al. 2014; Rolland et al. 2014). These global studies have provided foundational resources and important insights into basic principles governing transcriptional regulatory networks. These include the identification of recurring motifs of regulatory interactions (Lee et al. 2002; Alon 2007; Davidson 2010; Stergachis et al. 2014) and of groups of genes that participate in common biological processes (Bar-Joseph et al. 2003; Dutkowski et al. 2013). However, these network maps do not generally capture the notion that key control information flows from a small number of core TFs. Recent studies have revealed that core TFs bind clusters of enhancers called super-enhancers and that the super-enhancer (SE)-associated genes include those encoding the core TFs themselves (Hnisz et al. 2013, 2015; Whyte et al. 2013). The ability to identify super-enhancer-associated TF genes, and thus candidate core TFs, should permit modeling of CRCs for all human cell types for which super-enhancer data are available.
Here we describe a method to reconstruct cell-type–specific CRCs based on the properties of core TFs identified in ESCs and several other cell types: They are encoded by genes whose expression is driven by super-enhancers, and they bind to each other's super-enhancers in an interconnected auto-regulatory loop. We report CRC models for 75 cell and tissue types throughout the human body. These models recapitulate and expand on previously described CRCs for well-studied cell types and provide core circuitry models for a broad range of human cell types that can serve as a first step to further mapping of cell-type–specific gene expression control pathways.
Results
Models of core regulatory circuitry
To construct core regulatory circuitry models of human cell types, we used the logic outlined in Figure 1. Detailed studies of the transcriptional control of cell identity in ESCs and a few other cell types have shown that core TFs have three properties. Core TFs are encoded by genes associated with super-enhancers (Hnisz et al. 2013; Whyte et al. 2013), bind the SEs associated with their own gene (Whyte et al. 2013), and form fully interconnected auto-regulatory loops with the other core TFs by binding enhancers together with the other core TFs (Fig. 1A; Odom et al. 2004, 2006; Boyer et al. 2005; Sanda et al. 2012). Candidate core TFs were predicted for multiple cell and tissue types using these three criteria, as described below.
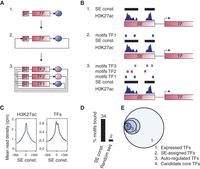
A method to build core regulatory circuitry. (A) Graphical description of the method used to create core regulatory circuitry (CRC) models. 1. Identification of SE-assigned expressed TFs. 2. Identification of the TFs that are predicted to bind their own SE, considered as auto-regulated. 3. CRCs are assembled as fully interconnected loops of auto-regulated TFs. (B) Cartoon showing: 1. TF-assigned SE constituents defined by H3K27ac ChIP-seq peak signals; 2. TFs having at least three DNA-binding sequence motif instances in their SE constituents are considered auto-regulated; 3. TFs with SEs having at least three DNA-binding sequence motif instances for each of the other predicted auto-regulated TFs together form an interconnected auto-regulatory loop. (C) Metagenes for the ChIP-seq signal for H3K27ac (left) and for the average ChIP-seq signal for POU5F1, SOX2, and NANOG (right) in H1 hESCs in the region ±5 kb around the center of the SE constituents. (D) Average percentage of DNA-binding motifs that are actually bound by the TFs from ChIP-seq data for POU5F1, SOX2, and NANOG in H1 hESCs, in either SE constituents or sets of random genomic sequences of the same size. (E) Venn diagram showing the average numbers, across 84 samples, of: 1. TFs having motifs that are expressed (445 TFs); 2. TFs having motifs that are expressed and assigned to a SE (61 TFs); 3. TFs having motifs that are expressed and assigned to a SE and that are predicted to bind their own SE (39 TFs); 4. TFs that are part of the CRC model (15 TFs).
For 75 human cell and tissue types, we first identified the set of active genes that encode TFs that are proximal to SEs (Fig. 1B, step 1). SEs have high levels of signal density for H3K27ac and were identified from H3K27ac ChIP-seq data compiled from multiple laboratories (Supplemental Table S1), as previously described (Hnisz et al. 2013). Recent chromatin conformation data indicate that SEs generally interact with the proximal active gene (Dowen et al. 2014), so the proximal active gene, identified through H3K27ac density at its TSS (see Methods), was assigned as the regulatory target of each SE.
Previous studies have shown that core TFs bind their own super-enhancers (Hnisz et al. 2013; Whyte et al. 2013), so we next identified the set of SE-assigned TF genes whose products are predicted to bind their own SEs (Fig. 1B, step 2). Binding was predicted by searching SE constituents for DNA sequence motifs corresponding to the TF product of the gene assigned to that SE. We compiled DNA-binding sequence motifs for 695 TFs from multiple published sources (Supplemental Table S2; Berger et al. 2008; Wei et al. 2010; Robasky and Bulyk 2011; Jolma et al. 2013; Mathelier et al. 2014) and scanned SE constituent sequences for the presence of the TF binding motifs, using the FIMO software package from the MEME suite (Grant et al. 2011). SE constituents were used for the motif search, as TF binding distributions peak on the SE constituent sequences defined by H3K27ac ChIP-seq peak signal (Fig. 1C). Furthermore, the presence of multiple DNA sequence motifs at SE constituents is predictive of the binding of a TF, whereas this is not the case, on average, across the genome (Fig. 1D). This confirms previous observations of better TF binding prediction in open chromatin sequences compared to other regions of the genome (Pique-Regi et al. 2011; Zhong et al. 2013). We considered the SE-assigned TF genes that were predicted to bind their own SE as auto-regulated, as prior evidence in ESCs indicates that such genes do regulate their own expression (Tomioka et al. 2002; Okumura-Nakanishi et al. 2005; Navarro et al. 2012).
To identify the SE-assigned TFs able to form an interconnected auto-regulatory loop by binding to each other's super-enhancers, we next identified, from the set of TFs considered auto-regulated, the TFs that are predicted to bind the SE of the other auto-regulated TFs, through a motif analysis in SE constituent sequences (Fig. 1B, step 3). We assembled interconnected auto-regulatory loops for each cell or tissue type (Fig. 1A, step 3) and selected the loop containing the set of TFs most often represented across the set of loops as the representative model of CRC (Supplemental Fig. S1). On average, across 75 cell types, 15% of the genes considered expressed and encoding TFs were assigned to an SE, 9% were predicted to be auto-regulated, and 3% were identified as candidate core TFs (Fig. 1E; Supplemental Table S3).
hESC core regulatory circuitry
The model of CRC predicted for human H1 ESCs (Fig. 2A, left panel) indicates that the approach described here captures the previously described core TFs and CRC for ESCs and suggests that additional TFs contribute to this core circuitry. The H1 ESC CRC contains three factors—POU5F1, SOX2, and NANOG—that are considered the foundation of the CRC in ESCs (Jaenisch and Young 2008; Young 2011). All three factors are essential for the pluripotent state (Nichols et al. 1998; Niwa et al. 2000; Avilion et al. 2003; Chambers et al. 2003; Mitsui et al. 2003; Masui et al. 2007; Silva et al. 2009; Theunissen et al. 2011), regulate their own genes and those encoding the other two factors (Tomioka et al. 2002; Catena et al. 2004; Boyer et al. 2005; Chew et al. 2005; Kuroda et al. 2005; Okumura-Nakanishi et al. 2005; Rodda et al. 2005; Loh et al. 2006), and can be used to reprogram fibroblasts to an induced pluripotent state (Takahashi and Yamanaka 2006; Yu et al. 2007).

H1 ESC core and extended regulatory circuitry. (A) (Left) CRC model for H1 human embryonic stem cells. The role of each TF in ESC pluripotency and self-renewal is listed in Table 1. (Right) H1 hESC extended regulatory circuit. Examples of SE-assigned genes that are predicted to be bound by each of the TFs in the CRC. The role of these factors in ESC pluripotency and self-renewal is listed in Supplemental Table S5. (B) ChIP-seq data for H3K27ac, POU5F1, SOX2, and NANOG showing binding of the TFs to each of the SEs of the SE-assigned TFs in the hESC CRC. SE genomic locations are depicted by red lines on top of the tracks. (C) Pie charts showing the percentages of SE-assigned genes (top row) or all expressed genes (bottom row) whose regulatory sequences are predicted to be bound by increasing numbers of hESC candidate core TFs. (D) Diagram showing putative transcriptional regulation of miR-371a on SOX2 expression in hESCs.
The results of the algorithm we describe suggest that seven additional TFs contribute to the ESC CRC (Fig. 2A, left panel). Most of these factors have previously been implicated in control of the stem cell state, and there is ChIP-seq evidence indicating that their super-enhancers are bound by POU5F1, SOX2, and NANOG (Fig. 2B). FOXO1 and ZIC3 have previously been shown to be essential for the maintenance of pluripotency (Lim et al. 2007; Zhang et al. 2011; Declercq et al. 2013). In hESCs, FOXO1 regulates POU5F1 and SOX2 expression (Zhang et al. 2011). ZIC3 directly activates Nanog expression in mouse ESCs (mESCs) and can contribute to reprogramming of human fibroblasts into an induced pluripotent state (Lim et al. 2007; Declercq et al. 2013). NR5A1 (also known as SF1) and RARG can influence the pluripotent state (Guo and Smith 2010; Wang et al. 2011), and both bind to regulatory regions of the POU5F1 gene and regulate its expression (Barnea and Bergman 2000; Yang et al. 2007; Guo and Smith 2010). The other three TFs—MYB, RORA, and SOX21—are best known for their roles in other stem cells. MYB and RORA have roles in establishing or maintaining self-renewing populations of hematopoietic cells (White and Weston 2000; Lieu and Reddy 2009; Cheasley et al. 2011; Zuber et al. 2011; Doulatov et al. 2013), while SOX21 is involved in regulating pluripotency in intestinal stem cells, where its expression is influenced by SOX2 (Kuzmichev et al. 2012). Thus, there are multiple lines of evidence, summarized in Table 1, that support the inclusion of POU5F1, SOX2, NANOG, FOXO1, ZIC3, NR5A1, RARG, MYB, RORA, and SOX21 in a model of hESC CRC.
Role of hESC candidate core TFs in ESC identity and in the regulation of other candidate core TF expression
In ESCs, loss of cell identity can be assayed by measuring POU5F1 protein levels, where reduced levels are associated with loss of pluripotency, and by counting cell nuclei, where reduced numbers can reflect loss of self-renewal (Chia et al. 2010; Kagey et al. 2010). To test whether the candidate core TFs play roles in control of ESC identity, we analyzed POU5F1 expression changes and cell nuclei number changes after depletion of each of these TFs using data from a genome-wide siRNA screen in H1 hESCs (Chia et al. 2010). These data confirm that the candidate core TFs contribute to pluripotency and/or survival and proliferation of hESCs (Supplemental Fig. S2A). Gene-set enrichment analysis (GSEA) of the set of candidate core TFs shows these factors are encoded by genes that are among those whose knock-down most impacts POU5F1 expression and cell nuclei count (Supplemental Fig. S2B,C). These functional assays in H1 hESCs thus provide supporting evidence for a functional role of the candidate core TFs in control of hESC identity.
Extended hESC regulatory circuitry
POU5F1, SOX2, and NANOG contribute to the formation of SEs at hundreds of active ESC genes that play prominent roles in cell identity (Whyte et al. 2013), suggesting that a simple extended model of regulatory information can be constructed to include these additional SE-assigned genes downstream from the core TFs. We identified the SE-assigned genes whose enhancers and promoters are predicted to be bound by the candidate core TFs in order to construct a model of extended hESC regulatory circuitry (Fig. 2A, right panel). Analysis shows the regulatory sequences of the SE-assigned genes are predicted to be bound by a greater number of candidate core TFs than the regulatory sequences of all expressed genes (Fig. 2C). Sixty-eight percent of the SE-assigned genes are predicted to be bound by each of the core TFs. Experimental evidence (Kunarso et al. 2010) shows that POU5F1 contributes to the regulation of at least 30% (z-test P-value <2.2 × 10−16) of these downstream SE-assigned target genes (Supplemental Table S4). Thus, in the model of extended hESC regulatory circuitry, the core TFs co-occupy and likely regulate the expression of a large portion of SE-assigned genes.
The model of extended hESC regulatory circuitry contains many genes that are known to play prominent roles in ESC biology (Young 2011). These include the TFs PRDM14, SALL4, and ZNF281, the chromatin regulators DNMT3B, JARID2, and SETDB1, and the miRNA miR-371a, all of which have established roles in pluripotency, self-renewal, or differentiation (detailed functions and associated references in Supplemental Table S5). Among the SE-assigned genes, some transcriptional regulators may create feedforward or feedback loops of regulation with the genes in the extended CRC to modulate the direct effect of core TFs. For example, miR-371a, the human homolog of miR-290 which is essential for mESC survival (Medeiros et al. 2011), may fine-tune SOX2 expression in hESCs (Fig. 2D). SOX2 is identified as a highly probable target of miR-371a by multiple miRNA target predictor algorithms, including TargetScan (Lewis et al. 2005), miRDB (Wong and Wang 2015), and PITA (Kertesz et al. 2007) (Supplemental Table S6), and recent functional assays in human cancer cells (Li et al. 2015) support a role for miR-371a in direct regulation of SOX2 expression. We therefore suggest that the ESC gene expression program is controlled by a CRC consisting of ten key TFs that (1) bind the SEs of their own genes and regulate their own expression, and (2) co-bind the SEs of many other genes important for ESC identity and regulate their expression.
CRC and extended regulatory circuitry for many cell types
We next developed models of CRC and extended regulatory circuitry for each of 75 human cell and tissue types (Fig. 3; Supplemental Table S3). The predicted CRCs contain key transcriptional regulators of cell identity that have been previously identified (Supplemental Table S7). This includes, for example, TBX5 in the heart (left ventricle) CRC (Ieda et al. 2010; Song et al. 2012; Nam et al. 2014), PDX1 in the pancreas CRC (Jonsson et al. 1994; Horb et al. 2003; Zhou et al. 2008), and SOX2 in the brain (hippocampus middle) CRC (Graham et al. 2003; Ferri 2004; Sisodiya et al. 2006; Lujan et al. 2012). They also contain well-characterized proto-oncogenes of cancer subtypes represented by cancer cell lines, such as ESR1 and GATA3 in MCF-7 breast cancer cells (Usary et al. 2004; Holst et al. 2007) and TCF7L2 and SMAD3 in HCT-116 colon cancer cells (Supplemental Fig. S3; Zhu et al. 1998; Tuupanen et al. 2009). Importantly, our approach recapitulates the oncogenic circuitry that had been previously identified in T-cell acute lymphoblastic leukemia (T-ALL) Jurkat cells (Sanda et al. 2012; Mansour et al. 2014), as one of the CRCs for Jurkat cells contains the four oncogenic TFs—GATA3, MYB, RUNX1, and TAL1—previously characterized as core TFs in this cell line (Fig. 4A). Together, these data indicate that the CRC models capture much existing knowledge of TFs that play key roles in control of cell identity across diverse cell and tissue types.

Core and extended regulatory circuitry for multiple cells and tissue types. Core and extended circuitry for (A) brain (hippocampus middle), (B) adipocytes (adipose nuclei), (C) heart (left ventricle), and (D) pancreas. The number of SE-assigned genes predicted to be co-occupied by each of the candidate core TFs and 30 examples of those are displayed on the right part of the maps.

Experimental validation for T-ALL Jurkat cell circuitry. (A) Core regulatory circuit containing GATA3, MYB, RUNX1, and TAL1 for T-ALL Jurkat cells. (B) ChIP-seq data for H3K27ac, MYB, RUNX1, TAL1, and GATA3 showing binding of the TF to each of the SEs in the T-ALL Jurkat cell core circuit. SE genomic locations are depicted by red lines on top of the tracks. (C) Boxplots showing fold change (FC) in expression for Jurkat cells transfected with the indicated shRNAs vs. control shRNAs, for either the set of candidate core TFs displayed in A (red) or the full set of TFs considered expressed in Jurkat cells (blue). P-values quantifying the difference between the two sets were calculated using a Wilcoxon test.
We used experimental data to test the accuracy of our predictions in newly identified CRCs. The binding of the core TFs to the super-enhancer sequences of the other predicted TFs in the core is supported by ChIP-seq data for core TFs in T-ALL Jurkat cells (Fig. 4B). Available ChIP-seq data for TFs in the CRCs for other cell types were also analyzed and lend functional support for the predicted binding interactions in the CRCs (Supplemental Fig. S3). To test the mutual regulation of the TFs in the core, we investigated the effects of shRNA depletion of MYB, RUNX1, TAL1, and GATA3 on expression of candidate core TF-encoding genes in T-ALL Jurkat cells (Fig. 4C). Analysis of the data shows that when a core TF is depleted, the expression of the TFs in the core is significantly down-regulated compared to the set of TFs considered expressed in the cell. This observation is in agreement with a direct effect of the core TFs on the expression of the other TFs in the core.
The candidate core TFs identified across a wide range of cell types show features of core TFs that have previously been described (Lee and Young 2013). Analysis of the candidate core TFs across samples shows that these are cell-type–specific or lineage-specific: 34% of the core TFs identified across cell types are predicted to be core TFs in only one cell type, and 77% are predicted to be core TFs in less than five cell types (Fig. 5A). DNA-binding domain structures can provide insight on the functional roles of TFs (Vaquerizas et al. 2009), so we compared the frequency of different DNA-binding domains in candidate core TFs to those in ubiquitously expressed housekeeping TFs. Compared to housekeeping TFs, candidate core TFs are depleted in the most common type of TFs—zinc finger domain-containing TFs—and enriched in various classes of TFs that have been associated with developmental processes, such as homeodomain-containing TFs (Fig. 5B). Analysis of expression data shows that candidate core TFs exhibit higher transcript levels when compared to the full set of TFs considered expressed in the cell (Fig. 5C). The candidate core TFs are thus cell-type– or lineage-specific, enriched for functional association with development processes, and show a relatively high level of expression compared to other TFs expressed in the cell.

Features of candidate core TFs. (A) Percentages of TFs identified as candidate core TFs in a given number of cell or tissue types. The number of cell or tissue types in which a TF is identified as a candidate core TF is displayed with boxes on the right. A representative sample of each cell and tissue type is used when multiple samples from the same cell or tissue type are present in the data set. (B) DNA-binding domains that are significantly differentially represented in the set of candidate core TFs and housekeeping TFs. (C) Transcript levels for the set of candidate core TFs and for the full set of TFs considered expressed in each sample. P-values quantifying the difference between the two sets were calculated using a Wilcoxon test.
Analyzing CRCs across cell types, we identified features of CRCs that should help guide further experiments to better understand the transcriptional pathways involved in development and disease. We observed that a substantial fraction of candidate core TFs is expressed in multiple cell types, typically within a lineage. This feature of shared core TFs within lineages is evident through hierarchical clustering of candidate core TFs across all data sets (Fig. 6A). It suggests that specific combinations of TFs may be required to control complementary aspects of cell identity and that circuitries may be rewired through ectopic expression of a few TFs between similar cell types. We also found that, compared to other TFs, candidate core TFs are found significantly more often in the set of genes associated with diseases or traits via genome-wide association studies (GWAS), which suggests their involvement in cell identity and disease development (Fig. 6B; Supplemental Fig. S4). Previous studies have shown that disease-associated SNPs are enriched in SEs (Hnisz et al. 2013; Parker et al. 2013), and there are multiple examples of noncoding disease-associated SNPs overlapping the super-enhancers associated with TFs in the CRC (Supplemental Fig. S4).
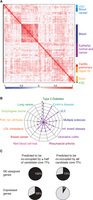
Properties of CRCs of multiple human cell and tissue types. (A) CRCs cluster according to cell type similarity. Hierarchical clustering of candidate core TFs for 80 human samples. The matrix of correlation based on Pearson coefficients identifies specific clusters for hematopoietic stem cells (HSC), blood cancer cells, blood cells, epithelial normal and cancer cells, cardio-pulmonary system cells, upper gastrointestinal system, and brain cells. Correlation values range from −1 to 1 and are colored from blue to red according to the color scale. (B) Radar plot showing the enrichment of candidate core TFs, compared to noncore TFs, in GWAS list of genes for multiple diseases or traits. P-values were calculated using a z-test, and 1/P-values are plotted for the diseases or traits that showed an enrichment P-value <5 × 10−2 of candidate core TFs. (C) Pie charts showing the average percentages for all samples of SE-assigned genes (top row) or of all expressed genes (bottom row) whose regulatory sequences are predicted to be co-occupied by more than half or by all the TFs in the CRC.
We generated models of extended regulatory circuitry for 75 cell and tissue types using the same process described above for the hESC extended regulatory circuitry (Fig. 3). The features of these extended circuitries are consistent with those observed for hESCs. On average, across samples, 73% of the SE-assigned genes are predicted to be co-occupied by each of the candidate core TFs (Fig. 6C), and these SE-assigned target genes of the CRC play prominent roles in specific cell identities (Fig. 3).
Discussion
There have been tremendous advances in our understanding of the general mechanisms involved in control of gene transcription (Roeder 2005; Rajapakse et al. 2009; Bonasio et al. 2010; Conaway and Conaway 2011; Novershtern et al. 2011; Adelman and Lis 2012; Peter et al. 2012; Spitz and Furlong 2012; Zhou et al. 2012; de Wit et al. 2013; Gifford et al. 2013; Kumar et al. 2014; Ziller et al. 2014; Dixon et al. 2015; Tsankov et al. 2015), but the pathways by which a small set of core TFs control gene expression programs have yet to be mapped in most cells. We describe here models of core transcriptional regulatory circuitry for 75 human cell and tissue types. These models show significant percentages of overlap between the TF-TF binding interactions we predict in the CRCs and the TF-TF interactions identified in previous high-throughput analyses, for similar cell types (Supplemental Table S8; Neph et al. 2012). The CRC models we provide include known core TFs and reprogramming TFs that have been previously identified in a few cell types but add a large list of candidate cell identity regulators. These include ubiquitous and signaling TFs that should work together with the minimal set of TFs required to reprogram cells from one state to another, to establish and maintain cellular identity. These models provide the foundation for future studies of the transcriptional pathways that control cell identity in these diverse cell types of the human body.
Key target genes of the CRC were identified in a first step toward understanding how the information flows from the core TFs to all expressed genes. Across all cell and tissue types, the candidate core TFs were predicted to preferentially co-occupy SE-assigned genes, compared to all expressed genes. As SE-assigned genes are typically key for cell identity (Chapuy et al. 2013; Hnisz et al. 2013; Parker et al. 2013; Whyte et al. 2013; Loft et al. 2015), this shows that the concerted action of candidate core TFs may be preferentially targeted to those key cell identity genes. This led us to envision a model whereby the core TFs promote hallmarks of cell identities through co-binding the SEs of their own genes and regulating their own expression, and co-binding the SEs of many other genes important for cell identity and regulating their expression. The maps of CRC were thus extended to include the SE-assigned target genes of the CRC. These maps of extended regulatory circuitry are founding models for the description of more comprehensive networks that describe additional levels of regulation that should include signaling pathways, as super-enhancers serve as integrating platforms for signaling (Siersbæk et al. 2014b; Hnisz et al. 2015).
The approach presented here constitutes a first attempt to map CRCs in a wide range of cell types and harbors several limitations that should be considered when using the data. The analyses were restricted to TFs that were assigned to a SE in the data set and for which DNA-binding motifs are available. The CRC models also rely on data derived from cell lines, which do not necessarily reflect the state of cells in their normal niche, or from biopsies, which include mixed populations of cell types. Another consideration is the challenge of comprehensive experimental validation of the circuits, which would in principle require knock-out of individual core TFs and perhaps combinations of these TFs. Ongoing efforts to characterize DNA-binding motifs for TFs (Jolma et al. 2013; Mathelier et al. 2014; Hume et al. 2015), taking into account the influence of their TF partners, and the role of coactivators and chromatin regulators on their binding to regulatory sequences (Chen et al. 2008; Yan et al. 2013; Siersbæk et al. 2014a; Schmidt et al. 2015), and further experimental testing should help refine the description of the CRC models we provide here.
CRC models should provide guidance for reprogramming studies and may prove valuable for better understanding transcriptional dysregulation in disease. Candidate core TFs are enriched in the genes associated with multiple diseases or traits through GWAS, supporting their role in disease development. Furthermore, SEs are hotspots of noncoding disease-associated sequence variants (Maurano et al. 2012; Hnisz et al. 2013; Parker et al. 2013; Corradin et al. 2014; Farh et al. 2015). Some of these variants may modify the binding sites for core TFs, providing a mechanism for disease-associated transcriptional misregulation. This is the case, for example, of TFs we predict in CRCs such as TAL1 in T cells, TBX5 in cardiac cells, TCF7L2 in colorectal cancer cells, and ESR1 and GATA3 in breast cancer cells (Tuupanen et al. 2009; Cowper-Sal lari et al. 2012; Sur et al. 2012; Van den Boogaard et al. 2012; Bauer et al. 2013; French et al. 2013). Extended regulatory circuits integrating candidate core TFs and their SE-assigned target genes for many human cell types may thus help better understand disease-associated genetic variation, leading someday to circuitry-directed therapeutic interventions.
Methods
ChIP-seq data
H3K27ac ChIP-seq data were either downloaded from GEO (accession numbers in Supplemental Table S1) or generously shared by the NIH Roadmap Epigenome project (Bernstein et al. 2010). ChIP-seq data for H3K27ac (Kwiatkowski et al. 2014), MYB (Mansour et al. 2014) and TAL1 (Palii et al. 2011) in Jurkat cells and for POU5F1 (Kunarso et al. 2010), SOX2 (Hawkins et al. 2011), and NANOG (Kunarso et al. 2010) in H1 hESCs were downloaded from GEO. ChIP-seq data for CREB1, EBF1, ELF1, ETS1, PAX5, and POU2F2 in GM12878 lymphoblastoid B cells, for TCF7L2 in HCT-116 colon cancer cell line, and for ESR1 in T-47D breast cancer cell line were downloaded from ENCODE (Gertz et al. 2013).
CRC mapper
The algorithm we developed to identify core regulatory circuitry uses as input H3K27ac ChIP-seq reads aligned to the human genome, together with the ChIP-seq peaks identified by MACS (Zhang et al. 2008) and the enhancer table output from ROSE (https://bitbucket.org/young_computation/rose) (Lovén et al. 2013). SEs identified with ROSE are assigned to the closest transcript predicted to be expressed. For each SE-assigned TF, a motif analysis is carried out on the SE constituent sequences assigned to that TF using FIMO (Find Individual Motif Occurrences) from the MEME (Multiple Em for Motif Elicitation) suite (Grant et al. 2011). A database of DNA sequence motifs for 695 TFs was compiled from the TRANSFAC database of motifs (Matys et al. 2006), and from the MEME suite (January 23rd 2014 update), for the following collections: JASPAR CORE 2014 vertebrates (Mathelier et al. 2014), Jolma 2013 (Jolma et al. 2013), Homeodomains (Berger et al. 2008), mouse UniPROBE (Robasky and Bulyk 2011), and mouse and human ETS factors (Wei et al. 2010). For the motif search, the search space in SEs is restricted to extended SE constituents, as these are the regions that capture most of the TF binding in SEs (Fig. 1C). SE constituent DNA sequences are extracted, extended on each side (500 bp by default), and used for motif search with FIMO with a P-value threshold of 1 × 10−4. SE-assigned TFs whose set of constituents contains at least three DNA sequence motif instances for their own protein products are defined as auto-regulated TFs. From the set of auto-regulated TFs, the TFs predicted to bind to the SEs of other auto-regulated TFs, using the same criteria as described above, are identified. All possible fully interconnected auto-regulatory loops of TFs are then constructed through recursive identification. When multiple possibilities of fully interconnected auto-regulatory loops are found, the most representative fully interconnected auto-regulatory loop of TFs is selected as the model of CRC. This loop is defined as the loop containing the TFs that occur the most frequently across all possible loops. See Supplemental Methods for details.
Data access
ChIP-seq data for RUNX1 and GATA3 in Jurkat cells from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE76181. The CRC Mapper program is implemented in Python. It can be found in the Supplemental Material and is freely available for download at https://bitbucket.org/young_computation/crcmapper.
Competing interest statement
R.A.Y. and J.E.B. are founders of Syros Pharmaceuticals.
Acknowledgments
We thank members of the Young lab, especially Ana D'Alessio, Zi Peng Fan, and Jurian Schuijers for helpful discussions. This work was supported by the Foundation for the National Institutes of Health grant HG002668 (R.A.Y.). A.J.F. was funded by a National Science Foundation graduate fellowship, and B.J.A. is the Hope Funds for Cancer Research Grillo-Marxuach Family Fellow.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.197590.115.
- Received August 1, 2015.
- Accepted December 21, 2015.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








