
Genome-wide dynamics of alternative polyadenylation in rice
- Haihui Fu1,5,
- Dewei Yang2,5,
- Wenyue Su1,
- Liuyin Ma1,
- Yingjia Shen1,
- Guoli Ji3,
- Xinfu Ye2,
- Xiaohui Wu3 and
- Qingshun Q. Li1,2,4
- 1Key Laboratory of the Ministry of Education for Coastal and Wetland Ecosystems, College of the Environment and Ecology, Xiamen University, Xiamen, Fujian, China, 361102;
- 2Rice Research Institute, Fujian Academy of Agricultural Sciences, Fuzhou, Fujian, China, 350018;
- 3Department of Automation, Xiamen University, Xiamen, Fujian, China, 361005;
- 4Graduate College of Biomedical Sciences, Western University of Health Sciences, Pomona, California 91766, USA
- Corresponding authors: xhuister{at}xmu.edu.cn, yexinfu{at}126.com, qqli{at}westernu.edu
-
↵5 These authors contributed equally to this work.
Abstract
Alternative polyadenylation (APA), in which a transcript uses one of the poly(A) sites to define its 3′-end, is a common regulatory mechanism in eukaryotic gene expression. However, the potential of APA in determining crop agronomic traits remains elusive. This study systematically tallied poly(A) sites of 14 different rice tissues and developmental stages using the poly(A) tag sequencing (PAT-seq) approach. The results indicate significant involvement of APA in developmental and quantitative trait loci (QTL) gene expression. About 48% of all expressed genes use APA to generate transcriptomic and proteomic diversity. Some genes switch APA sites, allowing differentially expressed genes to use alternate 3′ UTRs. Interestingly, APA in mature pollen is distinct where differential expression levels of a set of poly(A) factors and different distributions of APA sites are found, indicating a unique mRNA 3′-end formation regulation during gametophyte development. Equally interesting, statistical analyses showed that QTL tends to use APA for regulation of gene expression of many agronomic traits, suggesting a potential important role of APA in rice production. These results provide thus far the most comprehensive and high-resolution resource for advanced analysis of APA in crops and shed light on how APA is associated with trait formation in eukaryotes.
Messenger RNA (mRNA) polyadenylation is a key step during gene expression in eukaryotes, which involves 3′-end cleavage of pre-mRNA and an addition of a poly(A) tail. Only mRNA with a poly(A) tail can be exported out of the nucleus to play out its roles in the cytoplasm, including mRNA stability, localization, and formation of a translational initiation complex (Millevoi and Vagner 2010).
The formation of a poly(A) tail at the correct location involves a complex and finely tuned biochemical process consisting of interactions among many polyadenylation factors and cis-elements, e.g., AAUAAA (Beaudoing et al. 2000). Before forming a poly(A) tail, a large number of poly(A) factors—including the cleavage and polyadenylation specificity factor (CPSF), poly(A) polymerase, cleavage stimulation factor subunits 1 and 2 (CSTF1 and CSTF2)—assemble near the 3′-end of pre-mRNA to achieve cleavage and poly(A) tail addition (Colgan and Manley 1997; Shi et al. 2009). In plants, a similar set of poly(A) factors were identified, albeit with some differences in the number of encoding genes and domain structures (Hunt et al. 2008, 2012). Genetic and biochemical analysis of these factors also revealed similar, but distinct, and sometimes unique, functions to their animal counterparts (Zhao et al. 2009, 2011; Thomas et al. 2012; Xing et al. 2013; Liu et al. 2014).
Previous studies showed that many eukaryotic genes harbor more than one poly(A) site (Ji and Tian 2009; Jan et al. 2011; Sherstnev et al. 2012), suggesting that the locations of poly(A) sites can be chosen in different regions of transcripts. This is termed alternative polyadenylation, or APA. For example, studies showed that ∼50% and 82% of genes in rice had more than one poly(A) site using the expressed sequence tags (EST) database, restriction enzyme digestion–mediated massively parallel signature sequencing (MPSS), or Illumina sequencing approaches, respectively (Shen et al. 2008, 2011). Using high-throughput sequencing, it was discovered that >70% of genes in Arabidopsis thaliana had two or more poly(A) sites, in which ∼17% was located within 5′ UTRs, coding sequences (CDSs), and introns (Wu et al. 2011). The number of APA genes in Arabidopsis was significantly more than that found in a previous study using a different genome-scale approach (Sherstnev et al. 2012).
The transcripts resulting from APA can be used to regulate gene expression, as demonstrated by previous studies showing that errors associated with polyadenylation could result in human diseases (Danckwardt et al. 2008; Mayr and Bartel 2009) or developmental defects in plants (Thomas et al. 2012). For example, FCA, a gene with both proximal and distal poly(A) sites, is associated with flowering time control in plants. In general, the distal poly(A) site is used, and it produces a long transcript. However, when the proximal poly(A) site is used, it results in a short transcript without function, thus affecting flowering time (Boss et al. 2004). CPSF30, encoding a poly(A) factor in Arabidopsis, acts on APA selection in a broad spectrum of genes since its mutant reflects changes in a large number of gene poly(A) sites and the use of cis-elements in 3′ UTRs (Thomas et al. 2012; Liu et al. 2014). Otherwise, APA is a common post-transcriptional regulatory mechanism in other eukaryotes, leading to transcripts with variable 3′ UTR lengths (Mangone et al. 2010; Ozsolak et al. 2010) and impacting physiological properties of proteins (Takagaki et al. 1996) and mRNA stability (Mayr and Bartel 2009).
Rice is one of the most important crops and also a model monocot. Although some APA analyses in rice were performed, the roles of APA in development and tissue specification remain to be elucidated. Previous work evaluated rice poly(A) to the nearest restriction enzyme site next to a poly(A) site, resulting in inaccuracy at the nucleotide level (Shen et al. 2011). Hence, we used a poly(A) tag sequencing approach (PAT-seq) to investigate the genome-wide landscape of APA in 14 different tissues and developmental stages of rice.
Results
Profiling of rice poly(A) sites
First, raw reads were processed and low-quality reads discarded, followed by mapping the remaining reads to the Oryza sativa japonica reference genome by Bowtie 2 (Supplemental Table S1; Langmead and Salzberg 2012). After removing potential internal priming (Wu et al. 2011), a total of 136,955,352 individual PATs and 68,220 poly(A) site clusters (PACs) were obtained (Supplemental Table S2A), which were dispersed in 28,032 genes (listed in Supplemental Table S2B). Among them, 13,419 genes (47.9%) had more than one PAC, indicating that the phenomenon of APA occurs extensively in rice transcriptome. As shown in Figure 1, the distributions of these PACs in different genic and intergenic regions of the genome were mostly in a similar order (3′ UTR > intergenic > intron > promoter > CDS > 5′ UTR) in different tissues except in pollen, where more PACs apparently are located in the intergenic regions and less in the 3′ UTR (for more details, see Supplemental Material).
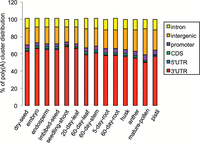
Distribution of PACs in genomic regions in different samples. (CDS) Coding sequences; (3′ UTR) three primer untranslated region; (5′ UTR) five primer untranslated region.
The verification of the poly(A) sites shows that 40,480 (70%) poly(A) sites of the EST data overlapped identical sites in the PAT-seq data (Shen et al. 2008), indicating a significant match of these two data sets (for more details, see Supplemental Material; Supplemental Fig. S1). Pearson correlation across tissues was 0.59–0.83 between log2(PAT) and log2(FPKM), suggesting the reasonable representation of relative gene expression level by PAT-seq (Supplemental Fig. S2). A Pearson correlation >0.9 showed that PACs had good reproducibility in three replicates (Supplemental Fig. S3).
Poly(A) sites in mature pollen are distinct
To gain more insight into PACs lying within different genomic regions across 14 tissues, single-nucleotide profiles ranging from 100 nucleotide (nt) upstream to 100 nt downstream from the cleavage sites were studied as previously described (Loke et al. 2005; Wu et al. 2011). The results of other tissues were very similar, except the introns of pollen (Fig. 2A). The content of A at the cleavage site (position “0”) of pollen was noticeably higher than that of leaf, indicating potential preferences in cis-element. At the same time, however, A content was distinctly lower in the NUE region of pollen introns compared with that of other samples (Fig. 2A). Such change hints at different poly(A) signals used in these introns. Therefore, we used MEME (Bailey et al. 2009) analyses to find different signals in pollen introns. As shown in Figure 2B, the A-rich motif was ranked at the top in the flanking region −35 bp to −10 bp of the cleavage site from leaf, while the T-rich motif was particularly enriched in the same region from the introns of pollen. Since polyadenylation requires the interaction of polyadenylation factors and cis-elements, the selection of certain poly(A) signals may reflect difference of expression levels of polyadenylation factors between pollen and leaf.
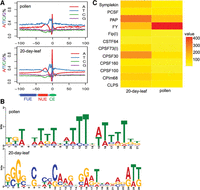
Motif around the poly(A) sites. (A) Single-nucleotide profiles around the poly(A) sites found in different genic regions. (NUE) Near upstream element; (CE) cleavage element; (FUE) far upstream element. (B) Motif analysis by MEME. (Top) Introns of the pollen; (bottom) leaf_20days. Letter heights indicate the frequency. (C) Relative expression levels of poly(A) factors (Hunt et al. 2008) between pollen and leaf_20days. PAP (LOC_Os06g21470), FY (LOC_Os01g72220), Symplekin (LOC_Os07g49320), PCSF (LOC_Os09g3927), Fip(I) (LOC_Os03g19570), CSTF64 (LOC_Os05g43780), CPSF73(I) (LOC_Os03g63590), CPSF30 (LOC_Os06g46400), CPSF160 (LOC_Os04g18010), CPSF100 (LOC_Os09g39590), CFIm68 (LOC_Os07g08960), and CLPS (LOC_Os02g12570). Color blocks indicate total PAT counts for each poly(A).
To examine this possibility, we used the PAT-seq transcriptome data collected herein to measure the expression level of many poly(A) factors, and the results showed that FY was significantly up-regulated in pollen compared with 20-d-old leaf, while CPSF30 showed an inverse trend (Fig. 2C). Interestingly, the orthologs of these two factors in human were recently revealed to recognize the poly(A) signals AAUAAA (NUE in plants) (Chan et al. 2014), and genetic evidence from Arabidopsis also demonstrated that CPSF30 is responsible for NUE recognition (Thomas et al. 2012). Taken together, these results indicate that the change of expression of FY and CPSF30 could be responsible for the difference of poly(A) site selection in pollen.
Significant portion of poly(A) clusters show tissue specificities
To further explore the possibility of tissue-specific PACs among all samples, we used two measures: a PAC only expressed in one tissue, but not in any other tissues tested herein, or a PAC in one tissue having significantly higher expression level (32-fold) than another tissue. Such tissue-specific PACs are rare across all 14 samples; pollen again showed the highest tissue specificity with 447 PACs (Fig. 3A). Of these, 47% were from the 3′ UTRs, while ∼44% were from intronic regions, as expected, based on the unique poly(A) signals described above. A distant second, 197 tissue-specific PACs were also found in 60-d-old root. Of these, 90% were mostly from 3′ UTRs, and only 9% were from introns, compared to pollen. Similarly, a previous study showed that tissue-specific PACs across different tissues in human were also very rare (Lianoglou et al. 2013). Pollen-specific isoforms were enriched in the development-related pathways, including energy, defense, and core protein biosynthesis (Supplemental Material; Supplemental Fig. S4). As another way to interrogate specificity, the degree of PAC distribution among the different samples was investigated (Supplemental Material; Supplemental Fig. S5).
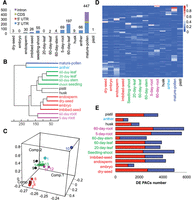
Expression patterns of PACs across 14 tissues. (A) Number of tissue-specific PACs in different samples. (B) The Euclidean distances between the samples as calculated from the regularized log transformation. (C) Principal component analysis of PAC expression pattern across 14 tissues: 1, anther; 2, dry seed; 3, embryo; 4, endosperm; 5, husk; 6, imbibed seed; 7, leaf_20days; 8, leaf_60days; 9, pistil; 10, pollen; 11, root_5days; 12, root_60days; 13, seedling shoot; 14, stem_60days. (D) PAT ratio of differentially expressed PACs across 14 tissues. DE PACs were identified by DEXSeq. For each DE PAC, the PAT ratio is calculated as the ratio of number of PATs in a respective tissue to the total number of PATs in all tissues. (E) The number of differentially expressed PACs between pollen and other tissues. The red bar represents up-regulated PACs, and the blue bar represents down-regulated PACs.
Expression patterns of PACs across 14 tissues
Unsupervised hierarchal clustering analysis and principal component analysis revealed that different tissues cluster in distinct branches (Fig. 3B,C). Furthermore, different developmental stages of the same tissues have similar expression patterns of transcript isoforms. However, cluster analyses showed that mature pollen differed from other tissues by exhibiting a distinct transcriptome pattern. PAT ratio of differentially expressed (DE) PACs among 14 tissues revealed that some PACs that were expressed across tissues were different from each other, especially those highly expressed in pollen (Fig. 3D). Therefore, when pollen was compared to other tissues, a large number of DE PACs were identified (│log2FoldChange│ > 2; P-adjust < 0.01) (Fig. 3E). These results showed the diversity of PAC expression and further showed that pollen is very special compared to other tissues.
3′ UTR length variation in rice
As shown in Supplemental Table S3, our results indicate that seed-related tissues had the shortest median 3′ UTR length among all 14 tissues. On the other hand, 20-d-old leaves had the longest 3′ UTRs; they were 17 nt longer than the shortest 3′ UTRs in APA genes. In particular, with the development of rice, the length of 3′ UTRs became incrementally longer (Supplemental Fig. S6). Even though the difference in median 3′ UTR length was <20 nt, statistical significance was reached (Supplemental Table S4).
The length of those genes with 3′ UTRs with a single PAC, whether differentially expressed or not, was shorter than the length of 3′ UTRs of APA genes (see Fig. 4A). However, among APA genes, in contrast, the 3′ UTR of differentially expressed genes was longer than that of APA genes not differentially expressed. This result indicated that differentially expressed genes tended to have longer 3′ UTRs, notwithstanding the use of proximal or distal PACs. Moreover, highly expressed APA genes tend to use shorter 3′ UTRs compared with those not highly expressed (Fig. 4B), and highly expressed APA genes used significantly more proximal PACs (Fig. 4C). Previous studies have also shown the use of shorter 3′ UTRs in highly expressed cancer cell genes (Ozsolak et al. 2010). Interestingly, the use of proximal PACs in highly expressed APA genes was reversed in mature pollen, where highly expressed genes used significantly fewer proximal PACs than genes with low expression. However, it was shown that shorter 3′ UTR length was used in highly expressed genes in testis (Ulitsky et al. 2012). The reason for such discrepancy in pollen requires further investigation.
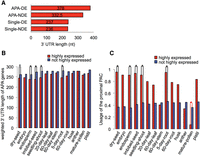
3′ UTR length analyses. (A) Median 3′ UTR length of distal PACs in different groups of genes. (APA-DE) Genes with at least one differentially expressed (DE) PAC; (APA-NDE) genes with multiple PACs but none is DE PAC; (single-DE) DE genes with single PAC; (single-NDE) genes with single PAC and not DE. The median length of proximal PACs for APA-NDE is 189 nt, and for APA-DE is 204 nt. (B) The weighted 3′ UTR median length of the highly and not highly expressed APA genes. (C) Usage of the proximal PAC in highly expressed genes of APA across tissues. The red line in mature pollen marks a reverse trend where highly expressed genes use less proximal PACs. Asterisk presents statistically significant differences (P-value <0.05).
APA site switching alters gene expression profile
APA site switching is a phenomenon whereby two or more APA sites of a gene change their usage frequency in different tissues or developmental stages (e.g., Fig. 5A). The heat-map showed that significant APA site switching occurs at different developmental stages/tissues (Fig. 5C,D). Most APA site switching genes (371–529) were observed between pollen and other tissues in pair-wise comparisons (Fig. 5C). Those with APA switching in non-3′ UTRs, other than 3′ UTRs, are still observed in significant amounts, but again, dominant in pollen (Fig. 5D). In addition, upon investigating APA site switching dynamics in pollen compared to other tissues (Fig. 5B), results showed that pollen possesses longer 3′ UTRs compared with embryo, endosperm, and dry seed but used shorter 3′ UTRs when compared with the rest of the tissues tested, except pistil, where they were almost equal. This is an example showing that the dynamic of 3′ UTR length control could be broader than previously anticipated.
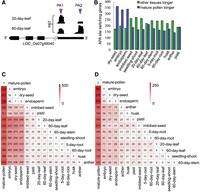
APA site switching genes and their distributions among different samples. (A) An example of a switching gene. Bottom is gene body, big black block is exon, and small black block is untranslated region; top indicates two poly(A) sites and middle is 3′ reads in the two tissues. (B) Switching genes between mature pollen and other tissues. (C) Switching genes with 3′ UTR PACs in pair-wise comparisons. (D) APA site switching genes with non-3′ UTR PACs in pair-wise comparisons.
To confirm the APA switching gene in different stages, we performed qRT-PCR for four 3′ UTR lengthening genes and one 3′ UTR shortening gene using primers that amplify a common region (PA1 + PA2) and the longer 3′ UTR (PA2). The qRT-PCR results match what were tallied from the PAT-seq large-scale data (Supplemental Fig. S7).
Intriguingly, with the development of rice, more genes used longer 3′ UTRs than shorter 3′ UTRs between two early stages (Supplemental Fig. S8), thus contributing to overall 3′ UTR lengthening (Supplemental Table S3). Accordingly, our analysis found that some proteins would obviously become shorter in varying degrees as a result of APA leading to potential functional changes (Supplemental Fig. S8). As predicted based on what was shown, there was a sharp change from anther to pollen, but the number of genes that used shorter 3′ UTRs was more than the number that used longer 3′ UTRs. This was similar, as described above, to the observation that many PACs were located within introns. Gene Ontology analysis of switching genes in 3′ UTRs showed that switching genes between anther and mature pollen mostly functions in biological processes, including single-organism signaling, cell communication, etc. (Supplemental Fig. S9). Switching genes between anther and mature pollens that are located in the non-3′ UTR areas, however, affects a different array of pathways such as carbohydrate metabolic processes and cellular processes (Supplemental Fig. S9). The above analysis illustrated that development of rice might be regulated by different APA switching patterns.
APA genes are highly enriched at quantitative trait loci
To explore if APA plays any role in regulating the expression of quantitative trait loci (QTL), we first examined whether APA genes or APA site switching genes are enriched in QTL regions compared with randomly selected genomic regions (see Methods). The coincidence of QTL regions with APA genes was significantly higher than expected (P = 2.36 × 10−12). Similar analyses were done on APA site switching genes, which were located in 150 of the 1078 QTL regions, but in this case, only 77 randomly selected regions possessed APA switching genes. Therefore, the coincidence of QTL regions with APA site switching genes was also significantly higher than expected (P = 3.58 × 10−7). We further subdivided QTL regions into different groups according to their trait categories to study which categories were enriched with APA genes. Results showed that 14 out of 17 kinds of QTLs had high statistical confidence, with three exceptions: biochemical, leaf senescence, and biotic stress (Supplemental Table S5). To rule out the possibility that such relationships derived from difference in the number of genes between QTL and random selected regions, we performed a control experiment by computing the total number of genes with or without APA located on the two defined genomic regions, with or without QTL. The results, however, showed no significant difference in gene numbers, with or without APA, in either genomic region. This result suggests that QTL tends to use APA genes, implying that APA genes may play an important role in determining agronomic traits of rice.
Next, we investigated the relationship between APA gene expression level and QTL across all 14 tissues. To this end, highly expressed APA genes of each tissue (Z-score of log2PAT > 2) were selected, and their genomic distributions were studied. As shown in Figure 6 (only top 30 shown; see full list in Supplemental Table S6), highly expressed APA genes were mainly enriched in two groups of QTL traits consistently on the top tier across all samples. The first group includes the top five: root dry weight, 1000-seed weight, days to heading, plant height, and spikelet number. The second group is slightly less enriched and includes leaf length, panicle number, biomass yield, total biomass yield, panicle length, grain yield, and tiller number. Importantly, most of these traits were also in the three main categories of QTL: yield, vigor, and anatomy, which were shown to be significantly relevant to APA and switching genes (Supplemental Table S7). These results further suggest that APA genes may play an essential role in the determination of agronomic traits in rice.
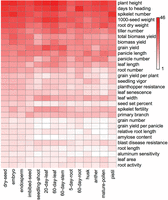
Number of highly expressed APA genes are enriched in QTLs (top 30) across 14 tissues. Each color block represents PAC amount (scale bar on the right) of QTL in each tissue.
Discussion
Rice transcriptomes have been extensively studied using high-throughput sequencing methods. Up to now, however, only a few reports have investigated 3′-end information on a genome-wide scale. The present work reports a comprehensive and high-resolution map of genome-scale poly(A) sites systematically characterizing the role of APA gene expression in 14 rice tissues that have agronomic importance, and the dynamic profiles of 3′-ends of genome-wide information at different stages of rice were also revealed.
Many poly(A) sites in the intergenic regions were identified. While their authenticity as potentially unannotated gene transcripts remains to be tested, their existence at such a scale (∼23%) requires further investigation. Similarly, previous studies also showed that there were many poly(A) sites in the intergenic regions (Lopez et al. 2006; Shepard et al. 2011; Derti et al. 2012). Concerning the rice data presented herein, additional evidence implies that these are authentic poly(A) sites. First, a large number of poly(A) sites of PAT-seq data were confirmed by previously collected rice EST data (often sequenced by low throughput and Sanger method). There are ∼70% of the intergenic PACs that are confirmed by EST. Second, single-nucleotide profiles of these intergenic PACs surrounding the poly(A) sites (Supplemental Fig. S10) are similar to the classical poly(A) signal profile in annotated 3′ UTRs, indicative of their authenticity to be regular poly(A) sites. However, the question of whether they are all from complete genes, or even coding for protein or RNA, is the subject for future studies because full-length transcript sequences are needed to reach a conclusion.
We also identified poly(A) sites located in regions other than 3′ UTRs. Their functions, as well as their potential in altering gene expression, are largely unknown. About 9%–13% of PACs were located within introns across 14 tissues, suggesting that many truncated transcripts might be generated. These transcripts might escape miRNA targeting to regulate gene expression at different stages of rice development, or the alternative form may lose function, as in the case of Arabidopsis FCA (McKnight et al. 2002). Of particular interest, APA sites in CDSs were also found (1.95%). This rate of APA in CDSs is different from that seen in Arabidopsis (11%), using a similar PAT-seq protocol (Wu et al. 2011). However, this figure was more similar to a previous study of Arabidopsis (1.52%), using a different method of direct RNA sequencing (Schurch et al. 2014). Two different results of APA in CDSs in Arabidopsis probably resulted from the use of different methods of library production, sequencing, and different ways of tallying APA. However, the difference between Arabidopsis and rice might stem from different species.
The rate of APA genes in the rice genome is similar to those of ESTs (Shen et al. 2008) but is less than that of MPSS-DGE and SBS-DGE (Shen et al. 2011). As pointed out by the investigators, MPSS-DGE and SBS-DGE could not provide exact cleavage sites information, and many poly(A) sites were double counted compared with EST data (Shen et al. 2011). The most tissue-specific PACs were in pollen, as seen in the testis of zebra fish. Relative to the control of gamete-specific gene expression, some intersection between plants and animals has been reported (Ulitsky et al. 2012). About 44% of PACs were from introns of pollen. Previous studies also showed that pollen-specific splicing patterns are common in Arabidopsis (Loraine et al. 2013). Many APA events were found that potentially produce truncated proteins in rice, meaning that some proteins lost function and thus changed the biological functions of some species.
The single-nucleotide profile of NUE regions for those intronic poly(A) sites found in pollen, particularly enriched in U (or T in DNA), was significantly different from the same region of other tissues, suggesting a different poly(A) signal in the introns of pollen. Recent studies have shown that several poly(A) factors have distinct preference in the choice of cis-element. For instance, AAUAAA hexamer was recognized by CPSF30 and WDR33 (plant homolog FY) but not by CPSF160, as suggested in the past (Chan et al. 2014). In this study, CPSF30 was down-regulated in pollen compared with leaf, while FY was distinctly up-regulated. This is in good agreement with the notion that reduced CPSF30 is accompanied with reduction of A-rich motif recognition (Thomas et al. 2012). Such an alteration of poly(A) factor expression levels is known to cause a change of APA site selections (Li et al. 2015).
It was shown that 3′ UTR length varies along the progression of development in animals (Ji et al. 2009). Here, we also found that seed-related tissues, especially in dry seeds, had the shortest average 3′ UTR length, suggesting that some important cis-elements, or miRNA target sites, may be lost from 3′ UTRs, indicating that some mRNAs were unable to stabilize by escaping from miRNA-mediated regulation or other regulatory elements in 3′ UTRs. Previous studies have shown that many mRNAs were stored in seed until sowing (Sano et al. 2015). Therefore, in order to maintain the stability of storage mRNA in dry seed, it is assumed that the shortest average 3′ UTR be used. Interestingly, with the development of rice, the average 3′ UTR length gradually becomes longer, potentially subject to additional miRNA regulation. Moreover, we found that 3′ UTR length in APA genes was longer than that of non-APA genes, suggesting the possible role of APA in regulating gene expression by increasing additional diversity and complexity.
In this study, we found many APA site switching genes among the different tissues or different developmental stages of rice, indicating that APA site switching events may play an important role during the growth of rice. Discrepancy among the expression levels of switching genes was higher than that of nonswitching genes. The fact that highly expressed PACs tend to use proximal poly(A) site is intriguing. Both of these imply that APA site switching is able to change the expression level, as previously identified by RNA-seq data and shown in Supplemental Table S8, and that proximal PAC usage may have the capacity to escape miRNA targeting or other regulatory mechanisms, thus maintaining high expression. For APA switching genes that involve switching between other genomic regions (CDS, introns, 5′ UTR), however, the impact could be greater and lead to the production of truncated proteins. Previous studies have also shown that miRNA was related to rice leaf senescence, and many miRNAs were identified in rice (Xu et al. 2014). We supposed that the APA site switching event may be a requirement for several genes in order to carry out further miRNA regulation during different developmental stages or tissues of rice. Consequently, we speculate that APA site switching events may regulate gene expression via two methods. One involves indirect regulation of gene expression level via miRNA. The other involves the production of null proteins or those with reduced function. Indeed, the functions of the switching genes may substantially affect rice growth and development. As listed in Supplemental Table S7, many genes reported to play an important role during the growth and development of rice were APA site switching genes also shown in this study. These cases indicated that APA genes may regulate many aspects during the development of rice, including, for example, photosynthesis and tillering, as well as salt and drought tolerance.
Furthermore, we provide evidence that QTLs tend to have APA genes, indicating that APA genes may participate in the regulation of some QTLs in rice. APA can generate several isoforms and increase the diversity of gene expression regulation, and this model may be able to meet the regulatory requirement of QTLs controlled by many genes. Particularly, APA regulation should be good for fine-tuning the expression of those genes with minor effects, just like QTLs. In addition, since rice has many QTLs, APA would be a good way to increase transcripts for QTL regulation but not increase gene number. We also found that some QTLs were enriched to specific tissues, illustrating that the choice of some QTLs occurred in these rice tissues. These results could therefore lay a theoretical foundation for understanding the formation of agronomic traits and may be helpful for molecular-assisted breeding in rice in the near future.
Methods
Plant materials
Rice (Oryza sativa L. subsp japonica cultivar Nipponbare) was grown in the experimental field of the Rice Research Institute, Fuzhou, Fujian Academy of Agricultural Sciences. Fourteen samples were collected from different developmental stages of rice (Supplemental Material; Supplemental Table S9).
PAT-seq library construction and sequencing
Total RNAs were first isolated by TRIzol reagent and used after DNase I digestion (Qiagen). The PAT-seq libraries were constructed as previously described (Liu et al. 2014; see Supplemental Material).
Data analyses
Poly(A) site analysis was performed as previously described (Wu et al. 2011). A strategy similar to that of a previous study was adopted to calculate the PAT-weighted 3′ UTR length of each gene (Wu et al. 2015). Of note, 3′ UTR switching and non-3′ UTR switching for each gene were detected as described previously (Mangone et al. 2010; Fu et al. 2011). Correlation analysis between PAT and RNA-seq was as previously described (Lianoglou et al. 2013). Details are in the Supplemental Material, and the custom scripts are included in the Perl_scripts.rar package also in the Supplemental Material.
APA genes and QTL analysis
The physical positions of 8217 rice QTLs were downloaded from Gramene (www.gramene.org), covering nine categories and 237 QTL traits. Only QTLs <500K nt were further analyzed, and 3468 QTLs were gained. To examine PAC distributions, PACs across tissues were mapped to these 3468 QTLs, and a total of 1590 QTLs with distinct information of chromosome, strand, and start and end coordinates were retained. To examine whether APA genes or APA site switching genes were enriched in QTL regions, we compared the number of studied genes contained in QTLs with that in control regions. First, overlapped QTLs were reduced, and then 1078 QTLs were obtained. Then the same number of control regions as that in QTL was randomly selected from genomic regions not hosting QTL but still required to preserve the same distributions of chromosomes and length as QTL. Ten trials were then run, each consisting of a random selection of control regions. Next, the number of studied genes in QTL and in control regions was calculated. The average value from 10 trials was used as a control. Then a two-way contingency table recording the numbers of studied genes in or outside of QTL and control regions was obtained. A Fisher's exact test was performed using the fisher.test function in R to test whether the difference was statistically significant. To study the relationship of APA and QTL across different tissues, highly expressed (Z-score of log2PAT > 2) APA genes of each tissue were selected. For each QTL trait, the number of PACs in the highly expressed APA genes for each tissue was calculated.
qRT-PCR analysis of APA switching gene
qRT-PCR was performed on an iCycler (Bio-Rad) using SYBR green PCR master mix. Primer sequences are shown in Supplemental Table S10. All data were normalized to actin.
Data access
The sequencing (PAT-seq) data from this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra/) under accession number SRP073467. PACs can be visualized (Wu et al. 2016) and their coordinates downloaded at http://bmi.xmu.edu.cn/plantapa/, and the poly(A) sequence DNA data sets can be downloaded from the Supplemental Data file Rice_PAC_datasets.zip.
Acknowledgments
We thank other laboratory members for their discussions and suggestions, Haidong Qu and Wenjia Lu for technical assistance, and David Martin for language editing. Funding support of the project was from the 100 Talent Plans of Fujian Province and Xiamen City, Xiamen University and in part from the National Science Foundation (IOS-154173), all to Q.Q.L.
Author contributions: Q.Q.L. conceived the ideas. D.Y. and X.Y. provided materials. H.F., W.S., and L.M. performed the wet experiments. Q.Q.L., X.W., H.F., G.J., and Y.S. contributed to data analysis. Q.Q.L., H.F., and X.W. wrote the manuscript. All authors reviewed and approved the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.210757.116.
- Received May 31, 2016.
- Accepted October 6, 2016.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








