
Loss of HP1 causes depletion of H3K27me3 from facultative heterochromatin and gain of H3K27me2 at constitutive heterochromatin
- Kirsty Jamieson1,3,
- Elizabeth T. Wiles1,
- Kevin J. McNaught1,
- Simone Sidoli2,
- Neena Leggett1,
- Yanchun Shao1,
- Benjamin A. Garcia2 and
- Eric U. Selker1
- 1Institute of Molecular Biology, University of Oregon, Eugene, Oregon 97403-1229, USA;
- 2Department of Biochemistry and Biophysics and the Epigenetics Program, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104-5157, USA
- Corresponding author: selker{at}uoregon.edu
Abstract
Methylated lysine 27 on histone H3 (H3K27me) marks repressed “facultative heterochromatin,” including developmentally regulated genes in plants and animals. The mechanisms responsible for localization of H3K27me are largely unknown, perhaps in part because of the complexity of epigenetic regulatory networks. We used a relatively simple model organism bearing both facultative and constitutive heterochromatin, Neurospora crassa, to explore possible interactions between elements of heterochromatin. In higher eukaryotes, reductions of H3K9me3 and DNA methylation in constitutive heterochromatin have been variously reported to cause redistribution of H3K27me3. In Neurospora, we found that elimination of any member of the DCDC H3K9 methylation complex caused massive changes in the distribution of H3K27me; regions of facultative heterochromatin lost H3K27me3, while regions that are normally marked by H3K9me3 became methylated at H3K27. Elimination of DNA methylation had no obvious effect on the distribution of H3K27me. Elimination of HP1, which “reads” H3K9me3, also caused major changes in the distribution of H3K27me, indicating that HP1 is important for normal localization of facultative heterochromatin. Because loss of HP1 caused redistribution of H3K27me2/3, but not H3K9me3, these normally nonoverlapping marks became superimposed. Indeed, mass spectrometry revealed substantial cohabitation of H3K9me3 and H3K27me2 on H3 molecules from an hpo strain. Loss of H3K27me machinery (e.g., the methyltransferase SET-7) did not impact constitutive heterochromatin but partially rescued the slow growth of the DCDC mutants, suggesting that the poor growth of these mutants is partly attributable to ectopic H3K27me. Altogether, our findings with Neurospora clarify interactions of facultative and constitutive heterochromatin in eukaryotes.
It has become increasingly clear that covalent modifications of chromatin, such as methylation of specific histone residues and methylation of DNA, can have profound effects on genome functions. In animals, even partial disruption of DNA methylation leads to developmental defects and disease states (Robertson 2005). Similarly, methylation of histone H3 lysine 27 (H3K27me) by the Polycomb Repressive Complex 2 (PRC2) is critical for normal development in flies, plants, and other systems (Schwartz and Pirrotta 2007), and recent work implicates perturbation of H3K27me in a high fraction of pediatric gliomas (Schwartzentruber et al. 2012; Sturm et al. 2012; Wu et al. 2012; Chan et al. 2013; Lewis et al. 2013). It is of obvious interest to understand the normal regulation of epigenetic features such as methylation of DNA and H3K27, which normally mark constitutive and facultative heterochromatin, respectively. Unfortunately, despite numerous studies in a variety of systems, little is understood about how these chromatin modifications are controlled.
Epigenetic marks frequently influence one another, confounding analyses. For example, Schmitges et al. (2011) demonstrated that the amino terminus of histone H3 is recognized by the Nurf55-Suz12 submodule of PRC2 and that this binding is blocked by marks of active chromatin, namely, K4me3 and K36me2/3. Although the mechanism of such “crosstalk” is sometimes reasonably obvious, more often it is not, as illustrated by observations of H3K27me3 redistribution in response to defects in constitutive heterochromatin. More than a decade ago, Peters et al. (2003) noticed that mouse cells defective in both of the SUV39H methyltransferases, which are responsible for the trimethylation of histone H3 lysine 9 (H3K9me3) characteristic of pericentric heterochromatin, show redistribution of H3K27me3; both cytological and molecular analyses suggested that this Polycomb mark “relocated” to the neighborhood abandoned by H3K9me3. DNA methylation typically colocalizes with H3K9me3 but not with H3K27me3 (Rose and Klose 2014). Because DNA methylation can depend on H3K9me, and vice versa (Tariq and Paszkowski 2004), it was of interest to determine whether loss of DNA methylation would also result in redistribution of H3K27me. In early studies with mouse embryonic stem cells, reduced DNA methylation resulting from mutation of either the maintenance methyltransferase gene dnmt1 or the de novo DNA methyltransferase genes dmnt3a and dnmt3b did not result in an obvious change in the distribution of H3K27me (Martens et al. 2005). However, subsequent studies with Arabidopsis (Mathieu et al. 2005; Deleris et al. 2012), mouse embryonic fibroblasts (Lindroth et al. 2008; Reddington et al. 2013), embryonic stem cells (Hagarman et al. 2013), and neural stem cells (Wu et al. 2010) revealed that loss of DNA methylation, caused by disruption of DNA methyltransferase genes or treatment with the demethylating agent 5-azacytidine, provided the most potent trigger of H3K27me3 redistribution.
Considering that DNA methylation has been reported to stimulate H3K9 methylation, in both plants (Tariq and Paszkowski 2004) and animals (Jin et al. 2011), and that both of these epigenetic marks are tied to additional nuclear processes, interpretation of these fascinating results is problematic. We took advantage of a relatively simple system to explore possible relationships between marks of constitutive and facultative heterochromatin. Specifically, we used the filamentous fungus Neurospora crassa, which unlike many other simple model eukaryotes (e.g., budding and fission yeasts, Drosophila and Caenorhabditis elegans) has both DNA methylation and H3K27me (Aramayo and Selker 2013; Jamieson et al. 2013). The pathway for formation of constitutive heterochromatin in Neurospora is relatively well understood and essentially unidirectional, as illustrated in Figure 1A. Constitutive heterochromatin, which is primarily in centromere regions, is characterized by AT-rich (GC-poor) DNA resulting from the action of the genome defense system RIP (repeat-induced point mutation) operating on transposable elements (Selker 1990; Aramayo and Selker 2013). DIM-5, in the DIM-5/DIM-7/DIM-9/DDB1/CUL4 complex (DCDC) (Fig. 1B), methylates H3K9 associated with RIP'd DNA (Lewis et al. 2010a,b). Heterochromatin Protein 1 (HP1) specifically binds the resulting H3K9me3 (Freitag et al. 2004) and recruits the DNA methyltransferase DIM-2 (Honda and Selker 2008). Consequently, the genomic distribution of 5mC, HP1, H3K9me3, AT-rich DNA, and repeated sequences correlate almost perfectly (Fig. 1A). Importantly, mutation of dim-2 does not affect the distributions of H3K9me3 and HP1, unlike the situation in plants (Tariq and Paszkowski 2004) and animals (Espada et al. 2004; Gilbert et al. 2007). Similarly, mutation of the gene encoding HP1 (hpo) has almost no effect on the distribution of H3K9me3 (Lewis et al. 2009). Although mutations in genes encoding DCDC proteins render the organism slow-growing and sensitive to certain drugs (Lewis et al. 2010a), neither the components of the DNA methylation/constitutive heterochromatin machinery nor the components of the H3K27me2/3/facultative heterochromatin machinery are essential for viability of the organism, allowing us to test knockouts of genes for components of these processes for possible epigenetic interactions.

Heterochromatin formation in Neurospora crassa. (A) An ∼500-kb region, including the centromere of LG III, is shown to illustrate the pathway leading to constitutive heterochromatin in Neurospora. During the sexual phase of the life cycle, the genome defense system RIP (repeat-induced point mutation) recognizes repeated DNA (e.g., transposons and other repeated DNA shown as black rectangles) and litters them with C:G-to-T:A mutations (Selker 1990). In vegetative cells, lysine 9 of histone H3 (H3K9) associated with G:C-poor DNA is methylated by DIM-5, generating H3K9me3 (orange track), which is bound by the HP1-DIM-2 complex (yellow track) and catalyzes DNA methylation (green track). Perturbation of any step in the pathway eliminates the downstream steps without significantly influencing earlier steps. (B) Key proteins required to form constitutive (left) or facultative (right) heterochromatin. Methylation of H3K9 by DIM-5 depends on all five members of the DCDC (DIM-5/-7/-9, CUL4/DDB1 complex) (Lewis et al. 2010a). HP1 directly binds to H3K9me3 (cluster of three red hexagons labeled “Me”) and recruits the DNA methyltransferase DIM-2 (Honda and Selker 2008), resulting in a genome-wide correlation between H3K9me3 and DNA methylation (orange hexagons labeled “Me”) (Lewis et al. 2009). HP1 is also involved in the HCHC deacetylase silencing complex (Honda et al. 2012) and the DMM (Honda et al. 2010) complex, which limits spreading of constitutive heterochromatin. Methylation of H3K27 (cluster of three blue hexagons labeled “Me”) is carried out by the PRC2 complex consisting of SET-7, EED, SU(Z)12, and NPF (Jamieson et al. 2013).
Results
Loss of H3K9 methylation, but not loss of DNA methylation, causes redistribution of H3K27me to regions with RIP'd DNA
Our previous chromatin immunoprecipitation (ChIP) analyses with antibodies recognizing predominantly H3K27me2, predominantly H3K27me3, or either mark gave equivalent results, suggesting these two forms of H3K27me marks are distributed similarly in Neurospora, and revealed that these marks together cover ∼7% of the genome, including hundreds of genes, all of which are essentially silent (Jamieson et al. 2013). The H3K27me2/3 domains do not overlap with regions of constitutive heterochromatin; indeed, most H3K27me2/3 is near the ends of chromosomes in N. crassa, whereas the marks of constitutive heterochromatin, H3K9me3 and DNA methylation, are mostly in centromere regions. Conspicuously, however, some chromosomal regions show directly interspersed tracts of H3K27me2/3 and H3K9me3/DNA methylation, raising the possibility that the marks are necessarily mutually exclusive, as seems to be generally the case in other organisms (Shi and Dawe 2006; Squazzo et al. 2006; Pauler et al. 2009; Ernst et al. 2011; Kharchenko et al. 2011; Roudier et al. 2011). This prompted us to test for crosstalk between the two forms of heterochromatin, that is, to test if elimination of features characteristic of constitutive heterochromatin would impact the distribution of H3K27me2/3 and, conversely, to test if elimination of H3K27me2/3 would impact the distribution of constitutive heterochromatin.
While recent observations in both plant and animal systems revealed that removal of DNA methylation can profoundly influence the distribution of H3K27me3 (Mathieu et al. 2005; Lindroth et al. 2008; Wu et al. 2010; Deleris et al. 2012; Hagarman et al. 2013; Reddington et al. 2013), the fact that DNA methylation can indirectly induce H3K9 methylation in plants (Tariq and Paszkowski 2004) and animals (Jin et al. 2011) raised the possibility that some, or all, of the observed effects were indirect. We therefore tested for possible effects of knocking out either the single N. crassa H3K9me3 methyltransferase dim-5 (Tamaru and Selker 2001; Tamaru et al. 2003) or the single N. crassa DNA methyltransferase gene dim-2 (Kouzminova and Selker 2001). We first compared the distribution of H3K27me2/3, assessed by ChIP-seq, in wild-type N. crassa with the distribution of this mark in a dim-5 deletion strain. Strikingly, we observed a global redistribution of H3K27me2/3 in the dim-5 strain (Fig. 2; Supplemental Fig. S1A). The vast majority of normal H3K27me2/3 domains were lost on each of the seven chromosomes of N. crassa, while regions that are normally constitutive heterochromatin gained H3K27me2/3. Representative ChIP-seq results were validated by ChIP-qPCR at two subtelomere domains on Linkage Group (LG) I and at two genes on LG VII (Supplemental Fig. S2A). To explore whether the H3K27me2/3 redistribution was stochastic, we performed biological replicates of ChIP-seq in the dim-5 strain and observed apparently identical H3K27me2/3 distributions (Supplemental Fig. S1A). In addition, we performed ChIP-seq on a dim-5 strain with a different allele (dim-5HT1, which bears a nonsense mutation in the SET domain) (Tamaru and Selker 2001) and found it, too, showed redistribution of H3K27me2/3, although with minor differences (Supplemental Fig. S1A).

Distribution of H3K27me2/3 is mostly unaffected by elimination of DNA methylation, but loss of H3K9me3 in the dim-5 mutant causes redistribution of H3K27me2/3 to constitutive heterochromatin. ChIP-seq tracks of H3K27me2/3 in wild-type, dim-5, and dim-2 strains are displayed in dark blue, and tracks of H3K9me3 are displayed in green (inverted; shown for the wild-type strain only). While genome-wide redistribution of H3K27me2/3 was observed in a dim-5 strain, the vast majority of H3K27me2/3 domains were unchanged in a dim-2 mutant, with the exception of three novel H3K27me2/3 domains on LG VI, one of which normally sports H3K9me3. The black, light blue, and red lines indicate tracks of H3K27me2/3 ChIP-seq performed in wild-type, dim-5, and dim-2 strains, respectively. Predicted genes are displayed as gray rectangles below the ChIP-seq tracks. The scale bar (500 kb) applies to all seven chromosomes.
We next considered the possibility that the redistribution of H3K27me2/3 observed in the dim-5 strains is a consequence of loss of DNA methylation, which is completely controlled by H3K9me3 in N. crassa (Tamaru and Selker 2001; Tamaru et al. 2003). By using two different dim-2 strains to eliminate DNA methylation, we found that the distribution of H3K27me2/3 was nearly identical to that in the wild-type strain, indicating that 5mC does not appreciably influence H3K27me2/3 (Fig. 2; for ChIP-seq data for all chromosomes, see Supplemental Fig. S1A). A detailed survey of ChIP-seq data across all seven N. crassa chromosomes revealed that more than 200 H3K27me2/3 domains were unchanged; only three novel H3K27me2/3 domains were found that were not present in the wild type (Supplemental Fig. S1B). We conclude that unlike elimination of H3K9me3, elimination of DNA methylation does not cause marked redistribution of H3K27me2/3.
We next tested our expectation that mutants defective in the other members of the DCDC—namely, DIM-7, DIM-8 (DDB1), DIM-9, and CUL4, all of which are essential for both H3K9me3 and DNA methylation (Lewis et al. 2010a)—would also show redistribution of H3K27me2/3. The normal distribution of H3K27me2/3 was lost in all of these mutants, as illustrated for two chromosomes (Fig. 3; Supplemental Fig. S1A), but interestingly, the various mutants showed minor differences. While dim-5 and dim-9 showed extremely similar distributions of H3K27me2/3, mutants defective in DDB1 (dim-8) or CUL4 (cul4) showed somewhat less H3K27me2/3 in certain domains, most notably in the centromere regions. Surprisingly, the dim-7 knockout strain obtained from the Neurospora gene knockout project (Colot et al. 2006) showed an apparently complete loss of H3K27me2/3 (Supplemental Figs. S1A, S2A) raising the possibility that DIM-7 is required for methylation of both H3K9 and H3K27. However, we considered the possibility that an extraneous mutation was responsible for the complete lack of these marks, and found evidence that this is the case. Introduction of a wild-type dim-7 gene with its native promoter at the csr-1 locus rescued the DNA methylation defect without rescuing the H3K27 methylation defect (Supplemental Fig. S3A,B). We therefore designate this strain dim-7, dfh-1 (defective in facultative heterochromatin-1). ChIP-seq experiments with an independent dim-7 strain with a nonsense mutation at position L448 showed redistribution of H3K27me2/3 comparable to that observed in the other DCDC mutants (Fig. 3; Supplemental Fig. S1A).
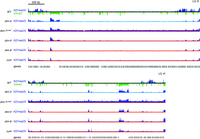
Effect of DCDC mutations on the distribution of H3K27me2/3. H3K27me2/3 ChIP-seq data are shown for two chromosomes of wild type (black line) and strains bearing deletions of dim-5 (light blue line), dim-7L448*, dim-9, dim-8, and cul4 (each indicated by red lines). All show redistribution of H3K27me2/3 to constitutive heterochromatin, with minor variations between mutants. The scale bar (500 kb) applies to both chromosomes. Data for all other chromosomes are presented in Supplemental Figure S1A.
Elimination of H3K27 methylation partially reverses defects of DCDC mutants
It is interesting to consider whether the abnormal presence of the facultative heterochromatin mark in regions that are normally marked by H3K9me3 might partially compensate for loss of this mark of constitutive heterochromatin. Alternatively, it was conceivable that abnormal H3K27me2/3 localization itself might lead to defects, for example, due to aberrant gene regulation or to effects on other genetic processes such as replication and chromosome segregation. These alternative possibilities make opposite predictions on the effect of knocking out H3K27 methylation machinery in a dim-5 background. Strains deficient in DIM-5 or other components of the DCDC show poor growth, abnormal chromosome behavior, and hypersensitivity to the alkylating agent methyl methanesulfonate (MMS) and modest sensitivity to the topoisomerase I inhibitor camptothecin (CPT), perhaps reflecting compromised kinetochore function and/or difficulties replicating damaged DNA (Lewis et al. 2010a). In contrast, strains with null mutations in set-7, which encodes the H3K27 methyltransferase of the PRC2 complex, appear to grow as well as wild-type strains (Jamieson et al. 2013) and show only modest sensitivity to MMS (Supplemental Fig. S4A,B).
We therefore asked if deletion of the set-7 gene in a dim-5 background would increase or decrease the growth defect and the drug sensitivity of dim-5 strains. Results of testing linear growth rate showed unambiguously that set-7; dim-5 double mutants are much more vigorous than dim-5 strains, though still slower growing than set-7 or wild-type strains (Fig. 4A). Similarly, introduction of set-7 into a dim-9 background increased its growth rate relative to the corresponding single mutant (Supplemental Fig. S5A). Consistent with this finding, introduction of set-7 into the dim-7 deletion strain bearing a yet-unidentified mutation that prevents H3K27 methylation, designated dfh-1, failed to rescue the growth defect of the dim-7 mutant (Supplemental Fig. S5B), whereas growth of dim-7 strains without dfh-1 (and thus having H3K27 methylation) was augmented by set-7 (Supplemental Fig. S5C). These findings suggest that abnormal distribution of H3K27me2/3 is partially responsible for the poor growth characteristic of strains defective in components of DCDC. We found no evidence that ectopic H3K27me2/3 functionally compensates for loss of the normal mark of constitutive heterochromatin. Mutation of set-7 partially alleviated the sensitivity of dim-5 to MMS (Supplemental Fig. S4A), but this may not simply be due to the loss of H3K27me2/3, per se because this effect was also found with the dim-7; dfh-1 double mutant that lacks H3K27me2/3 even with a wild-type set-7 gene (Supplemental Fig. S4A,B). Curiously, the set-7 DCDC double mutants showed increased sensitivity to CPT (Supplemental Fig. S4A,B).
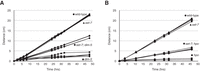
Phenotypic consequences of redistribution of H3K27me2/3 to constitutive heterochromatin. (A) The linear rate of growth was measured for sibling sets of wild-type, dim-5, set-7, and set-7; dim-5 strains. dim-5 strains (squares) show slow growth relative to wild-type (circles) and set-7 (triangles) strains, whereas strains defective in both set-7 and dim-5 (diamonds) show an intermediate rate of growth, suggesting that ectopic H3K27me2/3 is partly responsible for the poor growth of dim-5 strains. (B) Comparison of growth rates for wild-type, set-7, hpo, and set-7; hpo double-mutant strains. The slow growth phenotype of the hpo strain is partially relieved in the set-7; hpo double-mutant strain.
HP1 normally prevents methylation of H3K27 in constitutive heterochromatin
Our observation that loss of DNA methylation did not lead to the redistribution of H3K27me2/3 raised the possibility that another feature of constitutive heterochromatin downstream from H3K9me3 is key. H3K9me3 is recognized by HP1 in N. crassa, which forms at least three distinct protein complexes operating on heterochromatin: HP1/DIM-2 (Honda and Selker 2008); HP1/DMM (DNA Methylation Modulator) (Honda et al. 2010); and HCHC (HP1/CDP-2/HDA-1/CHAP) (Honda et al. 2012). Previous work showed that an hpo (heterochromatin protein one) mutant created by RIP (Freitag et al. 2004) has a nearly normal distribution of H3K9me3, at least on the chromosome examined in detail (LG VII) (Lewis et al. 2009), but is completely defective for DNA methylation. The hpoRIP2 strain is not a null allele so we mapped H3K9me3 by ChIP-seq in an hpo deletion strain and validated the results by ChIP-qPCR (Fig. 5A; Supplemental Fig. S6A). While some reduced H3K9me3 was observed, the vast majority of H3K9me3 was retained in the absence of HP1, which is consistent with our previous results for the hpoRIP2 allele and supports the model that heterochromatin formation is a largely unidirectional pathway in Neurospora (Fig. 1A).
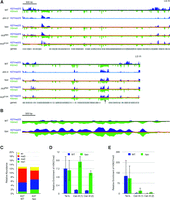
Loss of HP1 leads to H3K27me2/3 redistribution. (A) ChIP-seq tracks of H3K27me2/3 (dark blue) and H3K9me3 (inverted; green) are displayed for LG III and LG VI in a wild-type strain (black line), a dim-5 strain (light blue line), an hpo strain (red line), and two histone mutants, hh3R8A and hh3S10A (both indicated by red lines). H3K9me3 is relatively unaffected by a deletion of hpo, while H3K27me2/3 is redistributed to constitutive heterochromatin in an hpo strain, as in the dim-5 strain. Each of the histone mutants, which largely lose HP1 localization and have reduced H3K9me3, shows H3K27me2/3 redistribution similar to the dim-5 strain. The scale bar (500 kb) at the top left applies to both chromosomes. The black arrow indicates a centromere region on LG VI that is shown expanded 1000-fold below. (B) The distributions of H3K27me2/3 (blue) and H3K9me3 (inverted; green) were examined at a window size of 25 bp in the wild-type (black line) and hpo (red line) strains. At this scale (bar, 500 bp), the peaks of H3K9me3 and H3K27me2/3 in the hpo strain closely resemble one another, which is consistent with both histone marks occupying the same nucleosome. (C) The relative abundance of four modifications (mono- [me1], di- [me2], and trimethylation [me3] and acetylation [ac]) on the H3K27 residue as determined by LC-MS/MS is shown for wild-type and hpo strains. (D) ChIP-qPCR to detect the relative enrichment of H3K27me2 in wild-type and hpo strains at subtelomere IL (Tel IL) and two centromere III loci (Cen III). (E) The relative enrichment of H3K27me3 in wild-type and hpo strains at the same three loci as in D.
To explore the possibility that loss of heterochromatin machinery downstream from H3K9me3 was responsible for the H3K27me2/3 redistribution observed in the DCDC mutants, we performed ChIP for H3K27me2/3 on strains defective for HP1, HDA-1, CHAP, CDP-2, and DMM-1 (Supplemental Fig. S2B,C). Only the hpo mutant showed perturbed H3K27me2/3 comparable to that observed in dim-5 and other DCDC mutant strains (Fig. 5A; Supplemental Figs. S1A, S2B). H3K9me3 is bound by the chromodomain of HP1 (Freitag et al. 2004). We therefore tested a deletion of the chromodomain. H3K27me2/3 redistribution occurred, as in the hpo deletion strain (Supplemental Fig. S6B). This result suggests that HP1 binding to the H3K9me3 modification, but not the mark itself, directly or indirectly prevents H3K27me2/3 at constitutive heterochromatin. Consistent with the effect of elimination of H3K27me in DCDC mutants, introduction of set-7 into the hpo background also partially rescued its growth defect (Fig. 4B). This implies that ectopic H3K27me2/3 is detrimental even in the presence of H3K9me3.
Cohabitation of H3K9me3 and H3K27me2 in constitutive heterochromatin
Because loss of HP1 does not greatly affect the distribution of H3K9me3, the redistribution of H3K27me2/3 resulted in cohabitation of methylated H3K9 and H3K27, which are normally located in nonoverlapping domains (Fig. 5A; Supplemental Fig. S1A; Jamieson et al. 2013). Examination of the data at high resolution revealed that the peaks of H3K9me3 and H3K27me2/3 overlap, which is consistent with both marks occupying the same nucleosome (Fig. 5B). To investigate the possibility that individual H3 molecules were methylated at both residues, we performed middle-down hybrid chromatography/tandem mass spectrometry (LC-MS/MS) on H3 from wild-type and hpo strains and checked for co-occurrence of K9me3 and K27me2/3 (Schwämmle et al. 2014; Sidoli et al. 2014). Relevant data, illustrated in Table 1 and Figure 5C, showed that both strains have substantial levels of both H3K27me2 (∼4.4% of H3 in wild type and ∼4.9% of H3 in hpo) and H3K27me3 (∼6.6% of H3 in wild type and ∼4.1% of H3 in hpo). Most interestingly, the hpo strain, but not the wild-type strain, showed substantial levels of H3 with both H3K27me2 and H3K9me3; 2.2% of H3 showed both marks in the hpo strain, accounting for nearly half of the total genomic H3K27me2. In contrast, only ∼0.01% of H3 from the wild-type strain showed both marks. No significant cohabitation of H3K9me3 and H3K27me3 was observed in H3 from either strain. These findings indicate that individual H3 molecules can be methylated at both K9 and K27 and suggest that, at least in the case of the hpo strain, the novel H3K27 methylation in regions of constitutive heterochromatin is largely, if not exclusively, the dimethylated form. Consistent with this, no increase in H3K27me3 was found in H3 lacking modifications at K9 (Table 1; K9non).
Percentage of H3 with bivalent marks at K9 and K27
The MS results revealed that the hpo strain had a modest reduction in H3K27me3 and a corresponding increase in H3K27me2 (Fig. 5C). This, together with our finding of cohabitation of H3K9me3 and H3K27me2, but not H3K9me3 and H3K27me3, implied that the observed redistribution of H3K27me2/3 represents specific gain of H3K27me2 in regions of constitutive heterochromatin and loss of both forms of methylated H3K27 from facultative heterochromatin. To test this possibility directly, we carried out ChIP-qPCR using antibodies reported to be specific for the di- or trimethyl forms of H3K27 (Fig. 5D,E). We easily detected H3K27me2 at regions detected with the H3K27me2/3 antibodies (e.g., Telomere [Tel] IL, Centromere [Cen] III) in the hpo mutant and found background levels of signal in the Cen regions of the wild-type strain (Fig. 5D). Interestingly, whereas the H3K27me2-specific antibody showed approximately equal signals for the normal facultative heterochromatin (Tel IL) and the new H3K27me2/3 regions (Cen regions), the H3K27me3 antibody showed substantially lower signals at the new regions of H3K27me2/3 (Fig. 5E), consistent with the MS results indicating that the vast majority of methylated H3K27 in traditional regions of constitutive heterochromatin is dimethylated.
Redistribution of H3K27 methylation in histone H3 mutants
Our finding that mutation of either dim-5 or hpo, but not mutation of dim-2, hda-1, cdp-2, chap, or dmm-1, resulted in markedly abnormal localization of H3K27 methylation suggested that other strains defective in HP1 localization should also show abnormal localization of this mark. We tested two histone H3 mutants (hh3R8A and hh3S10A) that had previously been shown to have abnormal localization of HP1 (Adhvaryu and Selker 2008; Adhvaryu et al. 2011), perhaps due to their inhibitory effect on DIM-5 activity (Adhvaryu et al. 2011). ChIP-seq experiments with these strains showed that each had lost HP1 binding (Supplemental Fig. S1A), confirming our previous results, and that they both showed greatly reduced H3K9me3 and redistributed H3K27me2/3, much like the hpo mutant (Fig. 5A; Supplemental Figs. S1A, S7A,B). These results are consistent with our finding that HP1 prevents the deposition of H3K27me2 in constitutive heterochromatin.
H3K9me3 distribution is unaffected in PRC2 mutants
Given the redistribution of H3K27me2/3 upon loss of HP1 binding, we next investigated whether the loss of H3K27me2/3 alters H3K9me3. The N. crassa PRC2 complex is responsible for all H3K27me2/3, and deletion of any of the three core PRC2 members (set-7, eed, or suz12) abolishes H3K27me2/3 (Jamieson et al. 2013). We mapped H3K9me3 in strains bearing deletions of the gene for the PRC2 catalytic subunit SET-7 or the noncatalytic subunit EED. Neither ChIP-seq nor ChIP-qPCR experiments revealed altered H3K9me3 in either of the PRC2 deletion strains relative to wild type (Fig. 6; Supplemental Figs. S1A, S8), suggesting a polarized relationship between constitutive and facultative heterochromatin in Neurospora: Loss of H3K9me3 leads to redistribution of H3K27me2/3, while the reverse is not the case.
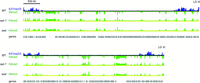
Constitutive heterochromatin is unaffected by loss of facultative heterochromatin. The distribution of H3K9me3 in the set-7 and eed strains (green tracks; distinguished by yellow lines) is not appreciably different from the distribution of H3K9me3 in the wild type (inverted green track; below black line). The distribution of H3K27me2/3 in the wild type is shown for reference (blue track; above black line). The scale bar (500 kb) applies to both chromosomes.
Discussion
Interplay of heterochromatin marks
Our understanding of mechanisms leading to the formation of heterochromatin, the densely staining chromatin commonly observed in eukaryotic cells, is still fragmentary. Eukaryotic organisms have two types of heterochromatin that serve largely distinct functions. Constitutive heterochromatin, which is mostly found in centromeric regions, is gene poor, typically marked by H3K9me3 and DNA methylation, and plays a role in chromosome segregation (Dernburg et al. 1996; Peters et al. 2001; Pidoux and Allshire 2004; Lewis et al. 2010a). In contrast, facultative heterochromatin is found in gene-rich chromosomal regions and is marked by H3K27me2/3 deposited by PRC2. Both types of heterochromatin are relatively transcriptionally inert, although as suggested by its name, facultative heterochromatin can become transcriptionally active under certain developmental conditions. Two important questions are as follows: (1) What controls the establishment of constitutive and facultative heterochromatin, and (2) do facultative and constitutive heterochromatin have partially interchangeable functions? We chose to investigate possible interactions between facultative and constitutive heterochromatin in N. crassa, which displays both types of silent chromatin but does not depend on either for viability (Lewis et al. 2010a; Jamieson et al. 2013). Although the majority of H3K27me2/3 in Neurospora is within subtelomeric regions, which also typically display H3K9me3 and DNA methylation, the marks of facultative and constitutive heterochromatin do not overlap in the wild-type genome.
Elimination of either of the two Neurospora PRC2 subunits required for H3K27me2/3, SET-7 and EED, had no effect on H3K9me3 and DNA methylation. This is interesting given the mixed reports from animal cells about whether loss of facultative heterochromatin impacts constitutive heterochromatin. In a study using both undifferentiated and differentiated mouse ES cells, knockdown of Suz12 resulted in loss of H3K9me3, but only in the differentiated cells (de la Cruz et al. 2007), while a separate study reported that Eed−/−-null, undifferentiated mouse ES cells showed both increases and decreases in 5mC at developmental genes (Hagarman et al. 2013).
Studies with animal cells and plants revealed changes in the distribution of H3K27me3 in response to altered H3K9me3 and/or DNA methylation (Peters et al. 2003; Martens et al. 2005; Mathieu et al. 2005; Lindroth et al. 2008; Wu et al. 2010; Deleris et al. 2012; Hagarman et al. 2013; Reddington et al. 2013). We considered the possibility that complex interactions among chromatin marks found in higher eukaryotes might be responsible for the apparently conflicting findings on the hierarchy of epigenetic marks in plants and animal cells. Conveniently, Neurospora has a relatively simple, nearly unidirectional pathway for constitutive heterochromatin formation (Fig. 1A). To explore the possible impact of H3K9me3 or DNA methylation on the localization of H3K27me2/3, we examined mutants that eliminate H3K9me3 (dim-5, dim-7, dim-8, dim-9, and cul4) or DNA methylation (dim-2). Strains with mutations in any of the genes required for H3K9me3 showed a gross redistribution of H3K27me2/3 on all seven chromosomes; most of the regions that lost H3K9me3 gained H3K27me2/3, while most of the regions that normally show H3K27me2/3 lost this mark (Figs. 2, 3). In contrast, elimination of DNA methylation did not influence the distribution of H3K27me2/3 (Fig. 2).
A new role for HP1
To gain insight into why loss of H3K9me3 led to the dramatic redistribution of H3K27me2/3 in all the DCDC mutants, we explored the possibility that a feature of constitutive heterochromatin downstream from H3K9me3 normally prevents deposition of H3K27me2/3 at constitutive heterochromatin. N. crassa harbors a handful of chromodomain proteins that should bind to methylated lysines, including one homolog of HP1 (Freitag et al. 2004). Results of ChIP-seq and in vitro binding assays revealed that Neurospora HP1 binds strongly to H3K9me2/3 but not to H3K27me2/3 (Freitag et al. 2004; ET Wiles and E Selker, unpubl.). Neurospora HP1 mediates DNA methylation by DIM-2 (Honda and Selker 2008) and is a member of the DMM complex, which controls the spread of heterochromatin (Honda et al. 2010), and the HCHC deacetylation complex, which silences heterochromatin (Honda and Selker 2008; Honda et al. 2010, 2012). We found that a null mutant for the HP1 gene (hpo) showed dramatic redistribution of H3K27me2/3, comparable to that observed in DCDC mutant strains (Fig. 5A). Deletion of the HP1 chromodomain also resulted in loss of H3K27me2/3 at facultative heterochromatin, suggesting that recognition of H3K9me3 is required to prevent methylation of H3K27 in constitutive heterochromatin (Supplemental Fig. S6B).
We employed mutants with amino acid substitutions adjacent to H3K9 to explore further the idea that HP1 binding limits the distribution of H3K27me2/3. ChIP-seq analyses were performed on strains with H3R8A and H3S10A mutations, which impact DNA methylation (Adhvaryu and Selker 2008; Adhvaryu et al. 2011) and had been reported to reduce binding of HP1 by factors of 20 and three, respectively (Jacobs and Khorasanizadeh 2002). Both the R8A and S10A strains showed reduced H3K9me3 and genome-wide loss of HP1 binding (Supplemental Fig. S1A), consistent with prior observations (Adhvaryu and Selker 2008; Adhvaryu et al. 2011). Importantly, redistribution of H3K27 methylation to regions of constitutive heterochromatin occurred in both R8A and S10A strains (Fig. 5A), consistent with the redistribution observed in the hpo deletion and chromodomain mutants.
We considered the possibility that a partner of HP1, other than the DNA methyltransferase, mediates the effect of HP1 loss on the distribution of H3K27me2/3. Since stability of the chromodomain protein CDP-2 depends on HP1 (Honda et al. 2012), it was conceivable that the H3K27me2/3 redistribution observed in the hpo strain was indirectly due to the loss of CDP-2. We found, however, that strains with deletions of cdp-2, hda-1, or chap do not phenocopy the DCDC mutants (Supplemental Fig. S2B).
It is not clear why regions that are normally constitutive heterochromatin become targets of PRC2. HP1 may directly interfere with methylation of H3K27 in these regions. Alternatively, an unidentified downstream feature of constitutive heterochromatin may be responsible for preventing methylation of regions that are normally constitutive heterochromatin. It will be interesting to learn whether loss of HP1 in other systems also leads to redistribution of H3K27me2/3, but there is already reason to suspect that HP1 binding, per se, is not directly responsible in all systems. Loss of the single Arabidopsis HP1 homolog TLF2/LHP1 does not cause redistribution of H3K27me2/3, but it should be noted that this HP1-like protein localizes to facultative, rather than constitutive, heterochromatin and appears to function rather differently than its homologs in animals and fungi (Mylne et al. 2006; Sung et al. 2006; Turck et al. 2007). It is also interesting that the fungus Cryptococcus neoformans, like N. crassa, has subtelomeric domains of methylated H3K27 but that mutation of the H3K9 methyltransferase (Clr4) does not cause redistribution of this mark (Dumesic et al. 2015). In contrast, loss of a noncanonical chromodomain-containing subunit of the C. neoformans PRC2 complex, Ccc1, leads to some redistribution of the H3K27me3. Loss of the apparent Neurospora homolog of Ccc1, TSH-1, which is not in the Neurospora PRC2 complex (Jamieson et al. 2013), does not influence the distribution of H3K27me2/3 (Supplemental Fig. S1A). Although much remains to be learned about the control of H3K27 methylation in eukaryotes, it appears fundamentally different from the mechanisms controlling H3K9 methylation.
Cohabitation of H3K9me3 and H3K27me2 in the absence of HP1
Features of facultative and constitutive heterochromatin do not normally overlap, but relocation of H3K27me2/3 in the hpo strain resulted in cohabitation of the facultative and constitutive heterochromatin marks. This was most obvious in centromere regions (Fig. 5A; Supplemental Fig. S1A). When examined in detail at 25-bp resolution, the ChIP-seq data showed regions where the two marks are directly superimposed (Fig. 5B). Results of middle-down hybrid LC-MS/MS showed that individual H3 molecules from the hpo strain harbor both H3K9me3 and H3K27me2, but not H3K27me3. Indeed, the fraction of H3 with both marks, ∼2%, appears high enough to account for all the new H3K27me2/3 observed by ChIP-seq. Normal facultative heterochromatin, which appears significantly reduced in the mutants defective in constitutive heterochromatin, harbors both H3K27me2 and H3K27me3 in Neurospora. Thus it is not surprising that the ratio of these two forms increases in the hpo strain (Fig. 5D). It will be interesting to learn whether the methylated K27 found in constitutive heterochromatin regions of DCDC mutants is also principally the dimethylated form. Perhaps the trimethylated form of H3K9 specifically prevents trimethylation, but not dimethylation, of H3K27 on the same molecule, consistent with middle-down analyses of the coincidence of various methylation states in mammalian cells (Schwämmle et al. 2014).
Genetic interactions of set-7 and constitutive heterochromatin mutants
Considering that dim-5 and other DCDC mutants show H3K27me2/3 in regions normally occupied by H3K9me3, we considered whether the ectopic H3K27me2/3 might compensate for loss of H3K9me3 and DNA methylation in centromere regions. We found, however, that elimination of H3K27me2/3 by deletion of set-7 in dim-5 and dim-9 mutants significantly promoted their growth (Fig. 4A; Supplemental Fig. S5A, respectively). Similarly, growth of hpo and dim-7 strains was improved by set-7 (Fig. 4B; Supplemental Fig. S5C, respectively). Interestingly, while set-7 did not increase the growth rate of the dim-7 deletion strain, which we found lacks H3K27me2/3 due to an extraneous mutation we denote dfh-1 (Supplemental Fig. S5B), it did decrease its sensitivity to MMS, as with dim-5 (Supplemental Fig. S4B). This suggests that the sensitivity of DCDC mutants to MMS is not simply due to the ectopic H3K27me2. Perhaps PRC2 itself, which has been reported to be recruited to sites of DNA damage (Campbell et al. 2013), is responsible for MMS sensitivity. Curiously, we found that set-7; dim-5 and set-7; dim-7 double mutants show greater sensitivity to the topoisomerase I inhibitor CPT than do the single mutants (Supplemental Fig. S4A,B). It may be relevant that telomeric repeats can be potent targets of topoisomerase I (Kang et al. 2004), and both H3K9me3 and H3K27me2/3 are normally present in subtelomeric regions. Perhaps methylation of H3K9 and K27 jointly safeguards the chromosome ends from CPT-mediated DNA damage. Regardless of the mechanism, our finding suggests that the H3K9me3 and H3K27me2/3 pathways are serving semi-redundant roles in protecting cells from inhibition of topoisomerase I.
Conclusions
In summary, loss of features of constitutive heterochromatin, namely, H3K9me3 and its binding partner HP1, radically affects the genome-wide distribution of H3K27me2/3 in Neurospora, whereas loss of DNA methylation, per se, does not, contrasting with reports on higher eukaryotes. Partially understood interactions among DNA methylation, H3K9 methylation, and HP1 binding, as well as technical limitations, complicate analyses in plants and animals. For example, in human cells, a knockout of DNMT1 results in loss of HP1 binding (Espada et al. 2004) raising the possibility that HP1 is responsible for the observed redistribution of H3K27me3 in animal cells engineered to lose DNA methylation. Similarly, in plants, loss of CG methylation leads to changes in H3K9me3; thus, the observed redistribution of H3K27me3 in a methylation mutant (Deleris et al. 2012) may be indirect. Considering that in the absence of HP1 binding PRC2 acts on regions that are normally constitutive heterochromatin, it seems possible that PRC2 utilizes some of the machinery that normally targets machinery for constitutive heterochromatin. Indeed, it is conceivable that PRC2 is normally associated with both facultative and constitutive heterochromatin and that PRC2 becomes activated at constitutive heterochromatin upon loss of HP1 binding.
Methods
N. crassa strains and methods
N. crassa strains were grown according to standard procedures (Davis 2000). Strains used in this study are listed in Supplemental Table S1.
ChIP and sequence analysis
ChIP was performed essentially as previously described (Tamaru et al. 2003), except mycelia were cross-linked for 10 min in 0.5% formaldehyde for histone modification ChIP or for 30 min in 1% formaldehyde for GFP ChIP. Tissue was disrupted by sonication for 30 pulses before chromatin was sheared using a bioruptor (Diagenode) for 15 min with a cycle of 30 sec on followed by 30 sec off, at high power. The following antibodies were used: anti-H3K27me2/3 (Active Motif, 39535), anti-H3K27me2 (Active Motif, 61435), anti-H3K27me3 (Millipore, 07-449), anti-H3K9me3 (Active Motif, 39161), and anti-GFP (Abcam, 6556). ChIP-qPCR was performed as previously described (Jamieson et al. 2013) using the SYBR FAST ABI Prism qPCR kit (KAPA, KK4605). For primers, see Supplemental Table S2. ChIP samples were normalized to antibody specific background levels at histone H4.
Sequence alignments were performed as previously described (Jamieson et al. 2013) except that reads were mapped to N. crassa OR74A (NC12) genome (Neurospora crassa Sequencing Project, Broad Institute of Harvard and MIT; http://www.broadinstitute.org/) and that read densities were averaged over 200-bp windows to generate all tiled data files with the exception of the tracks included in Figure 5B, in which read densities were averaged over 25 bp.
Preparation of ChIP-seq libraries
Approximately 10 ng of DNA was used to generate ChIP-seq libraries. Each library was prepared using the Illumina TruSeq kits A and B (Illumina, IP-202-1012 and IP-202-1024) according to the manufacturer's instructions. “Invisible” fragments between 250 and 400 bp were excised and purified using the MinElute gel extraction kit (Qiagen, 28606). Final libraries were PCR-amplified using one cycle for 30 sec at 98°C; 18 cycles for 10 sec at 98°C, for 30 sec at 60°C, and for 30 sec at 72°C; and a final extension for 5 min at 72°C.
Serial dilution spot tests
Serial dilutions of conidia were plated on medium with or without MMS (0.02%) or CPT (0.3 μg/mL), obtained from Sigma Aldrich, and grown at 32°C. Spot tests were typically scored at 72 h.
Histone H3 isolation and digestion
Total histones were isolated as previously described (Honda and Selker 2008) with minor modifications. Specifically, after the pellet was resuspended in 2 mL of CW buffer, 2 mL of 0.2 M sulfuric acid was added and incubated for 3 h at 4°C on a rotator. The sample was then centrifuged at 12,000 rpm for 10 min to pellet debris. The supernatant was mixed with 1/3 volume of 100% TCA and incubated overnight on ice to precipitate the histones. Purified total histones were resuspended in 0.1% trifluoroacetic acid and loaded onto a C18 column (Vydac, 218TP54) using an offline System Gold High Performance Liquid Chromatograph (Beckman Coulter) essentially as previously described (Young et al. 2009), with the following modifications to the buffers: buffer A, 0.1% trifluoroacetic acid; buffer B, 95% acetonitrile and 0.08% trifluoroacetic acid. Separation was performed using a gradient from 0% to 60% buffer B for 60 min, followed by a gradient from 60% to 100% buffer B for 10 min. Purified histone H3 was resuspended in 30 μL of 50 mM ammonium acetate (pH 4.0). Endoproteinase GluC was added to the sample at an enzyme:sample mass ratio of 1:20 and subjected to an overnight digestion at room temperature. The reaction was blocked by adding formic acid to a final concentration of 1% in preparation for liquid chromatography-mass spectrometry (LC-MS) analysis.
Middle-down nanoscale LC-MS/MS and data analysis
Samples were prepared as previously described with minor modifications (Sidoli et al. 2015). Samples were separated using an Eksigent 2D+ nanoUHPLC (Eksigent, part of ABSciex). The nanoLC was equipped with a two-column setup, a 2-cm precolumn (100 µm ID) in-house packed with C18 bulk material (ReproSil, Pur C18AQ 3 µm; Dr. Maisch), and a 12-cm analytical column (75 μm ID) packed with Polycat A resin (PolyLC, 1.9 µm particles, 1000 Å). The nanoLC buffers were as follows: Loading buffer was 0.1% formic acid (Merck Millipore) in water; buffers A and B were prepared according to the method described by Young et al. (2009). The gradient was delivered at a flow rate of 250 nL/min as follows: 5 min 100% buffer A, followed by a not linear gradient from 55% to 85% buffer B in 160 min and from 85% to 100% in 10 min. The flow rate for the analysis was 250 nL/min. LC was coupled with an Orbitrap fusion mass spectrometer (Thermo Fisher Scientific). The spray voltage was 2.3 kV, and the capillary temperature was 275°C. Data acquisition was performed at high resolution in the Orbitrap for both precursor and product ions, with a mass resolution of 60,000 for MS and 30,000 for MS/MS. The MS acquisition window was set at 660–720 m/z in order to include only peptides with charge state 8+ in the analysis. Dynamic exclusion was disabled. Isolation width was set at 2 m/z in order to exclude differentially methylated species during peptide isolation for MS/MS fragmentation. The five most-intense ions with MS signal higher than 5000 counts were isolated for fragmentation using ETD electron transfer dissociation (ETD) with an activation time of 20 msec. Three microscans were used for each MS/MS spectrum, and the AGC target was set to 2 × 10−5. Data processing was performed as previously described (Sidoli et al. 2014). Mascot v2.5 (Matrix Science) was used to search spectra for mono and dimethylation (KR), trimethylation (K), and acetylation (K) as dynamic modifications. Mascot result files were processed with isoScale slim (Sidoli et al. 2014; http://middle-down.github.io/Software) using a tolerance of 30 ppm.
Data access
The ChIP-seq data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE68897. The mass spectrometry data have been submitted to the Chorus database (https://chorusproject.org) under project number 988.
Acknowledgments
We thank the Genomics Core Facility at the University of Oregon for carrying out the high-throughput DNA sequencing, Michael Rountree for mapping one of the ChIP data sets for H3K27me3 in a dim-5 strain, Andy Klocko for providing an unpublished dim-7 mutant, Jeanne Selker and Vincent Bicocca for carrying out related exploratory experiments, and Diana Libuda and Jeanne Selker for comments on the manuscript. This work was supported by National Institutes of Health (NIH) grants to E.U.S. (GM093061 and GM035690), an NIH grant to B.A.G. (GM110174), and an American Heart Association (AHA) grant to E.T.W. (14POST20450071).
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.194555.115.
- Received May 15, 2015.
- Accepted November 4, 2015.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








