
Acute TNF-induced repression of cell identity genes is mediated by NFκB-directed redistribution of cofactors from super-enhancers
- Søren Fisker Schmidt1,3,
- Bjørk Ditlev Larsen1,3,
- Anne Loft1,
- Ronni Nielsen1,
- Jesper Grud Skat Madsen1,2 and
- Susanne Mandrup1
- 1Department of Biochemistry and Molecular Biology, University of Southern Denmark, 5230 Odense M, Denmark;
- 2The Novo Nordisk Foundation Center for Basic Metabolic Research, University of Copenhagen, DK-2200 Copenhagen, Denmark
- Corresponding author: s.mandrup{at}bmb.sdu.dk
-
↵3 These authors contributed equally to this work.
Abstract
The proinflammatory cytokine tumor necrosis factor (TNF) plays a central role in low-grade adipose tissue inflammation and development of insulin resistance during obesity. In this context, nuclear factor κ-light-chain-enhancer of activated B cells (NFκB) is directly involved and required for the acute activation of the inflammatory gene program. Here, we show that the major transactivating subunit of NFκB, v-rel avian reticuloendotheliosis viral oncogene homolog A (RELA), is also required for acute TNF-induced suppression of adipocyte genes. Notably, this repression does not involve RELA binding to the associated enhancers but rather loss of cofactors and enhancer RNA (eRNA) selectively from high-occupancy sites within super-enhancers. Based on these data, we have developed models that, with high accuracy, predict which enhancers and genes are repressed by TNF in adipocytes. We show that these models are applicable to other cell types where TNF represses genes associated with super-enhancers in a highly cell-type–specific manner. Our results propose a novel paradigm for NFκB-mediated repression, whereby NFκB selectively redistributes cofactors from high-occupancy enhancers, thereby specifically repressing super-enhancer-associated cell identity genes.
Obesity is associated with low-grade inflammation in the adipose tissue leading to impaired adipocyte differentiation and function (Guilherme et al. 2008). Proinflammatory signals originating primarily from the M1 macrophages lead to impaired insulin signaling in the adipocytes, thereby increasing lipolysis, decreasing lipid storage, and decreasing the release of adiponectin from adipocytes. The increase in plasma fatty acids from lipolysis and the decrease in adiponectin in turn contribute to the compromised insulin sensitivity in other tissues (Maury and Brichard 2010; Turer and Scherer 2012). In this context, tumor necrosis factor (TNF) released from the M1 macrophages is one of the most important proinflammatory cytokines, and targeted disruption of TNF or its receptors has been shown to protect against obesity-induced insulin resistance in mice (Uysal et al. 1997; Ventre et al. 1997). In addition to directly inhibiting the insulin signaling pathway (Cawthorn and Sethi 2008), TNF up-regulates expression of proinflammatory cytokines in the adipocytes, thereby leading to a feed-forward activation of the inflammatory response. TNF stimulation has also been shown to lead to repression of the expression of many adipocyte-specific genes (Ruan et al. 2002; Lo et al. 2013), where decrease in expression and activity of the master regulator of adipogenesis, peroxisome proliferator activated receptor γ (PPARG) (Zhang et al. 1996; Tang et al. 2006), is likely to contribute to repression of these genes during long-term exposure to TNF (>24 h). However, the mechanisms underlying the acute (<2 h) gene repression by TNF are not fully understood (Ye 2008), and genome-wide insight into the transcriptional reprogramming of the genome in response to TNF is lacking.
TNF signaling activates several proinflammatory transcription factors including the master inflammatory transcription factors nuclear factor κ-light-chain-enhancer of activated B cells (NFκB), which appears to be invariably involved in the inflammatory response in many different cell types (Moynagh 2005) and required for the inflammatory response in adipocytes (Ruan et al. 2002). The major transactivating subunit of NFκB is v-rel avian reticuloendotheliosis viral oncogene homolog A (RELA; also known as p65), which, following inflammatory activation, translocates to the nucleus and binds DNA as a heterodimer with the mature product of NFKB1 (p50) to GGGRNYYYCC response elements in the genome (Karin and Ben-Neriah 2000). This appears to be highly dependent on the chromatin landscape shaped by lineage-determining factors (Jin et al. 2011); however, RELA can also direct recruitment of chromatin remodeling factors (Agalioti et al. 2000; Natoli 2009) and facilitate chromatin remodeling at a subset of its binding sites (Lo et al. 2013). In addition to chromatin remodeling factors, RELA recruits histone acetylases and epigenetic reader proteins like bromodomain-containing protein 4 (BRD4), ultimately leading to recruitment of basal transcription machinery, Mediator, RNAPII, and elongation factors (Barboric et al. 2001; Gao et al. 2005; Huang et al. 2009; Zhao et al. 2013).
In contrast to gene activation, the mechanisms underlying transcriptional repression by RELA, and signal-dependent transcription factors in general, are incompletely understood. It has been proposed that TNF-induced suppression of glucocorticoid-activated genes involves RELA tethering to the glucocorticoid receptor (Rao et al. 2011), and such a mechanism may also be involved in repression of, e.g., inflammatory gene expression by nuclear receptors (Jonat et al. 1990; Pascual et al. 2005). Furthermore, it has been suggested that transcription factors compete for a limited amount of coactivators in the cell, whereby activation of one transcription factor reduces the amount of cofactors available for other transcription factors (Meyer et al. 1989; Kamei et al. 1996). While this mechanism has been suggested by recent genomic studies to account for transcriptional repression following ligand activation of nuclear receptors (He et al. 2012; Guertin et al. 2014; Step et al. 2014), the fact that only a small subset of active enhancers and genes is repressed has remained poorly understood.
Here, we show that TNF stimulation of human Simpson-Golabi-Behmel syndrome (SGBS) adipocytes induces acute RELA-dependent redistribution of cofactors and enhancer transcription in adipocytes leading to induction of inflammatory gene programs at the cost of adipocyte genes. We show that this repression does not involve RELA binding to the associated enhancers but rather loss of cofactors and eRNA transcription selectively from high-occupancy sites within super-enhancers. We furthermore demonstrate that gene repression by TNF is highly cell-type specific, reflecting cell-type–specific cofactor loss from high-occupancy enhancers and suppression of super-enhancer-associated cell identity genes.
Results
Acute reprogramming of adipocyte genes and enhancers by TNF
Exposure of adipocytes to TNF is known to lead to acute activation of inflammatory genes as well as repression of adipocyte genes; however, the mechanisms underlying acute repression of adipocyte genes are not well understood. We performed time-course experiments in human SGBS adipocytes using a dose of 10 ng/mL previously demonstrated to be required for biologically relevant effects of TNF treatment such as increased basal lipolysis (Ryden et al. 2002; Zhang et al. 2002). These experiments showed that TNF-mediated repression of PPARG mRNA is manifested after 3 h of TNF treatment (Supplemental Fig. S1A, left) and that this repression is preceded by a pronounced and rapid (within 45–90 min) suppression at the nascent transcript level that coincides with the initial TNF-induced burst in inflammatory gene expression (Supplemental Fig. S1A, right). Subsequent dose-response experiments confirmed that 10 ng/mL is required for efficient activation of inflammatory gene expression as well as acute and chronic repression of PPARG expression (Supplemental Fig. S1B). Thus, for subsequent analyses, we treated adipocytes with 10 ng/mL TNF for 90 min.
To investigate the acute effects of TNF on genome-wide transcription in human adipocytes, we took advantage of our recently developed computational method, iRNA-seq, which determines changes in transcriptional activity based on unique intron reads from total RNA-seq (Madsen et al. 2015). Using this, we detected a pronounced acute transcriptional reprogramming of the adipocyte genome in response to TNF, with 1143 and 654 genes significantly induced and repressed, respectively (FDR < 0.01) (Fig. 1A). In line with our recent study (Madsen et al. 2015), concordant results were obtained when assessing transcriptional changes by RNAPII chromatin immunoprecipitation (ChIP)-seq (Supplemental Fig. S1D) and by qPCR using intron-targeting primers (Supplemental Fig. S1E,F). The genes analyzed by qPCR (n = 26) were selected based on their iRNA-seq-estimated fold changes to achieve coverage across the dynamic range of the TNF response. Pathway analyses revealed that TNF-induced genes, as expected, are enriched in pathways related to cytokine signaling (Fig. 1B; Supplemental Table S1A,B). In contrast, genes acutely repressed by TNF are enriched in pathways related to lipid metabolism, and repressed genes are typically up-regulated during adipogenesis (Fig. 1C; Supplemental Fig. S1G). Further inspection of the genes repressed by TNF (Fig. 1A) revealed that several well-known transcriptional activators of adipocyte differentiation and function are acutely repressed by TNF, and like for PPARG, this repression is manifested and sustained at the mRNA level 3–6 h after TNF treatment (Supplemental Fig. S1H). Thus, in addition to induction of inflammatory gene expression, TNF signaling leads to acute repression of a subset of adipocyte genes, including multiple adipogenic transcription factors, the down-regulation of which is likely to contribute to the long-term effects of TNF on adipocyte function.
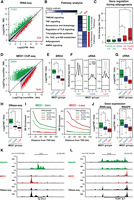
Acute reprogramming of adipocyte genes and enhancers by TNF. Following 10 d of differentiation, human SGBS adipocytes were treated with vehicle or TNF for 90 or 60 min before harvest of RNA for total RNA-seq and chromatin for ChIP-seq. In addition, total RNA-seq was performed in SGBS cells harvested at day 0 of differentiation. Changes in transcriptional activity and mRNA expression were determined using the iRNA-seq pipeline. (A) Scatter plot illustrating log2 normalized mean tag counts in introns of RefSeq gene bodies in control vs. TNF-stimulated SGBS adipocytes. Green and red dots represent genes that were determined to be induced and repressed, respectively (FDR < 0.01). Blue dots represent constitutive genes (FDR > 0.9; |log2FC| < 0.2). (B) Heat map representing −log(P-values) for the top four enriched pathways for genes induced and repressed by TNF. (C) Box plots representing fold change in mRNA expression (exon reads) during adipogenesis for TNF-regulated and constitutive gene groups defined in A and Supplemental Figure S1C. P-value: ¤ = 3.73 × 10−9,* < 1.99 × 10−15, Wilcoxon rank-sum test. (D) Scatter plot illustrating log2 normalized mean MED1 tag counts in MED1 sites in vehicle vs. TNF-stimulated SGBS adipocytes. Green and red dots represent sites with gain and loss of MED1, respectively, following TNF stimulation (FDR < 0.01). Blue dots represent sites with constant MED1 binding (FDR > 0.8; |log2FC| < 0.2). (E) Box plot showing log2 fold change in BRD4 tag counts for TNF-regulated and constant MED1 sites defined in D. P-value: * < 2.2 × 10−16, Wilcoxon rank-sum test. (F) Aggregate plots showing enhancer transcript levels in a 3-kb window around the center of intergenic TNF-regulated and constant MED1 sites defined in D. (G) Box plot representing log2 fold change in enhancer transcript levels at intergenic TNF-regulated and constitutive MED1 sites defined in D. Tags were counted in a region of 2 kb around the MED1 peak center. P-value: * < 2.2 × 10−16, Wilcoxon rank-sum test. (H) Box plot showing log2 fold change in DNase-seq tag counts for TNF-regulated and constant MED1 sites defined in D. P-value: * < 2.2 × 10−16, Wilcoxon rank-sum test. (I) Enrichment of TNF-regulated MED1 sites defined in D near TNF-regulated genes defined in A. Enrichment was determined as the number of binding sites per gene within different distances from the TSS (10–100 kb) of regulated genes relative to the number of binding sites per gene of constitutive genes. (J) Box plots representing log2 fold change (TNF/Veh) in iRNA-seq (left) and RNAPII (right) read counts for genes with TSS within 20 kb of TNF-regulated and constant MED1 sites defined in A. P-value: * < 2.2 × 10−16, Wilcoxon rank-sum test. (K,L) UCSC Genome Browser screenshots of RNAPII (green) and MED1 (red) occupancy and DNase-seq (black) tags at gene loci of a typical induced (CCL2) (K) and repressed (PLIN1) (L) gene.
To investigate the mechanisms underlying the acute TNF-induced transcriptional changes in human adipocytes, we determined the effect of TNF exposure for 60 min on enhancer activity. Using Mediator complex subunit 1 (MED1) ChIP-seq as a surrogate for enhancer activity (Heintzman et al. 2009; Kagey et al. 2010), we identified 5454 activated and 1840 repressed enhancers (FDR < 0.01) (Fig. 1D). For both activated and repressed enhancers, TNF-induced changes in MED1 occupancy are associated with corresponding changes in occupancy of BRD4 (Fig. 1E; Supplemental Fig. S1I), as well as other markers of enhancer activity such as RNAPII occupancy (Supplemental Fig. S1J,K; Bonn et al. 2012) and eRNA transcription (Fig. 1F,G; Kim et al. 2010). In contrast, TNF-induced changes in chromatin accessibility, assessed by DNase I hypersensitive (HS) sites sequencing (DNase-seq) (Crawford et al. 2006), primarily involves increased accessibility at TNF-activated enhancers, whereas TNF-induced loss of MED1 is associated with rather modest changes in chromatin accessibility (Fig. 1H; Supplemental Fig. S1L). Importantly, regions with a gain of MED1 are highly enriched in the vicinity of induced genes, whereas regions with a loss of MED1 are highly enriched near genes repressed by TNF (Fig. 1I). Conversely, the expression of genes with TSS within 20 kb of TNF-regulated enhancers tends to follow the activity of the enhancers (Fig. 1J). Thus, TNF treatment leads to acute and comprehensive reprogramming of the adipocyte genome through activation of enhancers associated with inflammatory genes such as CCL2 (Fig. 1K), as well as through repression of enhancers associated with many adipocyte genes like PLIN1 (Fig. 1L). Intriguingly, acute repression appears to occur largely independently of changes in the chromatin structure, suggesting that enhancers are not deconstructed during acute repression.
RELA mediates reprogramming of the adipocyte transcriptome through cofactor redistribution
To determine the role of RELA in the acute transcriptional reprogramming of human adipocytes by TNF, we knocked down RELA in SGBS adipocytes at day 6 of differentiation using lentiviral vectors. This resulted in robust knockdown of RELA and decreased TNF-mediated induction of well-known RELA target genes such as TNF and IL1A at day 10 of differentiation (Supplemental Fig. S2A). Total RNA-seq followed by iRNA-seq was then used to determine the genome-wide impact of RELA depletion on the acute transcriptional response to TNF. Interestingly, in addition to the expected decrease in TNF-mediated induction of inflammatory genes, RELA depletion also significantly diminished gene repression by TNF (Fig. 2A). In line with this, RELA depletion diminished the TNF-induced increase in eRNA and MED1 occupancy near TNF-activated genes as well as the loss of eRNA and MED1 on enhancers near TNF-repressed genes (Fig. 2B,C). Collectively, these results place RELA as a central mediator of acute TNF-induced transcriptional reprogramming through activation of inflammatory enhancers and genes as well as repression of adipocyte enhancers and genes.
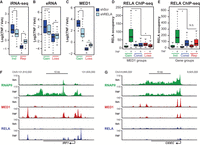
RELA mediates reprogramming of the adipocyte transcriptome through cofactor redistribution. (A–C) At day 6 of differentiation, SGBS adipocytes were transduced with lentiviruses expressing shRNA targeting RELA (shRELA, light blue) or scrambled control shRNA (shScr, dark blue). At day 10, the cells were treated with vehicle or TNF for 90 min before harvest of RNA for total RNA-seq and 60 min before harvest of chromatin for ChIP-qPCR. (A) Box plot representing log2 fold change (TNF/Veh) in nascent RNA for TNF-regulated genes (shScrV vs. shScrT, FDR < 0.01) in shScr- and shRELA-treated cells. P-value: * < 2.2 × 10−16, Wilcoxon rank-sum test. (B) Box plots representing log2 fold change in eRNA at intergenic TNF-regulated MED1 sites defined in Figure 1D in shScr and shRELA-treated SGBS adipocytes. P-value: * < 2.2 × 10−16, ¤ = 2.58 × 10−06, Wilcoxon rank-sum test. (C) Box plots representing log2 fold change in MED1 occupancy at a subset of TNF-regulated MED1 sites in shScr- and shRELA-treated SGBS adipocytes, n = 7–8. Data are representative of three independent experiments. P-value: * = 9.66 × 10−05, ¤ = 1.06 × 10−06, Student's t-test. (D) Box plots representing RELA occupancy at TNF-regulated and constant MED1 sites defined in Figure 1D. Differentiation and treatment of cells for RELA ChIP-seq was performed as described in Figure 1. P-value: * < 2.2 × 10−16, Wilcoxon rank-sum test. (E) Box plots representing summarized RELA occupancies at all RELA binding sites within 20 kb of the TSS of TNF-regulated genes defined in Figure 1A. P-value: * < 2.2 × 10−16, Wilcoxon rank-sum test. (F,G) UCSC Genome Browser screenshots of RNAPII (green), MED1 (red), and RELA (blue) occupancy at gene loci of a typical induced gene, IRF1 (F), and a typical repressed gene, CIDEC (G).
To investigate to what extent RELA is associated with TNF-regulated enhancers, we performed RELA ChIP-seq and analyzed RELA occupancy at sites which gain, lose, or have constant MED1 occupancy in response to TNF treatment. In line with RELA being a major driver of TNF-induced inflammatory gene expression, TNF-activated regions are highly enriched for RELA binding (Fig. 2D; Supplemental Fig. S2B). In contrast, repressed enhancers showed no enrichment of RELA compared with regions with unchanged MED1 occupancy (Fig. 2D; Supplemental Fig. S2B). This indicates that although RELA is required for TNF-induced repression of adipocyte enhancers, it does not bind repressed enhancers directly. Thus, these data do not support tethering of RELA to other transcription factors as a major mechanism involved in repression of adipocyte genes by TNF. In line with this, RELA binding is enriched within 20 kb of TNF-induced but not TNF-repressed genes relative to constitutive genes (Fig. 2E). Taken together, acute activation of inflammatory genes like IRF1 by TNF (Fig. 2F) involves direct RELA action at nearby enhancers, whereas RELA-mediated repression of adipocyte genes such as CIDEC (Fig. 2G) appears to be indirect. A possible mechanism for this indirect repression is redistribution of cofactors such as MED1 and BRD4 from adipogenic enhancers to RELA-activated enhancers near proinflammatory genes by mechanisms such as squelching (Meyer et al. 1989).
A model of selective redistribution of MED1 from high-occupancy sites predicts loss of MED1 and gene repression by TNF
A general problem with a cofactor redistribution model is that it does not explain why only a subset of active enhancers and genes are repressed by TNF. Indeed, when we explored the ability of RELA occupancy to predict loss of MED1 binding following TNF stimulation, we found that low RELA binding to an enhancer is not in itself a strong predictor of MED1 loss (Fig. 3A). Thus, despite the important role of RELA in acute repression of gene expression and enhancer activity by TNF, lack of RELA is not the sole determinant of enhancer repression. In contrast, and as expected from the enrichment of RELA binding at TNF-activated enhancers, RELA occupancy is a strong predictor of enhancer activation (Fig. 3A).
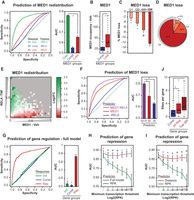
A selective redistribution model predicts gene repression by TNF. (A) Receiver operating characteristics (ROC)-curve for prediction of MED1 gain or loss (as defined in Fig. 1D) based on high and low RELA occupancy, respectively. Bar diagrams represent the AUC for each ROC-curve with error bars representing the 95% confidence interval of the AUC determined by DeLongs test. P-value: * < 2.2 × 10−16, determined by bootstrapping. (B) Box plot showing MED1 occupancy in vehicle-treated cells at sites that display constant occupancy or loss of MED1 upon TNF exposure. P-value: * < 2.2 × 10−16, Wilcoxon rank-sum test. (C) Bar diagrams representing mean % loss of MED1 following TNF exposure for MED1 sites divided into quartiles based on MED1 occupancies in vehicle-treated SGBS adipocytes. Error bars represent 95% confidence interval around the mean. P-value compared with Q4: * < 2.2 × 10−16, # = 5.28 × 10−12, Welch's t-test. (D) Pie chart representing the fraction of total genomic MED1 tags lost originating from the four different quartiles defined in C. (E) Heat map representing mean log2 fold change in MED1 occupancy following TNF treatment for MED1 sites binned by MED1 occupancy in vehicle-treated cells (x-axis) and by RELA occupancy in TNF-treated cells (y-axis). Bins with no data are plotted in gray. (F) ROC-curves and bar diagrams representing prediction of MED1 loss as defined in Figure 1D based on RELA occupancy in TNF-treated cells (blue), MED1 in vehicle-treated cells (red), or subtracted occupancies (purple). Bar diagrams represent the AUC for each ROC-curve with error bars representing the 95% confidence interval of the AUC determined by DeLongs test. P-value: * < 2.2 × 10−16, DeLongs test. (G) ROC-curves for prediction of the top 200 TNF-regulated and constitutive genes as defined in Supplemental Figure 1C based on summarized distance-adjusted occupancy scores as depicted. Bar diagrams represent the AUC for each ROC-curve with error bars representing the 95% confidence interval of the AUC determined by DeLongs test. P-value compared with repressed: * < 2.2 × 10−16, determined by bootstrapping. (H,I) Line plots showing the dependency between minimum transcription thresholds and ROC AUCs for the prediction of the top 200 repressed genes by TNF with the “full model” (purple) (H) defined in G and a “distance model” (red) (I) based solely on summarized distance scores for each gene. Green lines represent ROC AUCs for a model using intron read densities (RPK) for prediction of repression. RPK denotes reads per kb per 10 M reads. Error bars represent the 95% confidence interval of the AUC determined by DeLongs test. P-value compared with CPM: ^ < 5.0 × 10−2, ¤ < 5.0 × 10−4, # < 5.0 × 10−8, * < 5.0 × 10−12, DeLongs test. (J) Box plots representing the number of MED1 sites in vehicle-treated cells within 50 kb of the TSS for constitutive and repressed genes. P-value: * < 2.2 × 10−16, Wilcoxon rank-sum test.
Interestingly, we noticed that TNF-repressed enhancers are characterized by markedly higher occupancy of MED1 and BRD4 in unstimulated cells compared with constant enhancers (Fig. 3B; Supplemental Fig. S3A), indicating that sites with high cofactor binding are more sensitive to cofactor loss in response to inflammatory signals. In support of this, sites with high MED1 occupancy display a higher relative loss of MED1 following TNF stimulation compared with sites with low MED1 occupancy (Fig. 3C). Consequently, sites with high MED1 also display a markedly higher absolute loss of MED1 (Supplemental Fig. S3B), with 75% of the total MED1 loss occurring from the upper quartile of MED1 sites (Fig. 3D). This indicates that the combination of initial cofactor occupancy and the level of RELA binding after TNF stimulation determines whether cofactors are redistributed from or to a given site. To further investigate this, we grouped putative enhancers in unstimulated cells based on their basal MED1 occupancy and the level of RELA binding in TNF-stimulated cells and calculated the average fold change in MED1 binding for each group. Indeed, these analyses revealed that higher relative MED1 occupancy compared with RELA at a given site is associated with loss of MED1 and conversely sites with higher relative RELA occupancy are associated with gain of MED1 (Fig. 3E). Accordingly, by subtracting the two occupancies, we significantly improved the prediction of MED1 loss, compared with using either occupancy alone (Fig. 3F). Importantly, BRD4 occupancy substitutes well for MED1 occupancy to predict MED1 loss (Supplemental Fig. S3C), suggesting a general ability of high cofactor occupancy to predict enhancer repression.
Next, to predict gene regulation, we assigned enhancers to nearby genes and calculated distance scores for each enhancer-gene pair as previously described (Supplemental Fig. S3D; Tang et al. 2011), thereby giving higher weight to an enhancer the closer it is to the TSS. These distance scores were multiplied by occupancy scores for each enhancer before the distance-adjusted occupancy scores were summarized for each gene. Notably, this allowed us to predict TNF-mediated gene repression with similar accuracy as prediction of gene induction based on RELA binding (Fig. 3G; Supplemental Fig. S3E). Importantly, the performance of this model is not simply due to prediction of genes that are transcribed and hence has the potential to be repressed. Thus, when the minimum transcription threshold assessed as intron reads per kb (RPK) for included genes is increased, the area under the curve (AUC) for the receiver operator characteristics (ROC)-curve is relatively unaffected (full model). In contrast, the performance drops rapidly with increasing minimum transcription threshold when using transcription levels (RPK) as the predictor (Fig. 3H). Thus, the full model is much better than the actual level of transcription for prediction of which genes in a group of highly expressed genes will be repressed by TNF.
Interestingly, we noticed that a model considering only the summarized distance scores for all MED1 sites, thus disregarding the level of MED1 and RELA binding, is also a relatively strong predictor of gene repression (Fig. 3I), although not as strong as the full model (Supplemental Fig. S3F). This implies that adipocyte genes being repressed by TNF are characterized by having a high number of MED1 sites in the vicinity of the TSS, and indeed repressed genes generally have more MED1 sites within 50 kb of the TSS compared with constitutive genes (Fig. 3J). Taken together, RELA appears to mediate cofactor redistribution predominantly from sites with high cofactor occupancy, and this has the greatest impact on genes with multiple enhancers nearby. Importantly, our models predict enhancer and gene repression with similar accuracy as enhancer and gene activation by TNF.
Adipocyte super-enhancers and associated genes are sensitive to RELA-mediated redistribution
Recent results from the Young and Collins laboratories have demonstrated at the genome-wide level the importance of clusters of enhancers, termed super-enhancers or stretch-enhancers, for the regulation of cell identity genes (Hnisz et al. 2013; Parker et al. 2013; Whyte et al. 2013). It has been shown that constituent enhancers within super-enhancers display extremely high occupancies of master transcription factors and cofactors and that super-enhancers and their associated genes are highly sensitive to perturbation by knockdown of Mediator subunits or by chemical inhibition of bromodomains (Loven et al. 2013; Whyte et al. 2013).
To investigate the role of super-enhancers in repression of adipocyte-specific gene expression by RELA-mediated cofactor redistribution, we mapped super-enhancers in unstimulated cells essentially as previously described (Whyte et al. 2013). We thereby identified 933 basal super-enhancers (Fig. 4A), with markedly higher constituent levels of both MED1 and BRD4 than regular enhancers (Supplemental Fig. S4A,B). Interestingly, and in line with the results in Figure 3, super-enhancer constituents are more sensitive to MED1 and BRD4 redistribution than regular enhancers (Fig. 4B; Supplemental Fig. S4C,D). Furthermore, in contrast to regular enhancers, super-enhancers are highly enriched in the vicinity of genes repressed by TNF (Fig. 4C). Accordingly, super-enhancer–associated genes are more likely to be repressed by TNF than genes associated only with regular enhancers (Supplemental Fig. S4E,F). Interestingly, the basal super-enhancers identified in SGBS cells are enriched in the vicinity of genes suppressed in adipose tissue from obese human subjects compared with lean subjects in three independent studies (Supplemental Fig. S4G). This suggests that these super-enhancers may also be particularly sensitive to signals induced by the low-grade inflammation associated with obesity. Based on our work we hypothesize that this sensitivity includes cofactor redistribution.
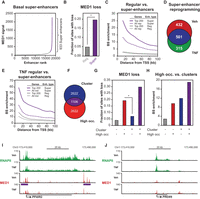
Adipocyte super-enhancers are sensitive to RELA-mediated MED1 redistribution. (A) Definition of basal super-enhancers. All identified MED1 (vehicle) binding sites (27,123) within 12.5 kb of each other were merged, resulting in 18,224 regions of which 933 were classified as super-enhancers in both MED1 replicates based on input-subtracted read counts. (B) Bar diagrams representing the fraction of basal regular and super-enhancer constituents with loss of MED1 in response to TNF exposure as defined in Figure 1D. P-value: * < 2.2 × 10−16, Pearson's χ2 test. (C) Enrichment of basal regular and super-enhancer constituents near TNF-repressed genes defined in Figure 1A. Enrichment was determined as the number of binding sites per gene within different distances from the TSS (10–100 kb) of repressed genes (top 200 and all repressed) relative to the number of binding sites per gene of constitutive genes. (D) Venn diagram representing the overlap between super-enhancers found in vehicle- and TNF-treated SGBS cells as defined in A and Supplemental Figure 4H, respectively. (E) Enrichment of TNF-activated enhancers (defined in Fig. 1D based on increase in MED1 occupancy) near TNF-induced genes defined in Figure 1A. TNF-activated enhancers are divided into two groups based on whether they overlap with a TNF-induced super-enhancer (defined in supplemental Fig. S4H) or not. (F) Venn diagram representing the overlap between enhancers defined as “cluster enhancers” based on their association with an enhancer cluster, or “high-occupancy enhancer” based on a high MED1 occupancy (defined in Supplemental Fig. S4K,L). (G) Bar diagrams representing the fraction of MED1 sites in the different groups defined in F that display loss of MED1 in response to TNF as defined in Figure 1D. P-value: * < 2.2 × 10−16, Pearson's χ2 test. (H) Enrichment of different types of MED1 sites defined in F near TNF-repressed genes defined in Figure 1A. Enrichment was determined as the number of binding sites per gene within 20 kb from the TSS relative to the number of binding sites per constitutive gene. (I,J) UCSC Genome Browser screenshots of RNAPII (green) and MED1 (red) occupancy at gene loci of a typical super-enhancer-associated gene repressed by TNF (PPARG) (I) and a regular enhancer-associated gene (PRDX6) not affected by TNF (J). The purple bars indicate the position of the super-enhancers.
The RELA-mediated redistribution of cofactors from super-enhancers leads to the loss of 432 of the 933 basal super-enhancers, whereas 315 super-enhancers are established de novo (Fig. 4D; Supplemental Fig. S4H). Interestingly, the 501 super-enhancers that are maintained following TNF treatment are characterized by higher occupancy of RELA compared with super-enhancers that are lost (Supplemental Fig. S4I,J), indicating that RELA recruitment contributes to stabilization/maintenance of these super-enhancers following TNF treatment. Notably, TNF-activated constituents within the 315 TNF-induced super-enhancers are highly enriched in the vicinity of TNF-induced genes compared with regular TNF-activated enhancers (Fig. 4E), suggesting that RELA-directed super-enhancer formation plays an important role in induction of inflammatory gene expression. These results are in line with recent studies from the Plutzky/Bradner and Evans laboratories demonstrating that inflammation induces de novo formation of RELA-bound super-enhancers at inflammatory genes in human umbilical vein endothelial cells (HUVECs) and macrophages, respectively (Brown et al. 2014; Hah et al. 2015).
Due to the assignment method, super-enhancers are identified based on both the number of constituents as well as the MED1 occupancy at these. To assess the importance of these two parameters individually, we defined clusters based solely on the number of constituent enhancers (Supplemental Fig. S4K), as well as high-occupancy enhancers based solely on MED1 occupancy at individual enhancers (Supplemental Fig. S4L). We then overlapped the constituents of these two groups (Fig. 4F) and investigated the sensitivity to RELA-mediated MED1 redistribution. Interestingly, while high MED1 occupancy alone appears to determine sensitivity to redistribution at the individual enhancer (Fig. 4G; Supplemental Fig. S4M), clustering of enhancers is an additional strong determinant of sensitivity to cofactor redistribution at the level of gene repression (Fig. 4H). Thus, RELA appears to mediate cofactor redistribution selectively from high-occupancy enhancers largely independent of their super-enhancer status, but the greatest effect on gene repression appears when these enhancers are part of an enhancer cluster. We propose that these two levels allow RELA to, in an indirect fashion, specifically repress super-enhancer–associated genes like PPARG (Fig. 4I), without affecting regular enhancer-associated genes like PRDX6 (Fig. 4J).
TNF-regulated genes are sensitive to repression by BRD4-inhibition
The above analyses indicated that repression of genes by TNF in adipocytes primarily is driven by indirect RELA-mediated cofactor depletion from high-occupancy enhancers. Thus, we hypothesized that the same set of high-occupancy enhancers and their associated genes would also display increased sensitivity to other general perturbations of cofactor recruitment, and treated adipocytes with the JQ1 chemical inhibitor of BRD4 (Fig. 5A; Filippakopoulos et al. 2010). Consistent with our predictions, iRNA-seq analyses showed that TNF-repressed genes are more sensitive than constitutive genes to transcriptional repression by JQ1 treatment (Fig. 5B), and adipocyte super-enhancers are highly enriched in the vicinity of JQ1-repressed genes (Supplemental Fig. S5A). Interestingly, in TNF-treated adipocytes, the TNF-repressed genes are not further repressed by the combined treatment with JQ1 (Fig. 5C; Supplemental Fig. S5B). In contrast, TNF-induced genes are now highly sensitive to JQ1 treatment (Fig. 5C; Supplemental Fig. S5C), indicating that inhibition of BRD4 now primarily interferes with TNF-induced super-enhancers near these genes. This is supported by enrichment of TNF-induced super-enhancers primarily in the vicinity of genes repressed by JQ1 in TNF-treated adipocytes (Fig. 5D), whereas lost super-enhancers are primarily enriched in the vicinity of genes repressed by JQ1 in vehicle-treated adipocytes. Accordingly, maintained super-enhancers are enriched in the vicinity of genes repressed by JQ1 in both conditions (Fig. 5D). Thus, inflammatory genes like SERPINE2 (Fig. 5E), which are associated with TNF-induced super-enhancers, become sensitive to BRD4 inhibition, while genes such as ADH1B (Fig. 5F) that are associated with lost super-enhancers become insensitive to further repression by BRD4. Genes such as ZEB2 (Supplemental Fig. S5D), which are associated with a maintained super-enhancer, are subject to repression by JQ1 in both vehicle and TNF-treated cells. In summary, these results further support our model that repression of specific gene programs can be mediated by nonspecific perturbations of cofactor activity and that sensitivity of genes to these perturbations seems to be defined at least in part by association with high levels of cofactor binding at super-enhancers.
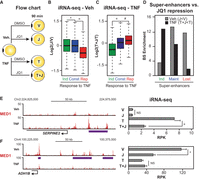
TNF-regulated genes are sensitive to repression by BRD4-inhibition. (A) Flow chart for treatment with JQ1. Human SGBS adipocytes were treated with vehicle or 10 ng/mL TNF in combination with DMSO or 500 nM JQ1 for 90 min before harvest of RNA for total RNA-seq. Changes in transcriptional activity were determined using the iRNA-seq pipeline. (B,C) Box plots illustrating log2 fold change in transcriptional activity following JQ1-treatment in vehicle- (B) and TNF-treated SGBS adipocytes (C). Green and red boxes represent genes that were determined to be induced and repressed, respectively, by TNF (V vs. T, FDR < 0.01). Blue boxes represent constitutive genes (FDR > 0.9; |log2FC < 0.2|). P-value: * < 2.2 × 10−16, ¤ = 7.10 × 10−4, # = 8.0 × 10−7, Wilcoxon rank-sum test. (D) Bar diagrams illustrating enrichment of super-enhancers that are induced, maintained, or lost (as defined in Fig. 4D) near genes repressed by JQ1 in vehicle- (V vs. J) or TNF-treated (T vs. T + J) adipocytes (FDR < 0.01). Enrichment was determined as the number of binding sites per gene within 20 kb of the TSS for repressed genes relative to the number of binding sites per constitutive gene. (FDR > 0.9; |log2FC| < 0.2). (E,F) (Left) UCSC Genome Browser screenshots of MED1 occupancy at gene loci of the gained super-enhancer-associated gene SERPINE2 (E) and the lost super-enhancer-associated gene ADH1B (F). The purple lines indicate position of super-enhancers. (Right) Bar diagrams illustrating transcription levels of SERPINE2 (E) and ADH1B (F) assessed as intron reads per kb per 10 M (RPK). FDR: * = 8.20 × 10−4, ¤ = 7.13 × 10−16, EdgeR.
Gene repression by TNF is highly cell-type specific
Previous studies have demonstrated that TNF stimulation in different cell types leads to induction of both cell-type–specific and shared gene programs, which is correlated with cell-type–specific and shared RELA binding, respectively (Jin et al. 2011). However, TNF-induced gene repression in this context has received little attention. Based on the central role of super-enhancers in gene repression by TNF in SGBS adipocytes, combined with results showing that super-enhancers are highly cell-type specific (Hnisz et al. 2013; Whyte et al. 2013; Loft et al. 2015), we hypothesized that TNF treatment should lead to acute repression of highly distinct gene programs in different cell types. To further investigate this, we analyzed publically available total RNA-seq and RNAPII ChIP-seq data from four additional human cell types (A549, IMR90, HeLa, and HUVEC) treated with vehicle or TNF for 1 h (Rao et al. 2011; Jin et al. 2013; Yang et al. 2013; Brown et al. 2014). iRNA-seq was used to determine acute changes in transcription based on total RNA-seq. We identified acutely induced and repressed genes in each cell type and determined the pathways enriched in these gene groups. Cross cell type comparison demonstrated that the gene programs and pathways repressed by TNF are indeed highly cell-type specific, whereas induction of inflammatory gene programs and pathways is more conserved across cell types (Fig. 6A,B; Supplemental Fig. S6A,B).
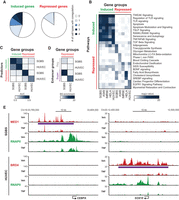
Gene repression by TNF is highly cell-type specific. (A,B) Publically available RNAPII ChIP-seq or total RNA-seq data from HUVEC, A549, IMR90, and HeLa cells treated with TNF or vehicle for 1 h were analyzed using iRNA-seq to assess ongoing transcription. The top 200/top 500 induced and repressed genes were identified based on the lowest P-value. (A) Pie chart representing the fraction of the top 200 genes induced (left) or repressed (right) by TNF treatment of SGBS adipocytes that are also among the top 200 induced and repressed genes in 1–4 additional cell types, respectively. P-value (Induced vs. Repressed): * = 1.06 × 10−14, Pearsons's χ2 test. (B) Heat map representing −log(P-values) for the top three enriched pathways for the top 500 genes induced and repressed by TNF in each cell type. Pathways among the top three enriched in more than one cell type are only represented once. (C,D) Publicly available BRD4 and RELA ChIP-seq data from HUVECs treated with TNF or vehicle for 1 h were analyzed using HOMER and compared with our SGBS data. (C) Heat map representing gene regulation by TNF in SGBS and HUVEC cell models based on RELA and cofactor occupancy. Induction models (green) are based on summarized distance-adjusted RELA occupancies as in Supplemental Figure S3E, and repression models (red) are based on summarized distance-adjusted occupancy scores as defined in Figure 3G and Supplemental Figure S6E. Models from both cell types were applied to predict the top 200 TNF-regulated genes in each cell type. (D) Heat map representing enrichment of regular enhancers and super-enhancers defined in SGBS and HUVEC cells within 20 kb of the TSS of genes repressed in either SGBS cells or HUVEC cells. Enrichment is relative to the number of sites within 20 kb of the TSS of constitutive genes (FDR/P > 0.9). (E) UCSC Genome Browser screenshots of RNAPII (green) and MED1 (red) occupancy at gene loci of two super-enhancer-associated genes, SOX18 and CEBPA, expressed and repressed by TNF in a cell-type–specific manner. The purple lines indicate the position of the super-enhancers.
To investigate to what extent our prediction models for repression apply to other cell types, we chose to further mine recently published data from TNF-induced changes in enhancer activity and RELA binding in HUVECs (Brown et al. 2014). We first identified regions with gain or loss of BRD4 binding following TNF stimulation of HUVECs and compared these with regions with gain or loss of MED1 occupancy following TNF stimulation of SGBS adipocytes. Interestingly, the cell-type–specific gene repression by TNF is paralleled by highly cell-type–specific loss of cofactor binding, whereas gain of cofactor binding is more conserved across the two cell types (Supplemental Fig. S6C). Furthermore, there is a relatively high enrichment of enhancers activated in HUVECs near genes induced by TNF in SGBS adipocytes and vice versa, whereas repressed enhancers are selectively enriched in the vicinity of genes that are specifically repressed in each cell type (Supplemental Fig. S6D). Importantly, prediction models based on RELA-binding following TNF treatment, and BRD4-binding in the basal state (i.e., similar to models defined by RELA and MED1 binding in SGBS cells [Fig. 3F,G]), are able to predict loss of BRD4 binding and gene repression by TNF in HUVEC cells (Supplemental Fig. S6E). Thus, our prediction models appear to be generally applicable. Interestingly, and in line with the results in Supplemental Figure S6D, RELA binding in SGBS cells can be used to predict gene activation in HUVECs and vice versa, whereas the repression models based on high cofactor and low RELA occupancy are highly cell-type specific (Fig. 6C). Importantly, basal super-enhancers are enriched specifically in the vicinity of genes repressed by TNF in each cell type (Fig. 6D), like CEBPA in adipocytes and SOX18 in HUVECs (Fig. 6E). Taken together, these results indicate that redistribution of cofactors is a general mechanism underlying specific repression of super-enhancer–associated cell identity genes by TNF.
Discussion
Here, we have extensively characterized the acute transcriptional response to TNF in human adipocytes and taken major steps toward understanding the mechanisms underlying acute gene repression by TNF in adipocytes as well as in other cell types. Using human SGBS adipocytes as a model system, we demonstrate that TNF induces a rapid genomic reprogramming leading to induction of inflammatory gene programs and repression of cell identity genes. This reprogramming, as evidenced by redistribution of MED1, BRD4, and enhancer transcription, appears to be driven by a prioritization of inflammatory enhancers at the expense of cell-type-specific enhancers. Interestingly, although RELA is required for both activation and repression of gene expression, RELA is not associated with repressed enhancers, demonstrating that repression is primarily indirect. Furthermore, in contrast to activation, repression occurs largely independent of changes in chromatin structure, suggesting that loss of cofactors precedes changes in chromatin accessibility at repressed enhancers.
The indirect repression by RELA appears to be mediated by redistribution of cofactors, such as MED1 and BRD4, from adipocyte enhancers to RELA-activated enhancers near proinflammatory genes. Similar mechanisms have recently been suggested by other studies to account for transcriptional repression following ligand activation of nuclear receptors (He et al. 2012; Guertin et al. 2014; Step et al. 2014), with redistribution of different coregulators like NCOA3, EP300, CREBBP, and MED1. Thus, nuclear receptors and RELA appear to share the ability to repress gene expression by eliciting a general redistribution of transcriptional coregulators. The fact that only a small subset of active enhancers and genes is repressed has so far remained poorly understood. Here, we show that cofactors are redistributed selectively from high-occupancy sites, thus providing a possible mechanism whereby RELA-mediated cofactor redistribution can elicit repression of a specific subset of active enhancers in adipocytes. In support of this mechanism, models based solely on the position of high-occupancy enhancers in the unstimulated state and RELA binding sites following acute TNF stimulation are able to predict the subset of enhancers and genes that are repressed by TNF. The accuracy in these predictions is similar to the accuracy with which RELA binding predicts acute enhancer and gene activation by TNF.
Interestingly, our results reveal that genes repressed by TNF are characterized by having a high number of MED1 enhancers in the vicinity of the TSS, which prompted us to investigate the role of super-enhancers in TNF-induced suppression of adipocyte genes. Super-enhancers are large clusters of enhancers with extremely high cofactor occupancy, and such enhancers have recently been described to be central drivers of gene programs that define cell identity (Hnisz et al. 2013; Whyte et al. 2013), thus extending previous work on the role of locus control regions in developmental- and cell-lineage–specific regulation of gene expression (Grosveld et al. 1987; Raich et al. 1990). While the basic mechanisms underlying the properties of these clusters of enhancers remain incompletely understood, super-enhancers have been shown to be more sensitive to perturbation by, e.g., knockdown of Mediator subunits or chemical inhibition of bromodomains than typical enhancers, possibly due to the cooperative and synergistic manner in which they recruit cofactors (Loven et al. 2013; Whyte et al. 2013). Interestingly, we find that super-enhancers and their associated genes are also more sensitive than regular enhancers to the perturbation that RELA-mediated cofactor redistribution constitutes. Importantly, we demonstrate that the sensitivity of super-enhancer-associated genes to perturbation is dual. First, cofactor loss is determined by the occupancy level at the individual enhancer largely independent of super-enhancer status; and second, gene repression is additionally determined by the clustering of high-occupancy enhancers into super-enhancers. We propose that these two levels reflect cooperative and synergistic recruitment of cofactors by multiple transcription factors at the individual enhancers and cooperative regulation of nearby genes by super-enhancer constituents, respectively. In line with this, we recently demonstrated that coactivator levels correlate with the number of transcription factors bound in specialized genomic loci designated transcription factor hotspots and that hotspots furthermore are enriched and appear to cooperate within super-enhancers (Siersbæk et al. 2014). We suggest that these two levels of cooperativity, and consequently also sensitivity to perturbation, allow super-enhancer-associated genes to be specifically and most dramatically repressed by RELA-mediated cofactor redistribution. Interestingly, and in line with recent studies by the Plutzky/Bradner and the Evans laboratories, TNF treatment in adipocytes also leads to de novo formation of RELA-bound super-enhancers, which appear to play an important role in the induction of the inflammatory gene program (Brown et al. 2014; Hah et al. 2015). The Plutzky/Bradner laboratories further demonstrated that inflammatory super-enhancers in endothelial cells are subject to repression by the BRD4 inhibitor JQ1 and that treatment with this inhibitor reduced TNF-induced atherogenic endothelial responses and atherosclerosis in vivo (Brown et al. 2014).
JQ1 has also been demonstrated to specifically target super-enhancer–associated genes driving cancer (Loven et al. 2013); thus, it presents as an attractive drug to target undesired transcriptional programs, but little attention has been given to how this affects the cell identity gene programs in normal cells in their basal state. We show here that the same set of cell identity genes that are repressed by TNF also display high sensitivity to JQ1 treatment, consistent with perturbation at the cofactor level as the major mechanism underlying gene repression by TNF. In TNF-treated adipocytes, JQ1 expectedly reduces inflammatory gene expression; however, TNF-repressed genes are not further affected by JQ1 treatment. Thus, while JQ1 effectively blocks inflammatory gene expression, it does not restore the expression of cell identity genes suppressed by inflammation.
Consistent with the selective repression of super-enhancer-associated adipocyte genes by TNF, we show that gene repression by TNF across five different human cell types is highly cell-type specific. As previously reported (Jin et al. 2011), gene induction by TNF is also to some extent cell-type specific; however, the overlap between TNF-activated genes in different cell types is much larger than the overlap between TNF-repressed genes. Furthermore, the induced genes are largely enriched in the same inflammatory pathways in all five cell types, whereas pathways enriched for repressed genes are highly cell-type specific. Further mining of comparable data from TNF-treated HUVECs, including application of our models, indicated that cell-type-specific gene repression by TNF is driven by cell-type-specific cofactor loss from high-occupancy enhancers not occupied by RELA. This suggests that selective cofactor redistribution from high-occupancy enhancers and the consequent repression of super-enhancer-associated genes is a general mechanism for gene repression by RELA. Our results indicate that this may contribute to repression of cell identity genes in adipocytes by the chronic low-grade inflammation associated with obesity. The molecular mechanisms underlying the selective cofactor redistribution in response to activation of RELA or activation of nuclear receptors is incompletely understood. We consider it likely that squelching (Meyer et al. 1989) of cofactors by these potent signal-dependent transcription factors plays a role; however, it is clear that formal proof of squelching requires more detailed molecular analyses. Furthermore, it remains to be understood why high-occupancy enhancers are significantly more sensitive to cofactor redistribution than low-occupancy enhancers.
In conclusion, our results propose a novel paradigm for NFκB-mediated repression, whereby NFκB selectively redistributes cofactors from high-occupancy enhancers, thereby specifically repressing super-enhancer-associated cell identity genes (Fig. 7). Recent studies have indicated that mRNAs compete for access to the ribosomes (Baumgartner et al. 2011), and gene repression associated with acute stress has been suggested to serve primarily to redirect translational capacity to newly synthesized mRNAs (Lee et al. 2011; Lackner et al. 2012). Thus, repression of cell identity genes in response to TNF may be beneficial in the context of fighting an infection by allowing different cell types to transiently decrease their respective specialized functions, thereby gaining translational capacity for, e.g., proinflammatory cytokines to support local professional immune cells in the defense response in the tissue. We further propose that cofactor redistribution selectively from high-occupancy enhancers is a general mechanism contributing to transcriptional repression associated with activation of signal-dependent transcription factors.
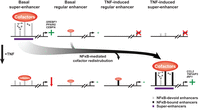
Model of transcriptional repression by NFκB-mediated cofactor redistribution. Following TNF treatment, cofactors redistribute to NFκB-activated enhancers, in particular, super-enhancers, near inflammatory genes. This redistribution occurs predominantly from the adipocyte basal super-enhancers that do not recruit RELA, thereby leading to specific repression of adipocyte genes associated with these super-enhancers.
Methods
Cell culture, differentiation, and treatment
SGBS cells were differentiated to adipocytes as previously described (Schmidt et al. 2011), and at day 10 of differentiation, cells were treated with 10 ng/mL human recombinant TNF (Life Technologies).
RNA extraction, cDNA synthesis, and quantitative real-time PCR
RNA extraction, cDNA synthesis, and quantitative real-time PCR (qPCR) were performed as previously described (Boergesen et al. 2011). Sequences of primers used for qPCR are included in Supplemental Table S2A.
RNA-seq
Following Isol-RNA lysis Reagent (5-Prime) extraction and EconoSpin (Epoc Life) column purification of total RNA, contaminant genomic DNA was removed by TURBO DNase digestion (Life Technologies), and ribosomal RNAs were removed using the Ribo-Zero Human/Mouse/Rat kit (Epicentre).
ChIP and ChIP-seq
ChIP experiments were performed according to standard protocols as described (Siersbæk et al. 2012). Antibodies used were RELA (C-20, sc-372, Santa Cruz), MED1 (M-255, sc-8998, Santa Cruz), BRD4 (A301-985A, Bethyl). Sequences of primers used for qPCR are included in Supplemental Table S2B.
DNase-seq
DNase-seq was performed on ∼10 million nuclei essentially as previously described (Siersbæk et al. 2011).
Library construction and sequencing
RNA-, DNase-, and ChIP-seq libraries were constructed according to the manufacturer's instructions (Illumina) as described (Nielsen and Mandrup 2014). Sequencing was performed on the Illumina HiSeq 1500 platform.
Lentiviral knockdown
shRNAi oligo DNA directed against RELA was cloned into pSicoR PGK Puro vectors (Addgene), and lentiviral particles were produced in Human Embryonic Kidney 293T cells as previously described (Nielsen et al. 2008). As a control, a shRNA nontargeting oligo (shScramble) was used. Sequences of shRNA oligos are included in Supplemental Table S2C. At day 6 of differentiation, SGBS adipocytes were transduced with shRNAi-expressing lentiviruses. Cells were used for further experiments on day 10 of differentiation.
Additional data sets
Total RNA-seq and RNAPII ChIP-seq data from TNF treatment of human SGBS adipocytes were previously published (Madsen et al. 2015) and are deposited to NCBI Gene Expression Omnibus (GEO) (accession GSE60462). An overview of accession numbers and experimental conditions for additional data sets used can be found in Supplemental Table S3.
Data analysis
Sequencing reads were mapped to the hg19 genome with STAR (Dobin et al. 2013) and further analyzed using iRNA-seq (Madsen et al. 2015) and HOMER (Heinz et al. 2010). Differential signal intensity in binding was determined using EdgeR (Robinson et al. 2010), and intersections between genomic position files were generated using BEDTools (Quinlan 2014). All ROC-curves were generated using the pROC package for R (Robin et al. 2011), and the UCSC Genome Browser (Kent et al. 2002) was used for data visualization. Further details about the data analyses are available in Supplemental Methods.
Data access
The RNA-seq, ChIP-seq, and DNase-seq data sets generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE64233.
Acknowledgments
We thank members of the Mandrup laboratory, in particular, Rasmus Siersbæk, for experimental support and fruitful discussions. Furthermore, we are grateful to Dr. Martin Wabitsch (University of Ulm, Germany) for providing SGBS cells and Per Sauerberg (Novo Nordisk A/S), for the kind gift of rosiglitazone. This work was supported by grants from the VILLUM Foundation to the VILLUM Center for Bioanalytical Sciences at the University of Southern Denmark, the Novo Nordisk Foundation, the Danish Independent Research Council | Natural Sciences, and the European Union Seventh Framework Programme (FP7/2007–2013) under grant agreement 311876: PATHWAY-27.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.188300.114.
- Received December 18, 2014.
- Accepted June 19, 2015.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








