
Genomic evidence for adaptation by gene duplication
- 1Department of Ecology and Evolutionary Biology, University of Michigan, Ann Arbor, Michigan 48109, USA;
- 2Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing 100101, China
- Corresponding author: jianzhi{at}umich.edu
Abstract
Gene duplication is widely believed to facilitate adaptation, but unambiguous evidence for this hypothesis has been found in only a small number of cases. Although gene duplication may increase the fitness of the involved organisms by doubling gene dosage or neofunctionalization, it may also result in a simple division of ancestral functions into daughter genes, which need not promote adaptation. Hence, the general validity of the adaptation by gene duplication hypothesis remains uncertain. Indeed, a genome-scale experiment found similar fitness effects of deleting pairs of duplicate genes and deleting individual singleton genes from the yeast genome, leading to the conclusion that duplication rarely results in adaptation. Here we contend that the above comparison is unfair because of a known duplication bias among genes with different fitness contributions. To rectify this problem, we compare homologous genes from the budding yeast Saccharomyces cerevisiae and the fission yeast Schizosaccharomyces pombe. We discover that simultaneously deleting a duplicate gene pair in S. cerevisiae reduces fitness significantly more than deleting their singleton counterpart in S. pombe, revealing post-duplication adaptation. The duplicates–singleton difference in fitness effect is not attributable to a potential increase in gene dose after duplication, suggesting that the adaptation is owing to neofunctionalization, which we find to be explicable by acquisitions of binary protein–protein interactions rather than gene expression changes. These results provide genomic evidence for the role of gene duplication in organismal adaptation and are important for understanding the genetic mechanisms of evolutionary innovation.
Despite the wide recognition of gene duplication as the primary source of new genes (Ohno 1970; Zhang 2003), the role of gene duplication in organismal adaptation is less clear (Zhang 2013). On the one hand, many believe that gene duplication facilitates adaptation, because the redundancy generated allows the evolution of new beneficial gene functions that are otherwise prohibited due to functional constraints (Ohno 1970; Crow and Wagner 2006; Flagel and Wendel 2009; Kondrashov 2012). On the other hand, unambiguous evidence for adaptation by gene duplication has been found in only a small number of cases (Zhang et al. 2002; Hittinger and Carroll 2007; Conant and Wolfe 2008; Deng et al. 2010; Nasvall et al. 2012; Ross et al. 2013). Furthermore, gene duplication may simply result in a division of ancestral functions into the daughter genes by complementary degenerative mutations, which need not promote adaptation (Force et al. 1999). The lack of unequivocal theoretical predictions and the paucity of existing empirical evidence warrant a genome-wide test of the adaptation by gene duplication hypothesis.
Let A1 and A2 be a pair of genes generated by the duplication of their progenitor gene, A, some time ago. If gene duplication facilitated adaptation, the fitness of the species that harbors the duplicates should have increased. Consequently, simultaneously deleting A1 and A2 from the species should cause a fitness drop that exceeds the drop caused by deleting A from an ancestral species prior to the duplication. Because it is impossible to directly analyze the ancestral species and the progenitor gene, one might use a randomly picked singleton gene in the extant species as a substitute for the progenitor gene of the duplicates (Dean et al. 2008). A recent analysis in the budding yeast Saccharomyces cerevisiae with this strategy found that the average fitness effect of deleting a pair of duplicate genes is similar to that of deleting a singleton gene, leading to the conclusion that gene duplication does not promote adaptation (Dean et al. 2008). However, because less important genes duplicate more often (i.e., genes with small fitness effects upon deletion duplicate more often than genes with large fitness effects) (He and Zhang 2006; Woods et al. 2013), the progenitors of the duplicate genes are expected to have smaller fitness effects than the singletons in S. cerevisiae, biasing the comparison.
To rectify this problem, we compare homologous genes from two species instead of unrelated duplicates and singletons from the same species. That is, in addition to S. cerevisiae, we used the fission yeast Schizosaccharomyces pombe, which diverged from S. cerevisiae ∼800 million years ago (Hedges et al. 2006), long enough for the occurrence of many gene duplication events in each lineage but not too long to render gene orthology inference unreliable. For each S. cerevisiae-specific gene duplication event, we used the single-copy S. pombe gene that is orthologous to both S. cerevisiae duplicates as a substitute for their progenitor. These trios (two S. cerevisiae genes and one S. pombe gene) are referred to as two-to-one orthologs. We similarly analyzed S. pombe-specific duplicate genes and their S. cerevisiae orthologs, which are referred to as one-to-two orthologs.
Results
A pair of duplicates has a higher fitness value than their singleton ortholog
Between S. cerevisiae and S. pombe, we identified 2487 one-to-one, 357 two-to-one, and 167 one-to-two orthologs, respectively. To directly compare the fitness effects of gene deletion between the two species requires that one-to-one orthologs should have similar fitness effects in the two yeasts (Fig. 1A), which is true on average (cf. I and V in Fig. 1E). Furthermore, these fitness effects are strongly correlated between the two yeasts (Pearson’s correlation r = 0.79; P < 10−300) (Supplemental Fig. S1).
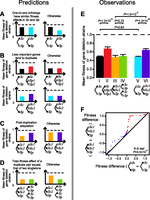
Predicted and observed fitness values of various gene deletion strains of S. cerevisiae (Sc) and S. pombe (Sp). (A–D) Predicted fitness values based on various hypotheses. (E,F) Observed fitness values. In all panels, one-to-one, two-to-one, and one-to-two orthologs between Sc and Sp are indicated by gene trees. A solid circle indicates the deleted gene(s) in the strain whose fitness is presented. The dashed line shows the corresponding wild-type fitness. (A) Expected fitness relationships when deleting one-to-one orthologs in Sc and Sp have similar fitness effects (left) and when they have different effects (right). (B) Expected fitness relationships when less important genes tend to duplicate (left) and when gene importance does not impact the probability of gene duplication (right). (C) The expected fitness relationship when gene duplication facilitates adaptation (left) and when it does not promote adaptation (right). (D) The expected fitness relationship under the theoretically maximal fitness gain after gene duplication (left) and that under a smaller fitness gain (right). (E) Observed mean fitness values of various gene deletion strains. The six columns contain 1620, 135, 156, 156, 1620, and 253 genes or gene pairs (left to right). There are fewer gene pairs in bar III compared with bar VI because the fitness values of some double deletion strains are unavailable. Error bars show one standard error and P-values are from Mann-Whitney U tests. (F) Quantile–quantile plot of fitness gains in Sc, relative to Sp. The x-axis shows the quantiles of the fitness difference between Sp and Sc strains lacking one-to-one orthologs, whereas the y-axis shows the corresponding quantiles of the fitness difference between Sp and Sc strains lacking two-to-one orthologs. Each dot represents a two-to-one orthologous trio (y-axis) and the corresponding one-to-one orthologous pair (x-axis) that has the closest quantile to the two-to-one trio. Red dots are duplicates with fitness gains (DWFG), whereas blue dots are duplicates without fitness gains (DWOFG).
The contention that less important genes tend to duplicate predicts that deleting a one-to-one ortholog from S. cerevisiae is more deleterious than deleting a one-to-two ortholog from S. cerevisiae (Fig. 1B, top), which is evident in our data (cf. I and II in Fig. 1E). A similar pattern is predicted (Fig. 1B, bottom) and observed (cf. V and VI in Fig. 1E) in S. pombe. Potential causes for this duplication bias were previously studied (He and Zhang 2006).
The hypothesis of adaptation by gene duplication predicts that simultaneously deleting a pair of duplicates in S. cerevisiae causes a larger fitness reduction than deleting their single-copy ortholog in S. pombe (Fig. 1C), which is indeed the case (cf. III and VI in Fig. 1E). This finding is robust to different computational criteria used (Supplemental Fig. S2). More rigorously, we constructed a quantile–quantile plot for the fitness differential of one-to-one orthologs and that of two-to-one orthologs (Fig. 1F). If the distribution of the fitness differential is identical between the two groups of genes, the dots should fall on the diagonal. The observed upward deviation from the diagonal suggests larger fitness gains after gene duplication than without gene duplication (Fig. 1F).
In theory, the maximal fitness contribution of a duplicate gene pair should on average equal that of two singleton genes that each approximates the progenitor of the duplicates in fitness contribution (Fig. 1D; see also Methods). This is indeed the case (P = 0.15, two-tail Kolmogorov-Smirnov test) (cf. III and IV in Fig. 1E), suggesting that the average amount of fitness gain after gene duplication approaches the theoretical maximum.
In principle, we could also test the hypothesis of adaptation by gene duplication in S. pombe using one-to-two orthologs. However, because of the paucity of genetic interaction data between S. pombe duplicates (Frost et al. 2012), we could not test the hypothesis at the genomic scale. Nevertheless, we found some individual cases that support the hypothesis of adaptation by gene duplication. Specifically, if a pair of S. pombe duplicates are synthetically lethal but their orthologous singleton gene in S. cerevisiae is nonessential, adaptation after duplication may be inferred. For example, S. pombe duplicates wee1 and mik1 are synthetically lethal (Lundgren et al. 1991; Yamaguchi et al. 1997; Katayama et al. 2002), whereas their S. cerevisiae singleton ortholog SWE1 is nonessential. These genes all encode protein kinases that function in cell cycle regulation. Similarly, S. pombe duplicates hrp1 and hrp3 are synthetically lethal (Walfridsson et al. 2005; Pointner et al. 2012), whereas their S. cerevisiae singleton ortholog CHD1 is nonessential. All three genes encode DNA helicases, functioning in the regulation of transcription elongation.
Neofunctionalization underlies post-duplication adaptation
There are four potential fates of duplicate genes (Zhang 2013): pseudogenization (Nei 1969; Ohno 1970; Zhang 2003), subfunctionalization (Ohno 1970; Hughes 1994; Force et al. 1999), neofunctionalization (Ohno 1970; Hughes 1994; He and Zhang 2005; Nasvall et al. 2012), and functional conservation (when an increased gene dose is beneficial) (Zhang 2003). In this study, subfunctionalization refers to a division of ancestral functions into daughter genes without functional improvement (He and Zhang 2005), whereas neofunctionalization includes acquisition of a new function or enhancement of an existing function, which may (Hughes 1994; He and Zhang 2005; Hittinger and Carroll 2007) or may not (Ohno 1970) occur in combination with subfunctionalization. The first two fates result in no improvement of organismal fitness, whereas the latter two could. If the fourth potential fate is the primary cause of our observation of duplication-associated adaptation (Kondrashov 2012), the gain of total expression for a S. cerevisiae duplicate pair (compared with their progenitor gene) should be larger for those duplicates with fitness gains (DWFG) (Fig. 1F, red dots; Supplemental Table S1) than for those without fitness gains (DWOFG) (Fig. 1F, blue dots; Supplemental Table S2). Using the orthologous S. pombe gene expression level as a proxy for the progenitor gene expression level (Qian et al. 2010), we calculated the ratio between the total expression level of a S. cerevisiae duplicate pair and that of its single-copy progenitor gene. As previously noted (Qian et al. 2010), this ratio is on average close to 1 instead of 2 (Fig. 2A), indicating that the expression level per gene generally halves after duplication. Further, we found no significant difference in this ratio between DWFG and DWOFG (Fig. 2A), suggesting that a gene-dose increase cannot be the primary reason for the observed fitness gain after duplication. The above comparison is appropriate because DWFG and DWOFG have no significant difference in their distributions among gene ontology (GO) categories. Thus, neofunctionalization remains the only possible prominent mechanism underlying the observed duplication-associated adaptation.
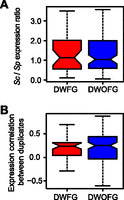
Comparison of gene expressions between duplicates with (DWFG) and without (DWOFG) fitness gains. (A) The ratio between the total expression level of a pair of S. cerevisiae (Sc) duplicates and the expression level of their S. pombe (Sp) ortholog is similar between DWFG and DWOFG (P = 0.79, two-tail Mann-Whitney U test). (B) Expression-level correlation between Sc paralogs across conditions and biological processes is similar between DWFG and DWOFG (P = 0.42, two-tail Mann-Whitney U test). In both panels, the bottom and top of each box are the first and third quartiles, and the band inside the box shows the median. The whiskers extend to the most extreme data within 1.5 times the interquartile range below the first quartile and above the third quartile.
Neofunctionalization depends on the effective gene age
Neofunctionalization may occur at the moment of gene duplication due to partial duplication of a gene or insertion of a duplicate gene into another locus that generates chimeric genes (Katju and Lynch 2003; Kaessmann et al. 2009). Alternatively, neofunctionalization can be a prolonged process that occurs gradually after gene duplication (He and Zhang 2005). To distinguish between these two scenarios, we examined the S. cerevisiae duplicates generated in different branches of the fungal species tree (Fig. 3A; Wapinski et al. 2007). We found the fitness gain of a duplicate pair to be independent of its phylogenetic age (Fig. 3B). Nevertheless, when we calculated the number of synonymous substitutions per synonymous site (dS) between a duplicate pair and compared dS between DWFG and DWOFG, we found DWFG to have significantly greater dS (Fig. 3C). Because the expression levels of DWFG and DWOFG are similar (Supplemental Fig. S3), the difference in dS is unlikely due to differential selections for preferred synonymous codons. Rather, because of potential among-gene variation in the frequency of gene conversion that homogenizes DNA sequences between duplicate genes, gene ages inferred from dS reflect the divergence time of a pair of duplicates better than those inferred from the phylogenetic tree. The greater dS for DWFG than DWOFG implies that neofunctionalization depends on the effective gene age, which is the time since the last gene conversion event rather than the absolute gene age.
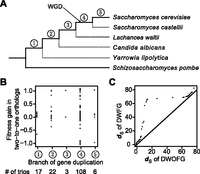
Gene duplication-associated adaptation tends to occur in duplicates with higher synonymous distances (dS). (A) A phylogeny of fungi allowing the separation of S. cerevisiae duplicates into five age groups. WGD indicates the timing of a known whole-genome duplication that occurred in the budding yeast lineage. (B) Fitness gain (shown by the y-axis of Fig. 1F) in two-to-one orthologs is independent of the phylogenetic timing of the duplication (Pearson’s correlation r = 0.03, P = 0.71; Spearman’s rank correlation ρ = 0.01, P = 0.91). Each dot represents a two-to-one trio. (C) Quantile–quantile plot of dS between S. cerevisiae duplicates of the DWFG group and that of the DWOFG group (P = 0.05, one-tail Kolmogorov-Smirnov test; P = 0.01, one-tail Mann-Whitney U test).
Gains of protein–protein interactions could explain the neofunctionalization
Neofunctionalization may be achieved by acquiring a new protein function or a new gene expression pattern. It was previously reported that duplicate genes evolve rapidly in expression pattern (Li et al. 2005; Thompson et al. 2013). If the inferred neofunctionalization after gene duplication is attributable to gains of new expression patterns, duplicates with fitness gains are expected to have larger expression differences and smaller expression correlations across cell cycle stages or media compared with those without fitness gains. This, however, was not observed (Fig. 2B). We also failed to find a larger number of unshared transcriptional factors regulating duplicate genes with fitness gains than those without fitness gains (P = 0.44, two-tail Mann-Whitney U test) (Supplemental Fig. S4). These observations suggest that neofunctionalization after gene duplication is not generally achieved by expression changes but by gains of new protein functions.
A potentially important mechanism of acquiring new protein functions is by establishing new protein–protein interactions (PPIs). We estimated the number of PPI gains after gene duplication by subtracting from the total number of distinct PPIs of a pair of S. cerevisiae duplicates the number of PPIs of their progenitor gene, which was assumed to equal the number of PPIs of the S. pombe ortholog. We found that the number of PPI gains is significantly greater for DWFG than for DWOFG (Fig. 4A, left). Because the PPI data are sparser in S. pombe than in S. cerevisiae, the presently known number of PPIs in S. pombe may not be a good estimate of that for the progenitor gene. We thus identified unshared PPIs between a pair of S. cerevisiae duplicates to infer the total number of subfunctionalized and neofunctionalized PPIs (Fig. 4A, middle) and used shared PPIs to infer the number of conserved ancestral PPIs (Fig. 4A, right). DWFG and DWOFG possess similar numbers of conserved ancestral PPIs (Fig. 4A, right); but, the total number of subfunctionalized and neofunctionalized PPIs is significantly greater for DWFG than for DWOFG (Fig. 4A, middle), suggesting that the gain of unshared PPIs is likely to be the underlying basis of the duplication-associated adaptation. Because subfunctionalization does not improve fitness, gain of new PPIs is most likely the molecular mechanism involved.
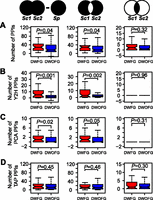
Gene duplication-associated adaptation is correlated with gains of binary protein–protein interactions (PPIs). (A) DWFG and DWOFG are compared for the difference between the total number of unique PPIs of a pair of S. cerevisiae (Sc) duplicates and that of the S. pombe (Sp) ortholog (left), the total number of unshared PPIs between a Sc duplicate pair (middle), and the number of shared PPIs between a Sc duplicate pair (right). Sc1 and Sc2 refer to the duplicate genes in S. cerevisiae, and Sp refers to their single-copy ortholog in Sp. The bottom and top of each box are the first and third quartiles, and the band inside the box shows the median. The whiskers extend to the most extreme data within 1.5 times the interquartile range below the first quartile and above the third quartile. (B–D) Same as A except that only those PPIs detected by yeast two-hybrid (Y2H) (B), protein-fragment complementation (PCA) (C), and tandem affinity purification (TAP) methods (D) are considered.
To further delineate the type of PPI gains underlying the duplication-associated adaptation, we divided all PPIs into three categories based on their method of detection: yeast two-hybrid (Y2H) (Uetz et al. 2000), protein-fragment complementation (PCA) (Tarassov et al. 2008), and tandem affinity purification (TAP) (Gavin et al. 2002). The first two methods identify binary PPIs, whereas the third identifies protein complex components. Compared with DWOFG, DWFG had significantly greater gains of PPIs and unshared PPIs identified by Y2H (Fig. 4B, left and middle) and PCA (Fig. 4C, left and middle), but not by TAP (Fig. 4D, left and middle). That both the in vitro Y2H method and the in vivo PCA method provide similar results strongly suggests that the gain of binary PPIs rather than protein complex membership is the underlying mechanism of the observed adaptation. Consistently, we found DWFG and DWOFG proteins to participate in similar numbers of unshared protein complexes (P = 0.47, two-tail Mann-Whitney U test) (Supplemental Fig. S5). Interestingly, there is no significant difference between DWFG and DWOFG in the number of nonsynonymous substitutions per nonsynonymous site (dN) or dN/dS between duplicates (Supplemental Fig. S6), suggesting that post-duplication neofunctionalization and adaptation may often involve a small number of nonsynonymous substitutions. Analysis of proteome data revealed no difference in phosphorylation (Supplemental Fig. S7) or glycosylation (Supplemental Fig. S8) gains between DWFG and DWOFG.
Similar adaptation patterns from different modes of duplication
The S. cerevisiae lineage experienced a whole-genome duplication (WGD) ∼100 million years ago, after the separation from the S. pombe lineage (Wolfe and Shields 1997). Some genes are strongly required to be balanced with some other genes in the genome in terms of gene dosage. Genes that require dosage balance tend not to duplicate successfully via individual gene duplication (IGD), but can be duplicated via WGD, because only the latter does not disrupt dosage balance. Given these considerations, one may predict that duplicates generated from WGD have a different adaptation pattern from duplicates generated from IGD (Papp et al. 2003; Wapinski et al. 2007; Qian and Zhang 2008). We compared the gains of fitness contribution from duplicates generated by WGD and those generated by IGD. Duplicates from WGD were obtained from Kellis et al. (2004), whereas the other duplicates were classified as IGD. We found no significant difference in the gain of fitness contribution between WGD and IGD duplicates (P = 0.84, two-tail Mann-Whitney U test). Further, WGD and IGD are not significantly different in terms of the gain of PPI (P = 0.16, two-tail Mann-Whitney U test), gain of protein complex membership (P = 0.49, two-tail Mann-Whitney U test), expression reduction (P = 0.91, two-tail Mann-Whitney U test), expression correlation between duplicates (P = 0.93, two-tail Mann-Whitney U test), gain of phosphorylation sites (P = 0.08, two-tail Mann-Whitney U test), and gain of glycosylation sites (P = 0.27, two-tail Mann-Whitney U test). Although these observations suggest that genes duplicated via WGD and via IGD do not differ in the patterns of adaptation and neofunctionalization, it remains possible that they are truly different, but the differences are undetectable here due to the limited sample sizes.
Discussion
In summary, using various functional and evolutionary genomic data, we discovered that simultaneously deleting a duplicate gene pair in S. cerevisiae reduces organismal fitness significantly more than deleting their singleton counterpart in S. pombe, providing unambiguous evidence for the importance of gene duplication in adaptation. We discovered that the duplicates–singleton difference in fitness effect is attributable to neither the gene dose increase nor expression pattern divergence after duplication. Rather, acquisitions of binary PPIs can explain the adaptation after gene duplication. Together, these findings establish, at the genomic scale, the role of gene duplication in organismal adaptation and its mechanistic basis.
Our findings, together with previous studies, suggest that duplicate genes that have been stably maintained in the genome for millions of years generally take the following evolutionary route. First, gene duplication occurs in an individual. Second, the duplication spreads in the population by genetic drift. Third, subfunctionalization by complementary degenerative mutations occurs relatively quickly such that both gene copies become necessary and hence are selectively maintained in the genome. Fourth, neofunctionalization gradually occurs, which improves organismal fitness. This scenario is consistent with what we previously suggested (He and Zhang 2005), although the earlier study had no data to establish the post-duplication adaptation.
We found that the duplication-associated adaptation often occurs by protein neofunctionalization, predominantly via acquiring new binary PPIs. Almost all cellular processes require PPIs, but new binary PPIs rarely emerge in the absence of gene duplication (Qian et al. 2011). Thus, gene duplication may have been disproportionately important in adaptations that need new PPIs compared with other adaptations, such as those requiring alterations of gene expression, which can occur by changing the modular cis-regulatory elements without gene duplication (Carroll 2008). Biophysical and evolutionary theories suggest that new PPIs begin as fortuitously beneficial, weak misinteractions that are gradually strengthened by positive selection (Kuriyan and Eisenberg 2007). It is likely that these weak interactions already existed prior to gene duplication but could not be strengthened due to antagonistic pleiotropy (Qian et al. 2012), which is subsequently resolved by gene duplication. The molecular evolutionary mechanism of this process is an important subject of future investigation.
S. cerevisiae and S. pombe diverged from each other ∼800 million years ago (Hedges et al. 2006) and are different in many biological aspects, such as cell division. These differences could have made the fitness incomparable between the two species. However, the fitness effects of gene deletion are strongly correlated between one-to-one orthologs of the two yeasts (Pearson’s r = 0.79, P < 10−300) (Supplemental Fig. S1). This high correlation is not due to those genes that have no fitness effect upon deletion in rich media, because the above correlation remains strong even measured by Spearman’s rank correlation (ρ = 0.71, P < 6 × 10−241). Further, although the biological network may have changed significantly between the two yeasts, we were able to test the hypothesis of adaptation by duplication without bias because of the use of one-to-one orthologs as a control (Fig. 1E,F). For instance, the hypothesis that the increased fitness contribution of S. cerevisiae duplicates is due to transfers of functions from singleton genes to the duplicates rather than neofunctionalization can be falsified because one-to-one orthologs between the two yeasts have similar fitness contributions.
One caveat of our analysis is that the fitness data of S. cerevisiae and S. pombe were compared for only one condition (rich media), due to data limitation. Consequently, we could detect only those duplicates whose fitness contributions under rich media increased; however, if this condition represents one randomly picked natural environment where yeast cells live, the result obtained here is unbiased. For example, we would also expect that when another randomly picked condition is examined, there is no significant difference between DWFG and DWOFG identified in that condition in terms of Sc/Sp expression ratio under that condition. Nevertheless, the total number of DWFG genes detected in at least one condition is likely to be substantially greater than that detected from the rich media only. Whether these predictions are correct and whether our findings made in two divergent unicellular fungi are generally true across eukaryotes and prokaryotes await further scrutiny.
Methods
Orthologous relations
We obtained one-to-one, two-to-one, and one-to-two orthologs between S. cerevisiae and S. pombe from the Fungal Orthogroups Repository (Wapinski et al. 2007). We also mapped duplication events onto the fungal species tree using the same resource.
Fitness values of gene deletion strains
It is a common practice in biology to compare the relative importance of a morphological, physiological, or behavioral characteristic between species. For example, sweet taste sensitivity is thought to be less important to carnivores than to herbivores (Zhao et al. 2010; Jiang et al. 2012), because of the scarcity of carbohydrates in meat. This statement compares the fitness effect of abolishing the sweet sensation in carnivores with that in herbivores. Similarly, the fitness effect of removing a gene from an organism can be compared between species under appropriate environments. We retrieved the fitness values of single gene deletion strains of S. cerevisiae from the data of Costanzo and colleagues (Costanzo et al. 2010). We obtained the fitness values of double gene deletion S. cerevisiae strains from a number of large-scale studies (Dean et al. 2008; DeLuna et al. 2008; Musso et al. 2008; Costanzo et al. 2010) and the Saccharomyces Genome Database (SGD; http://www.yeastgenome.org/). The detailed information about these data sets is provided in Supplemental Table S3. To make the fitness values comparable among different studies, we first normalized the fitness values among studies. Except for the data of Costanzo et al. (2010), we multiplied a constant for all the fitness values obtained in a study to make the average fitness of single gene deletion strains in the study identical to that of Costanzo et al. (2010). If a pair of duplicates is reported to be synthetically lethal, the double gene deletion strain has a fitness of 0. We retrieved the synthetic lethality data in S. pombe from BioGRID (http://thebiogrid.org/).
Because the fitness values of the S. cerevisiae gene deletion strains were quantified in glucose rich media (Supplemental Table S3), we analyzed the glucose rich medium
(YES) fitness data of S. pombe single gene deletion strains. Because virtually no double deletions of duplicate genes have been conducted in the systematic
survey of genetic interactions in S. pombe (Frost et al. 2012), we did not analyze S. pombe double deletion strains except for the aforementioned synthetic lethality data. The fitness values of all single gene deletion
strains of S. pombe were simultaneously measured by the Bar-seq method (Smith et al. 2009) using next-generation sequencing of the unique barcodes inserted into each deletion strain (Han et al. 2010). The numbers of barcode reads were recorded at six time points for both upstream barcodes and downstream barcodes (Han et al. 2010). If no read was observed for a strain at the starting time, we assumed 0.5 read in the calculation. Let Nij be the number of reads at time point i for strain j and 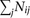 be the total number of reads for all strains at time point i. We then transformed the read number by
be the total number of reads for all strains at time point i. We then transformed the read number by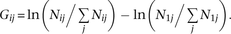 In theory, Gij should regress linearly with the number of generations of population growth, with the slope of the linear regression b = ln wj, where wj is the fitness of strain j relative to the population as a whole. We thus estimated wj. We further estimated the standard error of b (SEb). If
In theory, Gij should regress linearly with the number of generations of population growth, with the slope of the linear regression b = ln wj, where wj is the fitness of strain j relative to the population as a whole. We thus estimated wj. We further estimated the standard error of b (SEb). If 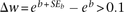 , the fitness estimate was considered unreliable and discarded. If the fitness estimates from the upstream and downstream
barcodes were both available, we used the average of them; if only one of them was available, we used the available one. Finally,
we included all essential genes and assigned a fitness of zero to each strain where an essential gene is deleted. Together,
we estimated the fitness effects of 3744 single gene deletions in S. pombe. We also changed the Δw cutoff to other values (0.05, 0.2, and 1) and found our conclusion to be qualitatively unaltered (Supplemental Fig. S2).
, the fitness estimate was considered unreliable and discarded. If the fitness estimates from the upstream and downstream
barcodes were both available, we used the average of them; if only one of them was available, we used the available one. Finally,
we included all essential genes and assigned a fitness of zero to each strain where an essential gene is deleted. Together,
we estimated the fitness effects of 3744 single gene deletions in S. pombe. We also changed the Δw cutoff to other values (0.05, 0.2, and 1) and found our conclusion to be qualitatively unaltered (Supplemental Fig. S2).
For each two-to-one orthologous trio, we calculated the fitness difference between the S. pombe strain in which the single-copy gene is deleted and the S. cerevisiae strain in which the corresponding pair of duplicates are simultaneously deleted. Consistent with the preceding cutoff used, we classified the trio into the group of duplicates with fitness gain (DWFG) when the fitness difference exceeds 0.1; otherwise, we classified it into the group of duplicates without fitness gain (DWOFG).
We examined whether the fitness contribution of an average pair of duplicate genes reaches its theoretical maximum. In theory, the fitness contribution of an average duplicate pair should not exceed that of two average singletons. Because the progenitors of duplicate genes have on average smaller fitness contributions than singletons, we need to identify pairs of singletons that have similar fitness contributions as the progenitors of the pair of duplicates. For each two-to-one orthologous trio, we found the fitness effect of deleting the single-copy gene in S. pombe (wd). We ranked all the one-to-one orthologs based on their fitness effects in S. pombe, and identified 10 S. pombe genes (five larger and five smaller than wd) whose fitness effects are the closest to wd. We randomly picked a pair among the 10 chosen S. pombe genes whose one-to-one orthologs in S. cerevisiae have been simultaneously deleted with an existing fitness effect estimate in the data of Costanzo et al. (2010). This fitness effect is the expected maximal fitness effect of the pair of S. cerevisiae duplicates after adaptive evolution.
Synonymous (dS) and nonsynonymous (dN) distances
For each two-to-one trio, we obtained the coding sequences of the S. cerevisiae duplicates from SGD. We aligned the DNA sequences following ClustalW alignment (Thompson et al. 1994) of their protein sequences. We used the program CODEML in the PAML package (Yang 2007) to estimate dS and dN as well as the dN/dS ratio between the S. cerevisiae duplicates. Note that estimates of dS typically have large estimation errors when they exceed 1, which makes it more difficult to detect a difference in dS between groups of genes. However, when a significant difference is detected, it is likely to be true.
Gene expression levels
We compared the gene expression levels between S. cerevisiae and S. pombe by following a previous study (Qian et al. 2010). We obtained the gene expression levels in S. cerevisiae and S. pombe measured by RNA-seq (Nagalakshmi et al. 2008; Wilhelm et al. 2008) and multiplied the numbers of reads in S. cerevisiae by 1.33 to make the mean expression levels of one-to-one orthologous genes of S. cerevisiae and S. pombe equal. Because genes with small numbers of reads tend to have large measurement errors, we excluded genes with fewer than 20 sequencing reads.
We further analyzed the S. cerevisiae transcriptomic data from 40 biological processes or conditions, including the time series of cell cycles, sporulation, and responses to various stresses. The transcriptomic data were obtained by microarrays and were retrieved from Kafri et al. (2005). We estimated the expression similarity between a pair of S. cerevisiae duplicates by calculating their Pearson’s correlation of expression levels in each process or condition and then averaging the correlation coefficients.
Yeast transcriptional regulation, protein–protein interaction (PPI), protein complex, post-translational modification, and gene ontology data
We retrieved transcriptional regulation data from MacIsaac et al. (2006) (orfs_by_factor_p0.001_cons0.txt), which reanalyzed the ChIP-chip data from Harbison et al. (2004). We obtained the PPI data from SGD (BIOGRID-ORGANISM-Saccharomyces_cerevisiae-3.1.72.tab2.txt). If a record is categorized as “physical” in “experimental system type,” it is considered a PPI. PPIs are further classified into yeast two-hybrid (Y2H), protein-fragment complementation assay (PCA), and tandem affinity purification (TAP), based on the term “experimental system.” We obtained protein complex data from SGD (go_protein_complex_slim.tab). We retrieved phosphorylation data from Data set S1 in Beltrao et al. (2009) and glycosylation data from Supplemental Table S1 in Zielinska et al. (2012). Gene ontology enrichment analyses were performed using FatiGO (Al-Shahrour et al. 2004).
Acknowledgments
We thank Xiaoshu Chen, Calum Maclean, Jian-Rong Yang, and three anonymous reviewers for valuable comments. This work was supported by research grants R01GM067030 and R01GM103232 from the US National Institutes of Health to J.Z.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.172098.114.
- Received January 6, 2014.
- Accepted April 23, 2014.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








