
Systematic mapping of occluded genes by cell fusion reveals prevalence and stability of cis-mediated silencing in somatic cells
- Timothy J. Looney1,9,
- Li Zhang1,9,
- Chih-Hsin Chen1,9,10,
- Jae Hyun Lee1,
- Sheila Chari1,
- Frank Fuxiang Mao1,2,
- Mattia Pelizzola3,
- Lu Zhang4,
- Ryan Lister3,
- Samuel W. Baker1,
- Croydon J. Fernandes1,
- Jedidiah Gaetz1,
- Kara M. Foshay1,
- Kayla L. Clift1,
- Zhenyu Zhang1,
- Wei-Qiang Li2,
- Eric J. Vallender5,
- Ulrich Wagner6,
- Jane Yuxia Qin1,
- Katelyn J. Michelini1,
- Branimir Bugarija1,
- Donghyun Park1,
- Emmanuel Aryee1,
- Thomas Stricker7,
- Jie Zhou7,
- Kevin P. White7,
- Bing Ren6,
- Gary P. Schroth4,
- Joseph R. Ecker3,
- Andy Peng Xiang2 and
- Bruce T. Lahn1,2,8,10
- 1Department of Human Genetics, University of Chicago, Howard Hughes Medical Institute, Chicago, Illinois 60637, USA;
- 2Center for Stem Cell Biology and Tissue Engineering, Sun Yat-sen University, Guangzhou 510080, China;
- 3Genomic Analysis Laboratory, The Salk Institute for Biological Studies, La Jolla, California 92037, USA;
- 4Illumina Inc., Hayward, California 94545, USA;
- 5New England Primate Research Center, Harvard Medical School, Southborough, Massachusetts 01772, USA;
- 6Department of Cellular and Molecular Medicine, University of California San Diego, La Jolla, California 92093, USA;
- 7Institute for Genomics and Systems Biology, Department of Human Genetics, University of Chicago and Argonne National Laboratory, Chicago, Illinois 60637, USA;
- 8Taicang Institute for Life Sciences Information, Taicang 215400, China
-
↵9 These authors contributed equally to this work.
Abstract
Both diffusible factors acting in trans and chromatin components acting in cis are implicated in gene regulation, but the extent to which either process causally determines a cell's transcriptional identity is unclear. We recently used cell fusion to define a class of silent genes termed “cis-silenced” (or “occluded”) genes, which remain silent even in the presence of trans-acting transcriptional activators. We further showed that occlusion of lineage-inappropriate genes plays a critical role in maintaining the transcriptional identities of somatic cells. Here, we present, for the first time, a comprehensive map of occluded genes in somatic cells. Specifically, we mapped occluded genes in mouse fibroblasts via fusion to a dozen different rat cell types followed by whole-transcriptome profiling. We found that occluded genes are highly prevalent and stable in somatic cells, representing a sizeable fraction of silent genes. Occluded genes are also highly enriched for important developmental regulators of alternative lineages, consistent with the role of occlusion in safeguarding cell identities. Alongside this map, we also present whole-genome maps of DNA methylation and eight other chromatin marks. These maps uncover a complex relationship between chromatin state and occlusion. Furthermore, we found that DNA methylation functions as the memory of occlusion in a subset of occluded genes, while histone deacetylation contributes to the implementation but not memory of occlusion. Our data suggest that the identities of individual cell types are defined largely by the occlusion status of their genomes. The comprehensive reference maps reported here provide the foundation for future studies aimed at understanding the role of occlusion in development and disease.
The hallmark of multicellular life is the presence of diverse cell types within a single organism that bear identical genomes but disparate gene expression patterns (Waddington 1940, 1957). A longstanding but unresolved question is how the same set of genetic instructions in the genome of an organism could give rise to such a wide variety of stable gene expression patterns found in different cell types. Emerging literature suggests the involvement of not only diffusible factors such as transcriptional activators and repressors that act in trans to regulate expression, but also chromatin marks such as DNA methylation and histone modifications that act in cis to modulate how genes respond to trans-acting factors (Blau and Baltimore 1991; Goldberg et al. 2007; Reik 2007).
However, trans-acting factors and cis-acting chromatin elements can influence each other in highly dynamic and complex ways, making it exceedingly difficult to determine which mechanism is causally responsible for a gene's expression status. For example, a chromatin mark could be consistently associated with the silent state of a gene, and when this mark is disrupted, the gene becomes active. This would appear to support a causal role of this mark in silencing the gene. However, an alternative scenario is that the chromatin mark is recruited by a transcriptional repressor to act as a downstream effector of silencing (Hublitz et al. 2009), such that upon disappearance of the repressor, the gene would become derepressed along with the erasure of the recruited chromatin mark (Melamed 2008; Perissi et al. 2010). In this scenario, even though the mark is involved in silencing, it is not the cause. Rather, the cause lies in the presence or absence of the repressor in trans.
Until recently, there had not been a robust method to assess whether the expression status of a gene is causally engendered by the cis-acting chromatin state of the gene or by the trans-acting cellular milieu surrounding the gene. To this end, we recently pioneered the use of cell fusion to probe whether the expression of a gene is mediated causally by cis or trans mechanisms (Lee et al. 2009). Specifically, we fused two different cell types and searched for genes that are silent in one fusion partner but active in the other partner, all within the same fused cell. Here, the active copies of the genes indicate the presence of trans-acting transcription factors for these genes in the fused cell. Logically then, the presence of both active and silent copies of the same genes, all exposed to the same trans milieu of the fused cell, indicates that they possess different chromatin states in cis, such that they respond differently to the transcription factors in the milieu—the active copy being responsive while the silent copy being refractory.
Two schemes can be considered here. First, certain self-perpetuating chromatin modifications are made to the silent copy during development, which render it refractory to transcription factors in the milieu. Second, it is the active copy, rather than the silent copy, that is subject to chromatin alterations that make it responsive to transcription factors. This could happen if initiating factors present transiently during development are required to turn on a gene by making its chromatin accessible to transcription factors, and the gene then remains active after the disappearance of the initiating factors. Existing data provide stronger support for the first scheme. Monoallelic silencing in mammals (e.g., X inactivation and imprinting) are classic examples of two copies of a gene showing differential expression within the same cellular environment. Current evidence has attributed the phenomenon to chromatin-mediated repression of the silent copy rather than the presence of initiating factors that specifically mark the active copy. Compelling evidence also comes from our recent studies. In one study (Foshay et al. 2012), we showed that in embryonic stem cells (ESCs), virtually all genes are responsive to heterologous transcription factors introduced by cell fusion (unlike somatic cells in which many genes are refractory to heterologously introduced transcription factors). This argues that the initial stage of development (as represented by ESCs) is characterized by global responsiveness to transcription factors, and some of the responsive genes would later become refractory as a consequence of differentiation. Such a scenario obviates the need for the transient presence of initiating factors. In a second study (Gaetz et al. 2012), we first used cell fusion to identify a set of genes in mouse fibroblasts that are refractory to transcription factors. We then introduced BACs containing some of these genes into the same mouse fibroblasts. We found that the majority of the BAC-carried genes were actively expressed while their endogenous counterparts remained silent. This argues that the silencing of the endogenous genes is due to the presence of a repressive mechanism acting in cis of these genes, rather than the absence of initiating factors for these genes in trans, because the latter scenario is not consistent with the BAC-carried genes being actively expressed. Based on the above arguments, we have come to refer to genes refractory to transcription factors as being “cis-silenced” or “occluded” to indicate the likelihood that repressive chromatin is the responsible agent (Lee et al. 2009). However, the possibility of initiating factors existing transiently during development to set some genes in a responsive state remains a formal possibility.
We also identified a class of genes that are silent before fusion but become activated after fusion. Our interpretation is that these genes are competent to respond to the activating milieu of the fused cell, and that their silencing prior to fusion is due to the lack of a transcriptionally supportive milieu in trans (i.e., absence of activators or presence of repressors). We termed these genes “trans-silenced” or “activatable” (Lee et al. 2009).
We hypothesized (Lahn 2011) and later demonstrated (Gaetz et al. 2012) that the occlusion of lineage-inappropriate genes in somatic cells plays an essential role in the restriction of somatic cell fate. Yet, our previous proof-of-concept studies were not performed on a sufficiently comprehensive scale to offer insight on genome-wide patterns of occlusion or its relationship to chromatin structure. Here, we report a systematic effort to map occluded genes in the genome. The resulting map, along with various whole-genome chromatin maps that we also generated, reveals the striking prevalence and stability of occluded genes, and sheds light on genome-wide genetic and chromatin patterns underlying occlusion.
Results
Prevalence of occluded genes in somatic cells
We sought to construct a map of occluded genes in a female mouse tail fibroblast line of 129 background (designated 129TF). To ascertain whether a silent gene in 129TF is occluded or activatable, we need to fuse 129TF to another cell type in which the gene is expressed. Given that only a small subset of silent genes in 129TF is expressed in another cell type, a systematic search for occluded genes in 129TF requires fusion to a wide variety of cell types that collectively express a large fraction of genes in the genome. To this end, we fused 129TF to 12 different rat cell types representing all three germ layers, including Schwann cells (designated S16), astrocytes (D1), proliferating neuroblastoma cells (B35), differentiated nonproliferating neuroblastoma cells (B35D), fibroblasts (R1A), skeletal myoblasts (L6), cardiomyoblasts (H9), osteosarcoma cells (UMR), chondrocytes (IRC), basophilic leukemia cells (RBL), intestinal epithelial cells (IEC), and hepatocytes (BRL). We chose to fuse mouse cells with rat cells because sequence divergence between these two species can be exploited to distinguish mouse and rat transcripts in fused cells. Furthermore, the evolutionary relatedness between mouse and rat minimizes potential incompatibilities of their regulatory networks.
Fusion was induced by polyethylene glycol (PEG). Fused cells were purified by drug selection, then cultured for 6–7 d before harvesting. Our previous work showed that gene expression patterns in fused cells should become stabilized in 3–4 d post-fusion (Lee et al. 2009). RNA-seq was then performed on fusion samples and non-fused parental cells, yielding 25 RNA-seq data sets corresponding to 129TF, 12 samples of non-fused rat cells, and 12 samples of fused cells. The quality of the RNA-seq data was confirmed by technical replicates and biological replicates (Supplemental Fig. S1).
We built a reference mRNA database of 16,053 mouse–rat orthologous gene pairs, and mapped RNA-seq data to this reference. For fused cells, we only mapped RNA-seq reads that uniquely aligned to one of the two species, taking advantage of mouse–rat sequence divergence. The number of reads matching each gene was then normalized to gene size and the total number of mapped reads in the RNA-seq data to produce a quantitative measure of expression level for the gene in the form of the number of transcripts per diploid genome (TPG). For fused cells, this measure corrects for the fusion ratio of the two partners. Based on several considerations (see Supplemental Text), we deemed a gene expressed if it produces ≥2 TPG, and silent if <0.2 TPG.
We defined the following categories of genes based on their expression levels in fused and non-fused cells. “Informative silent” genes in 129TF are defined as silent in 129TF, but expressed in at least one of the rat cells used to fuse with 129TF. The expression patterns of these genes in fused cells are likely to be informative regarding whether the 129TF copies are occluded or activatable. “Occluded” genes in 129TF are defined as silent in 129TF both before and after fusion, while expressed in the fusion partner both before and after fusion (Supplemental Table S1). “Activatable” genes in 129TF are defined as silent in 129TF before fusion but expressed after fusion, with the additional condition that the expression level from the 129TF genome in fused cells is >10% of the expression level from the fusion partner (Supplemental Table S1). “Extinguished” genes in 129TF are defined as expressed in 129TF before fusion but silent after fusion. Extinction is not a focus of our analysis. Similar criteria were used to define occluded, activatable, and extinguished genes in rat cells (Supplemental Table S1).
The 12 fusions between 129TF and rat cells each uncovered 85–246 occluded genes in 129TF (average 155), while the numbers of activatable and extinguished genes in 129TF are lower (Fig. 1A). A very small number of genes are occluded in some fusion(s) but activatable in other fusion(s) (Supplemental Table S1). These genes were removed from analysis because they likely represent noise in the assay (see Supplemental Text). In total, there are 790 occluded (Supplemental Table S2), 190 activatable (Supplemental Table S3), and 236 extinguished genes (Supplemental Table S4) in 129TF based on at least one of the fusions. By our definition, none of the occluded genes are activatable in any of the fusions, and similarly, none of the activatable genes are occluded in any fusion. We also tallied the numbers of occluded, activatable, and extinguished genes in rat cells (Fig. 1B). In total, 730 are occluded, 346 activatable, and 850 extinguished in at least one of the rat cell types as revealed by fusion with 129TF. We validate a subset of occluded and activatable genes in 129TF by the RT-PCR-seq assay (Supplemental Fig. S2). We also showed, using a bioinformatic chromosome loss analysis, that occlusion is not due to chromosome loss in fused cells (Supplemental Fig. S3).
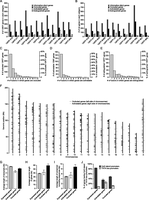
Identification of occluded genes and characterization of their structural features. (A,B) Number of informative silent, occluded, activatable, and extinguished genes in 129TF (A) or in rat cells (B) identified by each fusion. (C–E) Histograms of 129TF genes based on the number of fusions in which a given gene is occluded (C), activatable (D), or extinguished (E). (F) Genomic distribution of occluded and activatable genes in 129TF. (G–I) Average lengths of transcripts (G), average genomic lengths (H), and average lengths of conserved noncoding sequences (I) for expressed, occluded, and activatable genes. Genomic length is the distance between the annotated TSS to the annotated end of the gene body, per data provided by the UCSC Genome Browser. Noncoding sequence of a gene is the sequence from 2 kb upstream of TSS to the end of the gene body, minus all coding regions. (J) Percent of expressed, occluded, and activatable genes that possess either CpG island promoters or TATA box promoters.
For occluded genes in 129TF, 361 (45%) were shown to be occluded in more than one fusion (Fig. 1C). The same is true for 43 (22%) of activatable genes (Fig. 1D) and 110 (46%) of extinguished genes (Fig. 1E) in 129TF. The total number of informative silent genes in 129TF based on all the fusions is 1794, which means that ∼55% of informative silent genes are categorized as either occluded or activatable by the cell fusion assay. Given that RNA-seq revealed 6171 silent genes in 129TF, we estimate that the total number of occluded genes in 129TF could be in the range of 2000–3000, and similar numbers are estimated for each of the rat cell types. Taken together, our data show that occlusion is a highly prevalent phenomenon, affecting a sizeable fraction of silent genes across a wide variety of somatic cell types.
Structural and functional features of occluded genes
We examined the genome distribution of occluded and activatable genes in 129TF (Fig. 1F). They appear randomly distributed across the genome (Supplemental Fig. S4). We next carried out a number of comparisons among three categories of genes: expressed, activatable, and occluded (the latter two will sometimes be referred to as the silent categories). The average transcript lengths are similar across these three categories (Fig. 1G). For genomic length, defined as the genomic distance from the transcription start site (TSS) to the end of gene body, occluded genes are ∼50% longer than expressed or activatable genes (Fig. 1H). To explore whether longer genomic gene lengths correspond to more regulatory sequences, we examined conserved noncoding sequence (CNS) in each category, and found that occluded genes indeed have significantly longer CNSs than the other two categories (Fig. 1I). To the extent that CNSs are a proxy for cis-regulatory elements, this observation suggests that occluded genes tend to have more complex regulation than expressed or activatable genes, though there can be other interpretations.
It has been shown that different functional classes of genes tend to have different types of promoters. For example, housekeeping genes are highly enriched for CpG island promoters and rarely have TATA box promoters, whereas tissue-specific genes are more likely to have TATA box promoters (Sandelin et al. 2007; Mohn and Schubeler 2009). We computationally predicted whether the three categories of genes in 129TF possess CpG island promoters or TATA box promoters. The great majority of expressed genes have CpG island promoters, perhaps because a large fraction of expressed genes are housekeeping genes (Fig. 1J). Comparison between occluded and activatable categories showed that occluded genes are much more likely to have CpG island promoters than TATA box promoters whereas activatable genes are almost equally likely to have these two types of promoters (Fig. 1J). This observation points to systematic differences in promoter architecture between occluded and activatable genes.
Gene ontology (GO) analysis (Martin et al. 2004) showed that occluded genes in 129TF are highly enriched for regulatory functions, and include many key factors controlling important cellular and developmental processes as well as members of essentially every major signaling and transcription factor family (see Supplemental Text). In contrast, activatable genes are enriched for immune system functions, transport, response to stimulus, and various metabolic processes. That occluded genes are enriched for important developmental regulator is consistent with the role of occlusion in safeguarding the transcriptome identities of cells (Lahn 2011; Gaetz et al. 2012).
We scanned the proximal regulatory space (1 kb from TSS) of occluded, activatable, and expressed genes for the presence of putative transcription factor binding sites of ∼900 transcription factors in the form of position weight matrices obtained from the TRANSFAC database. A number of putative binding sites are differentially enriched between classes (Supplemental Table S5). Of particular note, activatable genes are enriched for binding sites of FOXO family members and FAC1 (BPTF) as compared with occluded genes (see Methods). FOXO factors are known to have roles in stress response, and are thought to act as “pioneering” factors due to their ability to bind targets within closed chromatin (Cirillo et al. 2002; Lee et al. 2005; Friedman and Kaestner 2006). FAC1 is a component of the nucleosome remodeling factor which catalyzes nucleosome displacement (Wysocka et al. 2006).
Robustness of occlusion
We examined the correlation of global gene expression profile between 129TF and its fusion partner, and between the same cell type before and after fusion. Several patterns emerged. First, in all the fusions, the correlation between 129TF and its rat partner is greater after fusion than before fusion (Fig. 2), indicating that the fusion has indeed equalized gene expression between the two cell types to some extent. Importantly, however, the correlation between 129TF before and after fusion, or between the same rat cell type before and after fusion, is greater than the correlation between 129TF and its rat partner after fusion (Fig. 2). This means that even in fused cells, the 129TF transcriptome is more similar to non-fused 129TF than to the rat transcriptome in the same fused cells. The high degree of transcriptome correlation for a cell type before and after fusion, coupled with the preponderance of occluded genes in all the somatic cell types examined, argues that the transcriptome identities of somatic cell types are substantially preserved despite fusion to other somatic cell types. It can thus be concluded that trans-acting factors expressed at near-physiological levels in somatic cells are insufficient to confer transcriptional identities of individual cell types. Rather, cis-acting mechanisms independent of trans-acting factors must play a major role.

Strong preservation of original gene expression patterns in fused cells. In 12 fusions between 129TF and rat cells examined at the population level (first 12 data groups) and one fusion between 129TF and R1A examined at the clone level (last data group), the correlation of global gene expression levels before and after fusion for either fusion partner is greater than the correlation between the two partners after fusion, indicating a strong effect of cis-mediated regulation on global gene expression. The average fusion ratio between 129TF and rat cells is indicated in parentheses for each fusion examined at the population level.
It has been suggested that the cell type contributing more nuclei in a fusion tends to dominate the final phenotype of fused cells (Palermo et al. 2009). This raises the possibility that occluded genes in the less numerous nuclei might become reprogrammed upon exposure to sufficient quantities of transcription factors originating from the more numerous nuclei. However, an alternate possibility is that occlusion in both partners is unchanged by cell fusion, and it is simply the ratio of the products produced by the two genomes that determine the final phenotype of the fused cell. Our investigation of the nuclear ratio in the various fusions supports the latter possibility (see Supplemental Text).
Thus far, all of the RNA-seq analyses were performed on populations of fused cells harvested at 6–7 d post-fusion. There are several questions to this approach. First, it is not known if genes found to be occluded at 6–7 d post-fusion would remain occluded over longer periods in culture. Second, given that many fused cells might not have undergone cell division, it is unclear if occlusion would survive DNA replication. Lastly, it is not clear whether the behavior of fused cells at the population level would be similar to that at the single cell level. To address these questions, we derived a clonal line from a single fused cell in the 129TF–R1A fusion. This line, designated 129TF × R1A (clone1), was cultured for ∼1 mo before being subjected to RNA-seq, at which time many rounds of cell divisions had taken place. Importantly, we found that in the clone, as in the population, the correlation of 129TF (or R1A) gene expression profile before and after fusion remained much greater than the correlation between the two partners in fused cells (Fig. 2, last data group). Additionally, the expression profile of 129TF (or R1A) in the clone is highly correlated with its expression profile in the population of fused cells (Spearman correlation is 0.97). We then examined whether any genes identified to be occluded in the population of fused cells became expressed in the clone. For 107 occluded 129TF genes (or 188 occluded R1A genes) in the population, only four (or four) became expressed in the clone. Thus, the 129TF × R1A (clone1) closely recapitulates the expression profiles and occlusion status of genes found in the population of 129TF–R1A fused cells.
We also analyzed an additional clone derived from the same original 129TR–R1A fusion as well as two subclones derived from the aforementioned 129TF × R1A (clone1), and found that these samples share highly similar expressions profiles (Supplemental Fig. S5). Of the 129TF genes found to be occluded in 129TF × R1A (clone1), all but two remained silent in all the other clones.
Taken together, these data demonstrate that occlusion as revealed by the cell fusion assay is a highly robust and temporally stable phenomenon irrespective of fusion ratio, post-fusion culture time, DNA replication, and whether fused cells are analyzed at the population level or clone level.
Whole-genome maps of chromatin marks
Many chromatin marks are highly correlated with gene activity (Goldberg et al. 2007), raising the possibility that they could play a role in the differential behavior between occluded and activatable genes. We investigated eight such marks, comprising six “active” marks generally associated with expression including RNA polymerase II (Pol II), H3K36 trimethylation (H3K36me3), H3K27 acetylation (H3K27ac), H3K9 acetylation (H3K9ac), H3K4 monomethylation (H3K4me1), and H3K4 trimethylation (H3K4me3), as well as two “silent” marks generally associated with lack of expression, including H3K27 trimethylation (H3K27me3) and H3K9 trimethylation (H3K9me3). We constructed whole-genome maps of these marks in 129TF cells using chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq).
We found that, as expected, expressed genes tend to have relatively homogenous chromatin properties in that they typically show enrichment for active marks and depletion for silent marks. They also typically lack promoter hypermethylation as described below. The two silent categories (i.e., activatable and occluded genes) are more variable in their chromatin properties (Supplemental Fig. S6 shows UCSC Genome Browser screenshots of ChIP-seq data for several representative expressed, activatable and occluded genes). We then compared the average chromatin profile of each mark over these three categories of genes, focusing on regions spanning from 2 kb upstream of TSS to the end of genes (Fig. 3). As expected, expressed genes are highly distinct from the two silent categories for all the marks, being enriched for active marks and depleted for silent marks. Of the two silent categories, occluded and activatable genes show difference for some of the marks, but to lesser degrees as compared with the difference between expressed and silent genes (Fig. 3).
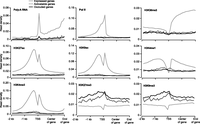
Chromatin profiles in promoter proximal regions of expressed, activatable, and occluded genes in 129TF. Profiles of poly(A) RNA are based on RNA-seq while the eight chromatin marks are based on ChIP-seq.
Base-resolution DNA methylome map
We explored the possible role of DNA methylation in occlusion given its association with gene silencing (Holliday and Pugh 1975; Riggs 1975; Reik 2007). We constructed a whole-genome cytosine methylation map for 129TF at single-base resolution using the Methyl-seq protocol (Lister et al. 2009). In total, we obtained sequences representing 9.3-fold coverage of each strand of the mouse genome. We detected 30,571,410 cytosines that are methylated at any level comprising 3.22% of the cytosines with sequence coverage. Of the methylcytosines, 99.98% are in CG context (referred to hereon as mCG). Among these mCG sites, the majority are highly methylated. For example, 53% of such sites show 80%–100% methylation (Supplemental Fig. S7A). Globally, there are large variations of mCG density on individual chromosomes (Supplemental Fig. S7B). Furthermore, promoter methylation levels of genes are on average anti-correlated with expression levels (Supplemental Fig. S7C). These results are qualitatively in line with our recent data on IMR-90 human fetal lung fibroblasts (Lister et al. 2009).
Before examining DNA methylation data, we first obtained the average CG dinucleotide density (defined as the number of CG on both strands per base pair) in expressed, occluded, and activatable 129TF genes. Similar to the analysis of chromatin marks, we focused on regions of genes from 2 kb upstream of the TSS to the end of the gene body. For all three categories of genes, CG density peaks in the vicinity of TSS (Fig. 4A). As expected, expressed genes have considerably higher CG density relative to the two silent categories. The CG density of occluded genes is lower than expressed genes but higher than activatable genes. These observations are consistent with data presented earlier (Fig. 1J) regarding the prevalence of CpG island promoters in the various categories of genes. We next obtained the average mCG density (defined as the frequency of mCG per base) as well as mCG density per CG (defined as the frequency of mCG per CG site) for each category of genes (Fig. 4B,C). In both cases, expressed genes showed depletion of methylation in the vicinity of TSS relative to the two silent categories. The mCG density around TSS for occluded genes is about twice that for activatable genes (Fig. 4B), which is partly due to occluded genes having greater CG density around TSS (Fig. 4A), and partly due to occluded genes having greater mCG density per CG in said regions (Fig. 4C).
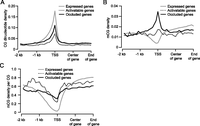
DNA methylation profiles in promoter proximal regions of expressed, activatable, and occluded genes in 129TF. (A–C) Profiles of CG dinucleotide density (A), mCG density (B), and mCG density per CG (C), with mCG density showing the greatest difference between occluded and activatable genes.
Chromatin signatures of occlusion
We sought to further analyze the chromatin correlates of different categories of genes. For each of the chromatin marks analyzed, we identified every genomic region where the mark is enriched. For all the marks except H3K36me3, we then examined whether any such enriched regions reside within the proximal regulatory space of genes (≤1 kb from TSS on either side). For H3K36me3, we examined whether the enriched regions reside within gene bodies (from TSS to transcriptional termination site) because this mark is predominantly associated with gene bodies (Kouzarides 2007). A gene is considered positive for a mark if enriched region(s) of the mark are present, and negative otherwise. For DNA methylation, we calculated the mCG density within the region spanning from 1 kb upstream of TSS to TSS; we did not consider sequences downstream from TSS to avoid tracking increased methylation in gene bodies of expressed genes (Suzuki and Bird 2008). Genes possessing mCG densities within the upper quintile of all genes were classified as hypermethylated, while those falling into the bottom quintile were classified as hypomethylated. Using these classifications, we determined the percentage of genes in each category that are positive for a given mark (Fig. 5A). As expected, a much higher percentage of expressed genes than the silent categories are positive for active marks and hypomethylation, whereas a much higher percentage of genes in the silent categories are positive for silent marks and hypermethylation. Of the two silent categories, activatable genes are significantly more likely to be positive for active marks and hypomethylation as compared with occluded genes, whereas occluded genes are significantly more likely to be hypermethylated. Occluded genes are also more likely to possess the two silent chromatin marks (H3K9me3 and H3K27me3) than activatable genes. Furthermore, of the occluded or activatable genes that are positive for the two silent marks, the enriched regions in occluded genes tend to have larger ChIP-seq read peaks (Fig. 5B).
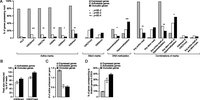
Chromatin signatures of different classes of genes. (A) Enrichment of specific chromatin marks or combination of marks (including DNA methylation) in expressed, activated, and occluded genes. Asterisks indicate that activatable and occluded genes are statistically different for a mark or combination of marks, with P-values calculated by two-tailed Fisher's exact test. (B) Occluded genes tend to have larger enrichment peaks than activatable genes for H3K9me3 and H3K27me3. (C) Expressed genes have a more average number of predicted active enhancers than silent genes. (D) More occluded than activatable genes correspond to genes possessing bivalent promoters in ESCs (Bernstein et al. 2006).
There is no simple correlation between the presence/absence of a single mark and whether a gene is occluded. Rather, there appears to be a complex relationship between chromatin and occlusion involving the interplay of multiple marks. To further explore this relationship, we analyzed all the chromatin marks in aggregate using a machine learning algorithm designed to iteratively evaluate combinations of chromatin mark values for their ability to classify silent genes as either occluded or activatable, using experimentally verified occluded and activatable genes as the training set. This algorithm produced composite chromatin signatures for occluded genes, and separately, signatures for activatable genes (see Methods). Of note is the fact that a subset of occluded genes is highly enriched around TSS for H3K9me3 and H3K27me3 along with DNA hypermethylation (Supplemental Fig. S6A). This signature most readily distinguishes this subset of occluded genes from activatable genes, which do not possess such a combination of marks. For activatable genes, a subset shows enrichment for multiple active marks and depletion for silent marks (Supplemental Fig. S6G). These activatable genes fit the classic definition of a transcriptionally poised gene and are readily distinguished from occluded genes which never possess multiple active marks near TSS.
To validate the chromatin signatures of occluded and activatable genes identified by machine learning, we incorporated them into a computational method for predicting occluded and activatable genes. We first applied the method to known occluded and activatable genes in 129TF. Using stringent criteria, we could predict occluded and activatable genes with high accuracy (10% and 1% false discover rate, respectively, for the genes where predictions could be made). We next applied our prediction algorithm to the set of 5133 silent genes in 129TF that had not yet been assigned to either the occluded or activatable category in the cell fusion studies. Using the same stringent criteria, we predicted 448 additional occluded genes (Supplemental Table S6) and 104 additional activatable genes (Supplemental Table S7). Predicted occluded genes are highly enriched for regulatory functions such as homeobox family members, transcription factors, embryonic morphogenesis, and cell fate commitment (P < 3.4 × 10−16, 9.9 × 10−18, 11.7 × 10−9, and 1.4 × 10−10, respectively), as were experimentally identified occluded genes. Predicted activatable genes were enriched for metabolic processes, including ATP binding, nucleoside binding, nucleotide binding, ribonucleotide binding, and ABC transporter (P-values of 1.8 × 10−2, 2.7 × 10−2, 5.1 × 10−2, 5.1 × 10−2, and 7.7 × 10−2, respectively), similar to experimentally identified activatable genes.
In addition to the composite analysis of all the chromatin marks, we also focused on particular combinations of subsets of marks that have previously been implicated in gene regulation. Active enhancers are shown to be characterized by the combination of H3K4me1 enrichment and H3K4me3 depletion (Heintzman et al. 2007). We used this combination to identify active enhancers and assigned them to nearest genes. We found that, as expected, expressed genes are highly enriched for active enhancers relative to the two silent categories, whereas activatable and occluded genes are not significantly different (Fig. 5C). It has been reported that mouse ESCs possess many “bivalent” domains where the active mark H3K4m3 and the silent mark H3K27me3 coexist (Bernstein et al. 2006). Bivalent domains tend to coincide with developmental genes that are lowly expressed in ESCs, leading to the suggestion that their function is to silence developmental genes in ESCs while keeping them poised for activation during differentiation. We examined how the various categories of genes in 129TF correspond to bivalent genes in ESCs. Of the two silent categories in 129TF, sizable fractions (30%–40%) correspond to bivalent genes in ESCs, whereas the same was true for a much smaller fraction (∼10%) of the expressed category (Fig. 5D). This is presumably due to the fact that expressed genes contain a high percentage of housekeeping genes. A larger fraction of genes in the occluded category than in the activatable category corresponds to bivalent genes in ESCs (Fig. 5D). This raises the possibility that genes undergoing occlusion in later stages of differentiation are more likely to be targeted by the kind of transcriptional regulation in ESCs that involves bivalent domains. In 129TF itself, the number of bivalent genes is rather small, and the fractions of genes that are bivalent show little difference between the occluded and activatable categories (Fig. 5A). The same is true for genes that are simultaneously positive for any active mark and any silent mark (Fig. 5A). This suggests that bivalency is no longer a prominent mode of gene regulation in this cell type.
In all, the above data show that, while the occluded or activatable status of a gene does not have a clean one-to-one correlation to a single mark, composite chromatin signatures can be gleaned across multiple marks. This suggests a complex chromatin basis involving the interplay of many marks that underlies whether a gene is occluded or activatable; it is also consistent with our data on the role of DNA methylation and histone deacetylation in occlusion (see below).
Role of DNA methylation in the memory of occlusion
To further explore the role of DNA methylation in occlusion, we examined the effect of the DNA methyltransferase inhibitor, 5-aza-2′-deoxycytidine (AdC), on the occluded state. The clonal line derived from the 129TF–R1A fusion, 129TF × R1A (clone1), was treated with AdC for one week and harvested either immediately (referred to as the AdC-treated sample), or with one additional week of recovery without the drug (the AdC-recovered sample). The durations of both treatment and recovery are long enough for multiple rounds of cell division to take place, which is necessary for AdC to take effect. We then obtained global gene expression profiles of the two samples by RNA-seq. Bisulfite sequencing of representative genes confirmed that demethlylation had taken place in AdC-treated cells, and that this demethylation persisted in the recovered cells (Supplemental Fig. S8).
In the untreated 129TF × R1A (clone1), a total of 101 129TF genes were found to be occluded. There is a notable increase in the aggregate expression of these genes in the AdC-treated sample as compared with the untreated sample (Fig. 6A). This effect is not uniform across all the occluded genes. Rather, the great majority of occluded genes were unaffected while a small minority were noticeably affected (Fig. 6B), with 11% of the genes being activated to levels over the ≥2 TPG threshold (Fig. 6C). This increased expression persisted in the AdC-recovered sample (Fig. 6A). Furthermore, the expression levels of individual occluded genes in the AdC-treated sample, including those activated by AdC, remained essentially the same in the AdC-recovered sample, with 8% of the genes remaining above the ≥2 TPG threshold (Fig. 6C), consistent with RT-PCR verification. Thus, occluded genes activated by AdC persisted in the active state even 1 wk after AdC removal. These results argue that DNA methylation contributes to the memory of occlusion for at least a subset of occluded genes. However, for many occluded genes, DNA methylation does not appear to play a memory role, suggesting the involvement of other chromatin mark(s). This is consistent with the observation that many occluded genes do not show promoter hypermethylation (Fig. 5; Supplemental Fig. S6). We also note that, because AdC does not lead to complete demethylation (Flotho et al. 2009), it is possible that DNA methylation has a greater role in occlusion than what our data suggest at face value.
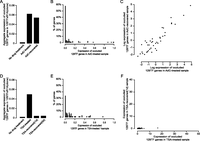
Permanent deocclusion of a subset of occluded genes in 129TF × R1A (clone1) by AdC but not TSA treatment. (A) AdC treatment caused a permanent increase in the expression of occluded 129TF genes in 129TF × R1A (clone1). Expression level is measured as the aggregate expression of occluded 129TF genes scaled to that of their R1A orthologs. (B) Histogram of occluded 129TF genes based on their expression levels in AdC-treated sample. Expression levels of individual 129TF genes are scaled to that of their R1A orthologs, and divided into 100 bins (expression levels >1 were combined into the last bin). Only genes for which the R1A copies are expressed at ≥2 TPG are considered. (C) Activation of occluded 129TF genes in 129TF × R1A (clone1) by AdC treatment is permanent. Expression levels of occluded 129TF genes in AdC-recovered sample are tightly correlated to that in AdC-treated sample, indicating that occluded 129TF genes activated by AdC treatment remained similarly active after seven days of recovery without the drug. Expression level is measured in TPG; logarithmic scales are used to allow better visualization of expression levels. (D) TSA treatment caused a transient increase in the expression of occluded 129TF genes in 129TF × R1A (clone1). Expression level is measured in the same way as in A. (E) Histogram of occluded 129TF genes based on their expression levels in TSA-treated sample. Expression level is measured in the same way as in B. (F) Activation of occluded 129TF genes in 129TF × R1A (clone1) by TSA treatment is transient. Activation of occluded 129TF genes in 129TF × R1A (clone1) by TSA treatment is reversed in TSA-recovered (1d) sample, indicating the highly transient nature of the activation. Expression level is measured in TPG.
Histone deacetylation involved in implementation but not memory of occlusion
To examine the possible role of histone acetylation in the memory of occlusion, we treated 129TF × R1A (clone1) with the histone deacetylase inhibitor trichostatin A (TSA), and harvested cells either immediately (referred to as the TSA-treated sample) or after 5 d of recovery without the drug (the TSA-recovered [5d] sample). We then obtained global gene expression levels of these two samples by RNA-seq.
We found that the expression level of occluded 129TF genes in the treated sample rose to a similar level as that found in the AdC-treated sample (Fig. 6D). TSA appears to activate occluded 129TF genes more evenly than AdC, but the effect is far from uniform (Fig. 6E), and similar to AdC, 10% of occluded 129TF genes are activated to above the ≥2 TPG threshold (Fig. 6F).
In the TSA-recovered (5d) sample, occluded 129TF genes no longer show a significant increase in expression relative to untreated cells, and no gene is above the ≥2 TPG threshold. This indicates that the activation of occluded genes by TSA is transient such that they revert back to the silent state once the drug is removed. To examine the kinetics of this reversal, we harvested TSA-treated cells after only one day of recovery, referred to as the TSA-recovered (1d) sample, and obtained global gene expression patterns by RNA-seq. We found that this sample was indistinguishable from the TSA-recovered(5d) sample in that occluded genes activated by TSA were no longer expressed (Fig. 6D,F). This indicates that the activating effect of TSA on occluded genes was rapidly reversed after drug removal. These results argue that histone deacetylation plays a role in implementing the silencing of at least a subset of occluded genes, but it exerts a highly transient effect and therefore appears unlikely to be involved in the memory of occlusion.
Lastly, we examined whether TSA and AdC tend to target the same set of genes. We plotted the expression levels of occluded 129TF genes in TSA-treated sample against that in AdC-treated sample, and found no correlation (data not shown), indicating that these two drugs target independent sets of occluded genes. This suggests that the repressive effect of DNA methylation and histone deacetylation are mechanistically independent.
Results from this and the previous section highlight the complexity in how chromatin marks might be involved in the regulation of occlusion. A mark could be involved in implementing the silencing of certain occluded genes such that removal of the mark leads to their activation. But that does not mean that the mark is a causal determinant of occlusion. For example, in the case of histone deacetylation, its removal only transiently activates affected genes rather than permanently erasing the occluded state. In order for a mark to serve as the causal determinant of occlusion, it needs to function as the memory of the occluded state, such that its removal will permanently erase occlusion.
Discussion
A major unresolved question in gene regulation is to what extent transcriptome identities of cells are determined by chromatin components acting in cis versus diffusible factors acting in trans. One prominent view posits that gene expression patterns characteristic of individual cell types are determined continuously by trans-acting factors (Blau and Baltimore 1991). Yet, this view seems incompatible with the notion in the epigenetic field that gene expression patterns are highly stable and heritable (Goldberg et al. 2007), which is likely due to cis-acting, chromatin-based regulation (Lahn 2011).
In this study, we showed that a large number of silent genes in somatic cells are in the occluded state whereby they are resistant to the activating effect of transcriptional activators. Indeed, gene occlusion is a widespread phenomenon, which we consistently observed in numerous somatic cell types across many different mammalian species beyond the cell types and species used in this study (Lee et al. 2009; Foshay et al. 2012; Gaetz et al. 2012) (unpubl.). We note that the cell lines we used are unlikely to fully recapitulate which specific genes are occluded in specific cell types in vivo. We also note that our study is based on synkaryons whereas many previous cell fusion studies relied on heterokaryons. Nevertheless, it is reassuring that different cell lines in our hands produced results that are in broad agreement with one another with regards to the general patterns of occlusion.
In another recent study, we demonstrated that the cellular milieu in somatic cell types actually supports the expression of most occluded genes in the cell—i.e., transcriptional activators for most occluded genes are already present in the cell even without fusion to other cell types (Gaetz et al. 2012). We further showed that occlusion of lineage-inappropriate genes indeed plays a critical role in safeguarding somatic cell fate (Gaetz et al. 2012). These studies shed light on the cis versus trans debate by arguing that the transcriptional milieu of different cell types is more similar than previously appreciated, and that the differential expression of many genes between different cell types is due largely to differential occlusion rather than differences in trans-acting factors. Indeed, we propose that at the most fundamental level, the identities of individual cell types are defined by which genes are occluded.
Somatic cells can be dedifferentiated to a fully pluripotent state by a number of procedures such as somatic cell nuclear transfer (SCNT) into oocytes (Gurdon 1962; Wilmut et al. 1997) and the use of defined transcription factors to create induced pluripotent stem cells (iPSCs) (Takahashi and Yamanaka 2006; Maherali et al. 2007; Okita et al. 2007; Wernig et al. 2007). Dedifferentiation is not inconsistent with occlusion if one assumes that pluripotent cells possess the capacity to erase occlusion across the genome, and that such “deocclusion” capacity is absent from somatic cells. The reprogramming of somatic cells to pluripotency can be achieved experimentally either by tapping into the existing deocclusion capacity of pluripotent cells (i.e., SCNT into oocytes), or by recapitulating key aspects of the deocclusion machinery (i.e., the creation of iPSCs by defined factors). This hypothesis is supported by our recent finding that embryonic stem cells (ESCs) are distinct from somatic cells in that they possess the capacity for global deocclusion (Foshay et al. 2012). Thus, the gene occlusion concept provides a simple and coherent framework for studying how the restriction of cell fate is brought about during development, erased naturally during reproduction or artificially by experimental manipulation, and possibly subverted in disease.
Methods
Cell culture
To obtain 129TF mouse tail fibroblasts (full name: 129TF-GPc1), cells were derived from a 3-wk-old female of 129 strain background according to published protocol (Xu 2005), transduced with a lentiviral vector carrying constitutively expressed EGFP driven by the human EEF1A1 promoter and puromycin resistance as described previously (Qin et al. 2010), followed by derivation of a clonal population. Cells were cultured in medium consisting of DMEM and 10% FBS. The origins of the rat cells are as follows: R1A (full name: R1A-RHcB) from Rat-1a embryonic fibroblasts (Stone et al. 1987) (gift from Shutsung Liao), IRC (full name: IRC-RHc17) from IRC chondrocytes (Horton et al. 1988) (gift from Walter Horton), L6 (full name: L6-RHc6) from L6 myoblasts (ATCC, cat# CRL-1458), RBL (full name: RBL-RHcC6) from RBL-2H3 basophilic leukemia (ATCC, cat# CRL-2256), H9 (full name: H9-RHcA10) from H9c2(2-1) cardiomyoblasts (ATCC, cat# CRL-1446), B35 (full name: B35-RHc4) from B35 neuroblastoma (ATCC, cat# CRL-2754), UMR (full name: UMR-RHc7) from UMR-106 osteosarcoma (ATCC, cat# CRL-1661), IEC (full name: IEC-RHc1) from IEC-18 intestinal epithelial cells (ATCC, cat# CRL-1589), S16 (full name: S16-RHc1) from S16 Schwann cells (ATCC, cat# CRL-2941), D1 (full name: D1-RHcB11) from D1 TNC1 type 1 astrocytes (ATCC, cat# CRL-2005), BRL (full name: BRL-RHc1) from BRL 3A hepatocytes (ATCC, cat# CRL-1442). Each of these rat cell types was a clonal population derived from cells transduced with a lentiviral vector carrying constitutively expressed DsRed-Express2 (Strack et al. 2008) or dTomato (Shaner et al. 2004) driven by the human EEF1A1 promoter and hygromycin resistance as described previously (Qin et al. 2010) and available through Cyagen Biosciences. They were cultured under conditions as published or recommended by the vendor.
Cell fusion
All fusions were performed following the same general protocol as follows. Cells were plated together for at least 2 h (or overnight). Prior to fusion, cells were washed with serum-free DMEM, and PEG 1500MW or 1000MW (50% w/v in serum-free DMEM) was added for 1 min. After removal of PEG, cells were washed three times with serum-free DMEM and allowed to recover for 2 h. Following this, cells were split to a lower density and plated in media containing both puromycin and hygromycin to select for double drug-resistant fused cells. Daily trypsinization aided in selection and purification of fused cells, with total RNA harvested between 6 and 7 d post-fusion. Variations in this protocol included cell to cell ratio, cell density, and concentrations of puromycin and hygromycin, all of which were determined empirically for each fusion. L6 was differentiated under low-serum condition prior to fusion. 129TF × R1A (clone1) and (clone4) were derived by FACS sorting of single fused cells into 96 well plates, while 129TF × R1A (clone1–2) and (clone1–4) were similarly derived by sorting 129TF × R1A (clone1) cells. Treatment of 129TF × R1A (clone1) with 5-aza-2′-deoxycytidine was carried out at 20 µM for 7 d and with trichostatin A at 1.5 µM for 1 d.
RNA-seq
About 10 µg of total RNA per sample was used for sequencing on an Illumina Genome Analyzer II following vendor's protocol, with 36 bases obtained per read.
Construction of mouse–rat ortholog reference database
Sequence libraries in FASTA format containing all annotated mouse and rat open reading frames (ORFs) were obtained from http://uswest.ensembl.org/index.html, and converted to protein sequence using blast2protein. Each mouse protein sequence was aligned to the library of rat protein sequences using BLAST. The output of this query was a ranking of rat sequences containing the highest homology with the query sequence. The rat protein sequence with the highest BLAST score was then aligned to the library of mouse protein sequences using an identical procedure. If the top scoring mouse protein sequence for this query was identical to the original mouse protein sequence, then the mouse and rat protein sequences were deemed reciprocal-best hits, and orthology was established. In this way we identified 47,663 pairs of orthologous ORFs representing 18,036 genes. Genes possessing multiple annotated transcripts were represented by multiple pairs of orthologous mouse–rat ORFs, some of which might correspond to transcripts whose biological function or prevalence have not been experimentally verified. For each gene represented by multiple pairs of ORFs, we chose the ORF pair whose mouse member most closely matched a human-verified RefSeq transcript. In the case where no RefSeq match was present, we chose the ORF pair whose mouse member had been present in the Ensembl database for the longest period of time. This resulted in a library of 18,036 genes, each represented by one pair of orthologous mouse–rat ORFs possessing a median DNA sequence conservation of 93% (Supplemental Fig. S9A). Our application requires that each member of an ortholog pair represents an equivalent portion of mouse or rat transcript. Misannotation of an ORF boundary for one ORF member (for example, by inappropriate extension or truncation of the ORF boundaries) would produce nonequivalent ORF pairs that would confound our ability to quantify the expression of each ortholog within the heterokaryon. To address this problem, we used a 12-base sliding window to assess local homology in each ORF pair. Windows containing six or more mismatched bases were redacted with Ns in the ORF library. This resulted in ORF pairs whose members were of identical or nearly identical size. We then removed genes of unknown genomic location in the mouse and genes for which the mouse–rat homology in the non-redacted portions of the orthologs is <80% and also genes with unknown genomic location in the mouse, which resulted in 16,326 genes. Then, genes for which >2% of RNA-seq reads from a non-fused cell type aligned perfectly and uniquely to the wrong species were removed (see below). This led to the final ortholog reference database containing 16,053 genes.
Alignment of RNA-seq reads to mouse–rat ortholog reference database
We used Maq software as described previously (Li et al. 2008) and stringent mapping settings (–n 3 –m 1) to align RNA-seq reads to reference sequences. RNA-seq data derived from non-fused mouse or rat cells were mapped to the reference of the corresponding species. RNA-seq data from fused cells were mapped to the mouse–rat ortholog reference. To eliminate reads of ambiguous genomic origin, we only retained reads that aligned perfectly to a single site within the database. We then examined the effect of these criteria on the total number of reads mapping to each gene. On average, selecting for perfectly and uniquely aligned reads reduced the total number of reads per ORF by about a third relative to the number of reads aligning to one or more sites with up to one mismatch per site (Supplemental Fig. S9B,C). To confirm that this reduction in mapped reads affected both orthologs in a pair equivalently, we calculated the correlation between the proportion of mouse reads lost and the proportion of rat reads lost for each gene. Indeed, read reduction was highly correlated between orthologs (Supplemental Fig. S9D), indicating that asymmetric read reduction would not be a major cause for the false identification of differentially expressed genes in fused cells. To establish species specificity of the read alignments, we counted the number of reads from non-fused cells of either species mapping to the opposite species perfectly and uniquely when aligned to the ortholog database containing both species. We saw that only 0.38% of mouse 129TF reads and 0.46% of rat B35 reads mapped perfectly and uniquely to rat and mouse genome, respectively, indicating that our procedure has high species specificity. To further reduce the effect of wrong species alignment, we counted the proportion of reads for each gene obtained from non-fused cells that mapped perfectly and uniquely to the wrong species. Genes with >2% of reads that do so were removed from analysis. For analyzing RNA-seq data from non-fused cells, reads were mapped to the portion of the ortholog reference database containing only sequences of the correct species, allowing one mismatch maximum and using default parameters.
Quantification of gene expression level
Expression level of each gene is calculated from RNA-seq data and expressed in the unit of TPG using the following equation: where E is the gene expression level in TPG, Q is the average quantity of poly(A) RNA produced per diploid genome in a cell, NA is Avogadro's constant, L is the average length of poly(A) RNA, W is the average molecular weight per base, d is the average transcript length of genes in reference database, r is the number of reads mapping to the gene, n is the total number of reads mapping to the genome, and k is the transcript length of the gene in reference database. We further assume that Q = 5 × 10−10 g (based on the fact that the average diploid genome in a cell produces ∼10 pg total RNA, of which ∼5% is poly-adenylated),
L = 2500 bases [including poly(A) tail], and d is 1636. Besides TPG, another commonly used unit of gene expression level is reads per kilobase per million mapped reads
(RPKM). We note that 1 RPKM equals ∼0.594 TPG. Silent genes are defined as <0.2 TPG, and active genes as ≥2 TPG. With these
cutoffs, the great majority of genes (∼90%) in a given cell type fall into either silent or expressed category (Supplemental
Fig. S9E shows a histogram of gene expression levels in 129TF). Assuming 20,000 genes in the genome, the average level of
gene expression across all the genes is ∼20 TPG.
where E is the gene expression level in TPG, Q is the average quantity of poly(A) RNA produced per diploid genome in a cell, NA is Avogadro's constant, L is the average length of poly(A) RNA, W is the average molecular weight per base, d is the average transcript length of genes in reference database, r is the number of reads mapping to the gene, n is the total number of reads mapping to the genome, and k is the transcript length of the gene in reference database. We further assume that Q = 5 × 10−10 g (based on the fact that the average diploid genome in a cell produces ∼10 pg total RNA, of which ∼5% is poly-adenylated),
L = 2500 bases [including poly(A) tail], and d is 1636. Besides TPG, another commonly used unit of gene expression level is reads per kilobase per million mapped reads
(RPKM). We note that 1 RPKM equals ∼0.594 TPG. Silent genes are defined as <0.2 TPG, and active genes as ≥2 TPG. With these
cutoffs, the great majority of genes (∼90%) in a given cell type fall into either silent or expressed category (Supplemental
Fig. S9E shows a histogram of gene expression levels in 129TF). Assuming 20,000 genes in the genome, the average level of
gene expression across all the genes is ∼20 TPG.
ChIP-seq
For all histone marks except H3K9me3 and H3K27me3, chromatin precipitation was performed following the published protocol (Hawkins et al. 2010) with the following modifications: Approximately 108 129TF cells were fixed in 1% formaldehyde for 10 min at room temperature, and sonicated on ice using a Sonic Dismembrator Model 500 and 1/8′′ diameter tapered microtip (Fisher Scientific). Sonication parameters were 50% amplitude and 32 cycles of 30 sec on and 3 min off. For H3K9me3 and H3K27me3, 129TF cells were treated with 0.75% formaldehyde for 10 min, then fixation was terminated with 0.125M glycine at room temperature. The isolated nuclei were incubated with micrococcal nuclease (1200 gel units per 10^6 nuclei, New England BioLabs) at 37°C for 40 min, then sonicated on ice under the aforementioned sonication conditions. Antibodies used for ChIP are as follows: H3K4me1 (Abcam, cat# ab8895), H3K4me3 (Millipore, cat# 17-614, part# CS200580), H3K9me3 (Abcam, cat# ab8898), H3K27me3 (Active Motif, cat# 39155), H3K27ac (Active Motif, cat# 39133), H3K9ac (Active Motif, cat# 39137), H3K36me3 (Abcam, cat# ab9050), Pol II (Covance, cat# MMS-126R). We used Dynabeads (Invitrogen) to bind antibody. We used 250–500 µg of chromatin for each ChIP, and every 100 µg of chromatin required 1 µg of antibody. Sequencing of ChIP material was performed on an Illumina Genome Analyzer II following vendor's protocol, with 36 bases obtained per read.
Analysis of ChIP-seq data
ChIP-seq reads were mapped to the July 2007 assembly of mouse genome, excluding sequences that were not finished or that have not been assembled with certainty (i.e., exclusion of sequences contained in the chrUn_random.fa and chrN_radom.fa files provided by the UCSC Genome Browser). Sequence alignment was accomplished using BWA (Banito et al. 2009) with default alignment parameters. For profiling ChIP-seq read densities in genes including promoters, we obtained promoter sequence defined as 2 kb upstream of the TSS from the Ensembl transcript IDs (hg18), plus the entire length of the gene body. Gene body was determined for each Ensembl transcript ID as the region spanning from the TSS to the end of transcription. Promoter and gene body were each divided into 20 bins each, and for each bin, the number of reads for which the BWA alignment position falls into the bin was tallied. Summary statistics for each gene category were generated by calculating the average read density per base of each bin for all group members. Prediction of active enhancers using H3K4me1 and H3K4me3 ChIP-seq data was performed with a previously described method (Heintzman et al. 2007). ChIP-seq data were input-corrected, binned, and normalized as described (Hawkins et al. 2010). Enhancers were predicted at a range of P-values, and P < 0.01 was used as a cutoff as it predicted a suitable number of enhancers. For evaluating H3K27me3 and H3K9me3 ChIP-seq peak sizes, read counts for peaks were determined using MACS (Zhang et al. 2008). To account for differences in sequence yield between ChIP-seq experiments, the average read count for each mark was scaled to the number expected upon processing 50 million uniquely mapped reads.
Methyl-seq
Two to five micrograms of genomic DNA in 50 µL EB buffer was sheared to ∼250-bp range with Covaris (Woburn). The ends of DNA were filled in and then an “A” base was added to the 3′ end of the DNA fragments according to Illumina standard end repair and add_A protocol (Illumina). Pre-annealed forked Illumina adaptor containing 5′-methyl-cytosine instead of cytosine was ligated to both ends of DNA fragments using standard Illumina adaptor ligation protocol (Illumina). Ligated fragments were then separated by 2% agarose gel. Two size ranges 275–350 and 350–475 bp (size including adaptor length) were selected from the gel. DNA from gel slices were purified and subsequently treated with EpiTect Bisulfite kit (Qiagen) with the following modification: The bisulfite conversion time was extended to ∼14 h by adding three cycles of denaturation at 95°C for 5 min and incubation at 60°C for 180 min. Then bisulfite-converted DNA was purified following the purification protocol for DNA isolated from FFPE tissue sample in the EpiTect Bisulfite kit, except for the following step. The desulfonation buffer was added to the spin column and incubated for 20 min at room temperature. Then bisulfite-treated DNA was purified one more time with MinElute PCR purification kit (Qiagen) and eluted into 15 µL EB buffer. Bisulfite-treated DNA fragments were enriched in the following PCR reaction: 7.5–15 µL of eluted DNA, 5 pmol of Illumina PCR primers, 62.5 nM of each dNTPs, and 2.5 U of PfuTurbo Cx hotstart DNA polymerase (Stratagene) in a total 50 µL volume, 2 min at 95°C, 30 sec at 98°C, then 4× (15 sec at 98°C, 30 sec at 60°C, 4 min at 72°C) followed by 10 min at 72°C. PCR reaction was purified by MinElute PCR purification Kit (Qiagen) and final libraries were eluted in 15 µL EB. The concentration of the final library was determined by the Agilent 2100 Bioanalyzer (Agilent) and subjected to paired-read sequencing of 75 bases per read on an Illumina GA sequencing machine.
Analysis of Methyl-seq data
Methyl-seq data were mapped and processed as described (Lister et al. 2009) with the following modifications to accommodate the paired-read data type. Both reads in a pair were trimmed of any adapter sequences at their 3′ ends, and reads were mapped to the NCBI m37 reference genome with Bowtie (Langmead et al. 2009) in paired-read mode, using the following parameters: –e 90 –l 20 –n 0 –k 10 –o 4 –I 0 –X 550 –pairtries 100 –nomaqround –solexa1.3-quals. Mapped reads in a read pair that overlapped were trimmed from their respective 3′ ends until the reads no longer overlapped, leaving a 1-bp gap. Mapped reads were filtered as follows: Any read with more than three mismatches was trimmed from the 3′ end to contain three mismatches, any read pair which contained a cytosine mapped to a reference sequence thymine was removed, and any read pairs that had more than three cytosines in the non-CG context within a single read was removed (possible non-conversion in bisulfite reaction). Read pairs were then collapsed to remove clonal reads potentially produced in the PCR amplification from the same template molecule, based on common start position of read 1. Methylcytosines were identified from the mapped and processed read data as described (Lister et al. 2009), including correction of any DNA methylation incorrectly categorized as non-CG due to SNPs in the sample versus reference genomes. This led to 195,165,520 uniquely mappable paired-ends reads totaling 23,982,322,166 bases. For profiling DNA methylation in genes including promoters, promoter and gene body sequences were obtained and divided into bins in the same way as the analysis of ChIP-seq data. The density of CG and absolute (mCG) and relative (mCG per CG) methylation were determined for each bin. Absolute methylation was computed as the average methylation level (methylated/[methylated + unmethylated] read counts) divided by the bin size in base pairs. Relative methylation was determined as the ratio between the absolute methylation and the CG density (CG/bp) in the same bin.
Examining the effect of 5-AdC treatment on select genes
For each select gene, PCR primers were designed to amplify orthologous mouse and rat regions falling within the promoter (1 kb upstream to TSS) when using bisulfite-treated DNA as a template. PCR products from multiple genes under each type of treatment were then pooled and deep-sequenced on the Illumina Genome Analyzer II. Methylated and unmethylated CGs were identified using the previously described protocol (Lister et al. 2009) modified to use Maq (Li et al. 2008) for sequence alignment. Sequencing depth averaged ∼100,000 reads/base pair.
Identification of conserved noncoding sequences
Evolutionarily CNSs were identified through multispecies comparison in ECRbase (Loots and Ovcharenko 2007). These data were further reduced to contain only regions possessing a minimal mouse–rat homology of 85%.
Identification of transcription factor binding motifs
We searched for transcription factor binding sites within 1 kb of the TSS of occluded, activatable, and expressed genes. In total, ∼900 transcription factor binding sites were evaluated in the form of position weight matrices obtained from the TRANSFAC database (Matys et al. 2003). To begin, we used the fimo function from the MEME suite (Bailey et al. 2009) and default settings to compile a list of all predicted binding sites for each transcription factor within the mouse genome. Next, the binding site positions from this list were cross-referenced with the locations of promoters of occluded, activatable, and expressed genes, and instances where the two overlapped were noted. In this way we obtained a total binding site count for each transcription factor within occluded, activatable, and expressed gene promoters. Finally, a two-sided Fisher's exact test was used to obtain a P-value for differential motif enrichment between gene classes. Detailed results of the analysis are presented in Supplemental Table S5.
Prediction of occluded and activatable genes based on chromatin signatures
We constructed a machine learning algorithm employing a supervised linear classification-based model to categorize unclassified silent genes as occluded or activatable. Such models have been applied to similar classification problems with success (Ramaswamy et al. 2003). To begin, we first built a database containing the chromatin mark values associated with distinct regulatory regions of each gene in the genome. These included the proximal promoter (1 kb upstream of the TSS to TSS), the TSS (1 kb upstream to 1 kb downstream of the TSS), and the gene body (TSS to transcription terminal site). For histone marks and Pol II, we incorporated into the database Boolean values indicating the presence or absence of signal peaks within each region as well as continuous values indicating the quantity of reads falling within each regulatory region. Peaks were identified by MACS using a ChIP-seq control data set from no-antibody pull-down and the set of ChIP-seq reads that mapped to the mouse genome with no mismatches and possessed a phred quality score ≥10. We next incorporated data describing DNA CG methylation and CG dinucleotide density into the same database. For these features, we surveyed the same regulatory regions, but used the number of mCGs or CGs falling into each region as a continuous data type, and whether that number was in the upper or lower quintile of values for all such regions in the genome as Boolean data types. In the second phase of prediction, these data were integrated into our classification model whose output was a score p(occluded) describing the relation of a gene's chromatin state to the chromatin state of experimentally identified occluded genes. The model iteratively evaluated data type and regulatory region for each chromatin mark to choose the combination that maximized predictive ability. It then exhaustively evaluated combinations of chromatin mark values to identify composite chromatin signatures that distinguished occluded genes from activatable genes. At the final step, a perceptron-based method (Rosenblatt 1958) was used to choose the best score threshold for the prediction of occluded and activatable genes.
Data access
All high-throughput sequencing data have been submitted to NCBI BioProject (http://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA213248. Accession numbers for individual data sets submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) are listed in Supplemental Table S8.
Acknowledgments
We thank the following individuals for technical assistance and/or critical discussions: Liyuan (James) Cao, Jacob Degner, Ryan Duggan, Agnieszka Grzegorzewska, Samantha Kuan, David Leclerc, Ying Luu, Michael Olson, Joseph Pickrell, Patrick Reed, Gregory Snyder, Ann Sperling, and Zhen Ye. This work was funded in part by the Chicago Biomedical Consortium with support from The Searle Funds at The Chicago Community Trust (B.T.L.), the Ellison Medical Research Foundation grant AG-SFS-2528-10 (B.T.L.), NIH graduate training grant T32GM007197 (T.J.L.), NIH postdoctoral fellowships F32HD061205 (K.M.F.) and F32HL922792 (J.G.), the National Natural Science Foundation of China grant 30971675,30928015 (A.P.X.), and the Key Scientific and Technological Projects of Guangdong Province grant 2007A032100003 (A.P.X.).
Footnotes
-
↵10 Corresponding authors
E-mail blahn{at}bsd.uchicago.edu
E-mail chchenew{at}yahoo.com
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.143891.112.
- Received May 27, 2012.
- Accepted September 4, 2013.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported), as described at http://creativecommons.org/licenses/by-nc/3.0/.








