
Genomic analysis of the causative agents of coccidiosis in domestic chickens
- Adam J. Reid1,
- Damer P. Blake2,3,
- Hifzur R. Ansari4,
- Karen Billington3,
- Hilary P. Browne1,
- Josephine Bryant1,
- Matt Dunn1,
- Stacy S. Hung5,
- Fumiya Kawahara6,
- Diego Miranda-Saavedra7,
- Tareq B. Malas4,
- Tobias Mourier8,
- Hardeep Naghra1,9,
- Mridul Nair4,
- Thomas D. Otto1,
- Neil D. Rawlings10,
- Pierre Rivailler3,11,
- Alejandro Sanchez-Flores12,
- Mandy Sanders1,
- Chandra Subramaniam3,
- Yea-Ling Tay13,14,
- Yong Woo4,
- Xikun Wu3,15,
- Bart Barrell1,21,
- Paul H. Dear16,
- Christian Doerig17,
- Arthur Gruber18,
- Alasdair C. Ivens19,
- John Parkinson5,
- Marie-Adèle Rajandream1,22,
- Martin W. Shirley20,
- Kiew-Lian Wan13,14,
- Matthew Berriman1,
- Fiona M. Tomley2,3 and
- Arnab Pain4
- 1Wellcome Trust Sanger Institute, Genome Campus, Hinxton, Cambridgeshire CB10 1SA, United Kingdom;
- 2Royal Veterinary College, North Mymms, Hertfordshire AL9 7TA, United Kingdom;
- 3The Pirbright Institute, Compton Laboratory, Newbury, Berkshire RG20 7NN, United Kingdom;
- 4Computational Bioscience Research Center, Biological Environmental Sciences and Engineering Division, King Abdullah University of Science and Technology, Thuwal, Jeddah 23955-6900, Kingdom of Saudi Arabia;
- 5Program in Molecular Structure and Function, Hospital for Sick Children and Departments of Biochemistry and Molecular Genetics, University of Toronto, Toronto, Ontario M5G 1X8, Canada;
- 6Nippon Institute for Biological Science, Ome, Tokyo 198-0024, Japan;
- 7Fibrosis Laboratories, Institute of Cellular Medicine, Newcastle University Medical School, Framlington Place, Newcastle upon Tyne NE2 4HH, United Kingdom;
- 8Centre for GeoGenetics, Natural History Museum of Denmark, University of Copenhagen, 1350 Copenhagen, Denmark;
- 9School of Life Sciences, Centre for Biomolecular Sciences, University of Nottingham, Nottingham NG7 2RD, United Kingdom;
- 10European Bioinformatics Institute, Genome Campus, Hinxton, Cambridgeshire CB10 1SA, United Kingdom;
- 11Division of Viral Diseases, Centers for Disease Control and Prevention, Atlanta, Georgia 30333, USA;
- 12Unidad Universitaria de Apoyo Bioinformático, Institute of Biotechnology, Universidad Nacional Autónoma de México, Cuernavaca, Morelos 62210, Mexico;
- 13School of Biosciences and Biotechnology, Faculty of Science and Technology, Universiti Kebangsaan Malaysia, 43600 UKM Bangi, Selangor DE, Malaysia;
- 14Malaysia Genome Institute, Jalan Bangi, 43000 Kajang, Selangor DE, Malaysia;
- 15Amgen Limited, Uxbridge UB8 1DH, United Kingdom;
- 16MRC Laboratory of Molecular Biology, Cambridge CB2 0QH, United Kingdom;
- 17Department of Microbiology, Monash University, Clayton VIC 3800, Australia;
- 18Departament of Parasitology, Institute of Biomedical Sciences, University of São Paulo, São Paulo, SP 05508-000, Brazil;
- 19Centre for Immunity, Infection and Evolution, Ashworth Laboratories, School of Biological Sciences, University of Edinburgh, Edinburgh EH9 3JT, United Kingdom;
- 20The Pirbright Institute, Pirbright Laboratory, Pirbright, Surrey GU24 0NF, United Kingdom
- Corresponding authors: ar11{at}sanger.ac.uk, ftomley{at}rvc.ac.uk, arnab.pain{at}kaust.edu.sa
Abstract
Global production of chickens has trebled in the past two decades and they are now the most important source of dietary animal protein worldwide. Chickens are subject to many infectious diseases that reduce their performance and productivity. Coccidiosis, caused by apicomplexan protozoa of the genus Eimeria, is one of the most important poultry diseases. Understanding the biology of Eimeria parasites underpins development of new drugs and vaccines needed to improve global food security. We have produced annotated genome sequences of all seven species of Eimeria that infect domestic chickens, which reveal the full extent of previously described repeat-rich and repeat-poor regions and show that these parasites possess the most repeat-rich proteomes ever described. Furthermore, while no other apicomplexan has been found to possess retrotransposons, Eimeria is home to a family of chromoviruses. Analysis of Eimeria genes involved in basic biology and host-parasite interaction highlights adaptations to a relatively simple developmental life cycle and a complex array of co-expressed surface proteins involved in host cell binding.
Chickens are the world’s most popular food animal, and the development of improved drugs and vaccines to combat poultry diseases are vital for worldwide food security. Protozoan parasites of the genus Eimeria cause coccidiosis, a ubiquitous intestinal disease of livestock that has major impacts on animal welfare and agro-economics. It is a particularly acute problem in poultry where infections can cause high mortality and are linked to poor performance and productivity. Eimeria belong to the phylum Apicomplexa, which includes thousands of parasitic protozoa such as Plasmodium species that cause malaria, and the widely disseminated zoonotic pathogen Toxoplasma gondii. Eimeria species have a direct oral-fecal life cycle that facilitates their rapid spread through susceptible hosts especially when these are housed at high densities (for review, see Chapman et al. 2013). Unsurprisingly, resistance to anticoccidial drugs can evolve rapidly under these conditions and there is a continuing need to develop novel therapies (Blake et al. 2011).
More than 1200 species of Eimeria are described (Chapman et al. 2013) and virtually all of these are restricted to a single host species. Domestic chickens (Gallus gallus domesticus) can be infected by seven Eimeria species, each of which colonizes a preferred region of the intestine, causing symptoms of differing severity (Table 1; Shirley et al. 2005). Five species induce gross pathological lesions and four of these are the most important in terms of global disease burden and economic impact (E. acervulina, E. maxima, E. necatrix, and E. tenella) (Williams 1998).
Eimeria species biology and genomic sag repertoire
Results
Genome sequences of the Eimeria species that infect domestic chickens
We generated annotated genome sequences of all seven species of Eimeria that infect domestic chickens. For E. tenella, a high quality reference genome incorporating annotation-directed manual improvements for targeted regions was produced for the Houghton strain (tier 1) (Supplemental Table S1) as well as Illumina genomic sequencing data for the Wisconsin and Nippon strains. For E. maxima, E. acervulina, and E. necatrix, we produced draft genomes with automated post-assembly improvements (tier 2) (Supplemental Table S1), and for E. brunetti, E. mitis, and E. praecox, we produced draft genomes alone (tier 3) (Supplemental Table S2). The 51.8-Mb E. tenella genome assembly corresponds well with a genomic map (described below), suggesting that it accurately reflects the true genome size (Supplemental Table S1). Furthermore the tier 1 and tier 2 assemblies are predicted to be 93%–99% complete with respect to the T. gondii genome sequence based on the presence of core eukaryotic genes (Supplemental Table S1; Parra et al. 2007).
To investigate chromosome structures, we used whole-genome mapping to improve contiguity of the tier 1 and 2 genomes and were able to place up to 46% of sequence data onto 15 or 16 optical maps for each genome (Supplemental Table S1), which is close to the haploid chromosome number of 14 (del Cacho et al. 2005). Although few sequence markers were available for each chromosome we were able to map unambiguous identities to six of 15 optical scaffolds in E. tenella (Supplemental Table S3).
Phylogeny and synteny between Eimeria species
Previous phylogenetic analyses using small numbers of sequences did not fully resolve relationships between Eimeria species of the chicken (Ogedengbe et al. 2011). Whole-genome phylogeny shows robust separation of E. tenella and E. necatrix from the other species, as well as separation of E. mitis and E. brunetti from E. praecox, E. maxima, and E. acervulina (Fig. 1A). In support of this phylogeny, there is extensive synteny between the genomes of E. tenella and E. necatrix (i.e., many orthologous genes in the same order across contigs, with only a small number of rearrangements) (Fig. 1B). There is notably less synteny between the genome of E. tenella and those of E. maxima and E. acervulina with much of the chromosome structure rearranged, although presumably retaining the same number of chromosomes. Synteny between the genomes of E. tenella and Toxoplasma gondii was nonexistent, with no more than three orthologs (ETH_00031645, ETH_00031660, and ETH_00031665) found in the same order.
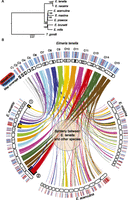
Whole-genome phylogeny and synteny between Eimeria species. (A) Maximum likelihood phylogeny showing the evolutionary relationships between Eimeria species based on alignment of 814 one-to-one orthologs shared with T. gondii. The scale is in substitutions per site. (B) Genomic scaffolds were placed onto optical maps. Black bands show map coverage. Coverage was noticeably better for E. tenella than the tier 2 species. E. tenella maps are named as chromosomes (e.g., C1) where it was possible to reliably identify that chromosome, otherwise they are given their optical map numbers (e.g., O4). Each E. tenella map has been assigned a color and ribbons highlight syntenic regions in the related genomes. E. necatrix is most closely related to E. tenella and correspondingly shows the greatest degree of synteny. The clearest exceptions are (1) O10, which is split between two optical contigs in E. necatrix, and (2) O8, which is similarly split. Map coverage is lower in E. acervulina and E. maxima and this gives the impression that there is a great deal of novel sequence in these species. However, this is largely the result of differential representation of the genomes in their respective maps. Each map is annotated with repeat-poor (blue) and repeat-rich (red) regions ≥30 kb. This highlights the barcode-like patterning across the whole of each genome.
Eimeria chromosomes display a banded pattern of repeat-poor and repeat-rich regions
Analysis of E. tenella chromosome 1 revealed alternating regions of repeat-poor (P) and repeat-rich (R) sequences (Ling et al. 2007). We now find this feature is conserved in all chromosomes of E. tenella and across the genomes of all Eimeria species examined (Fig. 1B). The short tandem repeat (STR) content of each genome shows a bimodal distribution with a high frequency peak close to zero and a broad, low frequency peak with a mean ∼20%–30% (Supplemental Fig. S1A). This confirms a bipartite structure for the genome. Any region of the genome is either repeat-rich (R; which we define as having a repeat density >5%) or essentially repeat-free (P). The precise repeat content differs between species, with E. tenella having fewer R regions than other species. Of note, E. necatrix is more repeat-rich in regions syntenic with E. tenella as well as across the genome generally (Supplemental Fig. S1B). Fifty-three point five percent of E. tenella genes were found in repeat-rich regions, suggesting that there is no preference for repeats to occur in gene-poor regions.
Eimeria protein-coding sequences are extremely rich in homopolymeric amino acid repeats (HAARs)
STRs can result in strings of single amino acids within predicted protein sequences. The extent of homopolymeric amino acid repeats (HAARs) is greater in Eimeria than in any other organism sequenced to date (Fig. 2A) and the distribution of homopolymer types is quite different from even closely related organisms such as T. gondii and P. falciparum. The most common STR in Eimeria species is the trinucleotide CAG (Fig. 2B), which occurs preferentially in coding sequences (Fig. 2C). CAG can potentially encode alanine (A), glutamine (Q), serine (S), cysteine (C), and leucine (L), but in E. tenella repeats are rarely translated as C or L. They are found preferentially as A or Q (Fig. 2D). HAARs of this type, encoding strings of at least seven amino acids, occur in 57% of E. tenella genes, with an average of 4.3 copies per gene. We confirmed that repeats are transcribed and translated in E. tenella, with the data predicting similar proportions of each HAAR type to that found in the genome (Fig. 2D).
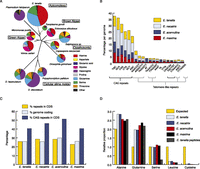
Characterization of HAARs in Eimeria protein sequences. (A) Eimeria tenella has a greater number of HAARs than any other genome sequenced and a distinct distribution of HAAR types compared with other repeat-rich genomes, including Plasmodium falciparum and the more closely related and not especially repeat-rich Toxoplasma. (B) The most common STRs in Eimeria genomes are variations on CAG. The second most common are variations on a telomere repeat which we call telomere-like repeats due to their locations throughout the genome. (C) CAG repeats occur in protein-coding regions of the genome more than expected. (D) CAG repeats can encode strings of one of five amino acids. In Eimeria they tend to encode alanine and glutamine more often than expected, serine as often as expected, and leucine and cysteine more rarely than expected. A very similar pattern is observed in a limited selection of E. tenella peptides derived from proteomics experiments.
An analysis of gene ontology terms showed that genes containing HAARs did not cluster in any particular functional class. However, those involved in information processing tasks such as translation, chromatin assembly, gene expression, and DNA metabolism had fewer HAARs than expected by chance (Supplemental Table S4). Indeed genes conserved across the eukaryotes had an overall lower than average repeat content (2.5% vs. 4.68% for all E. tenella genes). Proteins that are generally considered to be involved in host-parasite interactions such as SAGs, ROP kinases, and MICs had even fewer HAARs on average than those conserved across eukaryotes (0%, 1.88%, and 1.66%, respectively, vs. 2.5%).
We hypothesized that because HAARs are so common in Eimeria species, they are unlikely to interfere with protein structure and function. Indeed only 3.2% of E. tenella HAARs (687 of 21,191) occur in Pfam domains, which make up 12.4% of E. tenella protein sequences. By examining conserved proteins with known 3D structures, we found that serine and glutamine HAARs tend to be insertions in loop or turn regions with medium to high solvent accessibility, suggesting they do not affect protein folding (Supplemental Table S5). Alanine HAARs often align to helical regions and may result in very similar local structure (Perutz et al. 2002). Furthermore, homology modeling showed that HAARs tend to be located on the outside of proteins, away from regions involved in domain–domain interactions and active sites (Supplemental Fig. S2).
Comparative genomics of the Coccidia and wider Apicomplexa
Eight thousand, six hundred and three protein-coding genes are predicted in the E. tenella assembly, significantly more than the 7286 found in the related coccidian T. gondii (Supplemental Table S1), despite T. gondii having a nuclear genome that is ∼20% (10Mb) larger. By transcriptome sequencing we identified expression of 76% of predicted E. tenella genes (6700) across four developmental life stages (unsporulated oocyst, sporulated oocyst, sporozoite, and merozoite). The median sequence identity between E. tenella and T. gondii orthologous protein sequences was 39.7%, suggesting a large amount of sequence divergence between the two.
We identified several novel Eimeria-specific gene families (Supplemental Table S6; Supplemental Data set S1). We found that two of these families (esf1 and esf2) have higher Ka/Ks ratios than other genes (Supplemental Fig. S3; Supplemental Data set S2). This suggests that they may be under diversifying selection and could be important for host–parasite interactions.
The rhoptry organelles of T. gondii contain 30–50 kinases and pseudokinases (ROPKs) (Peixoto et al. 2010; Talevich and Kannan 2013), some of which are involved in remodeling the intermediate host cell and protecting the parasite against host defenses (Saeij et al. 2007; Fentress et al. 2010). Recent analysis showed that E. tenella has 28 ropk genes, including a subfamily not found in T. gondii (Talevich and Kannan 2013). We were able to identify orthologs of all these genes in E. necatrix; however, there is divergence in the more distantly related Eimeria species (Supplemental Table S7; Supplemental Data set S1). The overall protein kinase (PK) complement of Eimeria species (63–84 PK) is smaller than that of T. gondii (128 PK) (Peixoto et al. 2010) and Plasmodium species (85–99 PK) (Ward et al. 2004; Anamika et al. 2005; Miranda-Saavedra et al. 2012). This is not due solely to fewer ropks and fikks (an apicomplexan family highly expanded in Plasmodium) but also a reduction in CMGC kinases (the group which includes cyclin-dependent kinases) (Supplemental Table S7). It is proposed that CMGC kinases have evolved independently within the Apicomplexa to provide specialized functions related to lifecycle transitions (Talevich et al. 2011). Reduction of CMGC kinases in Eimeria, which has a simple life cycle and no intermediate host, may be an example of this specialization.
Metabolism is well conserved between Eimeria and Toxoplasma (Supplemental Fig. S4; Supplemental Data set S3), with the clearest difference being additional enzymes involved in Eimeria carbohydrate metabolism. Three of these catalyze reactions in the mannitol cycle, known to be essential for survival of Eimeria parasites and not present in other coccidian lineages (Schmatz et al. 1989; Liberator et al. 1998).
The apicoplast is a symbiotic plastid present in most apicomplexans and known to be essential for survival of T. gondii (He et al. 2001). Most ancestral plastid genes have moved into the nuclear genome but many of the gene products are post-translationally imported into the apicoplast. The mechanism of import to the apicoplast is poorly understood but two proteins, Tic20 and Tic22, are thought to mediate crossing of the innermost membrane (van Dooren et al. 2008; Lim and McFadden 2010). Genes encoding Tic20 and Tic22 are not found in the Eimeria species studied, suggesting either a distinct mechanism for crossing the apicoplast inner membrane or a change in apicoplast function in Eimeria.
Apicomplexan genomes have a paucity of common eukaryotic transcription factors (Coulson et al. 2004); instead, the major regulators of stage-specific gene expression are likely to be genes containing DNA-binding domains of the ApiAP2 family (Balaji et al. 2005; Campbell et al. 2010). In Eimeria the number of genes containing ApiAP2 domains was found to vary from 44 to 54 (Supplemental Fig. S5; Supplemental Data set S1). We clustered genes containing ApiAP2 DNA-binding domains from apicomplexans and representative outgroups and identified 121 orthologous groups (Supplemental Fig. S6). We found 21 Eimeria-specific ApiAP2 groups, 22 further groups shared by Eimeria and other Coccidia, and five pan-apicomplexan clusters (apiap2_og_336, apiap2_og _90, apiap2_og _1428, apiap2_og _546, apiap2_og _456) (Supplemental Data set S1).
We found a positive correlation between the number of ApiAP2 genes and genome size across the Apicomplexa (r2 = 0.92; Pearson) (Supplemental Fig. S5). This suggests that although Eimeria has a relatively simple lifecycle compared with some other genera, there is greater complexity in regulating its genome. Thus, we propose that across Apicomplexa, it is not the complexity of the developmental lifecycle that determines the complexity of regulation, but the amount of genome to be regulated.
Analysis of chromosome 1 of E. tenella identified retrotransposon-like elements (Ling et al. 2007) and we now confirm that these are related to long terminal repeat (LTR) retrotransposons from the group of chromoviruses (Supplemental Fig. S7). Chromoviruses are widespread among eukaryotic genomes but have not previously been identified in apicomplexans (Kordis 2005). With the exception of eimten1 in the E. tenella and E. necatrix genomes, the putative transposons are highly fragmented and diverged, indicating very little recent activity (Supplemental Figs. S8, S9; Supplemental Table S8). Retrotransposons cannot have been transferred horizontally from the host, because the chicken genome does not contain chromoviruses (Kordis 2005). Phylogenetic analysis of predicted reverse transcriptases from Eimeria and other species did not robustly support a closer relationship with either plant/algal or vertebrate/fungal lineages (Supplemental Fig. S7).
Eimeria-specific surface antigen genes
All apicomplexan genomes examined to date possess gene families encoding antigenic proteins that are expressed on the surface of invasive stages and thought to be important in interaction with the host immune system (Spence et al. 2013). The principal surface antigen gene family in E. tenella is sag, which encodes single domain, membrane-bound proteins tethered by GPI anchors to the surface of invasive sporozoites and merozoites (Tabares et al. 2004).
In E. tenella the majority of sag genes are tandemly arrayed in four clusters, each on a different chromosome (Fig. 3A). There are three subfamilies of sag genes: sagA is common to all species; sagB is restricted to E. tenella and E. necatrix; and sagC is restricted to the other species, being most expanded in E. brunetti and E. mitis (Table 1). All subfamilies encode signal peptides and addition sites for GPI anchors, but the sagC extracellular domain contains only four conserved cysteines whereas sagA and sagB have six. The sagB and sagC genes each have five exons, suggesting they may be more closely related to each other than to sagA genes, which have four exons. The presence of sagA genes in all the Eimeria species suggests that these provide a core function, while sagB and sagC genes may provide functions specific to the different clades.
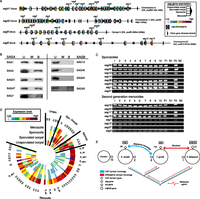
Analysis of the principle family of surface antigens in Eimeria spp. (A) The four loci of tandemly repeated sag genes in E. tenella are shown with each sag gene represented by bars describing the relative expression levels in four stages of the lifecycle. Gene names used previously in the literature are shown where appropriate. Arrows indicate direction of transcription. (B) We found that, of those tested, SAG proteins from subfamily A bound host cells, but those from family B did not. (U) Unwashed; (W) washed; (B) bound. (C) Expression of multiple sag genes in individual cells of E. tenella was detected using RT-PCR. Ten cells were analyzed for each of eight genes in both sporozoites and second-generation merozoites. (Lanes 1–10) Single-cell multiplex test RT-PCRs. Controls: (lane P1) positive, cDNA library multiplex RT-PCR; (lane N1) negative, no template multiplex RT-PCR; (lane P2) positive, single sporozoite, single target RT-PCR; (lane N2) negative, single sporozoite, single target PCR with no RT. (D) Expression values for all E. tenella sag genes were clustered and those clusters ordered by mean peak expression, showing that most genes peak in the second-generation merozoite. Where appropriate, genes are annotated with their genomic locus as defined in A. (E) Patterns of homology for Eimeria sag and Toxoplasma srs genes suggest that while Toxoplasma acquired the precursors to its key family of 6-cys surface antigens from a horizontal gene transfer of metazoan ephrin, Eimeria has derived them from the cysteine-rich secretory proteins, antigen 5, and pathogenesis-related 1 protein (CAP) family already found in Apicomplexa. Thin, single-headed arrows show phylogenetic paths for CAP- and ephrin-related domains, while bold arrows show the best evidence for the closest relatives of the sag and srs families. Numbers indicate gene frequencies for each family.
One core function of SAG proteins may be attachment to host cells prior to parasite invasion. In T. gondii the srs genes play a role in primary attachment and it is known that E. tenella SAG1 binds mammalian cells (Jahn et al. 2009). We found that multiple SAGA, but not SAGB proteins, were able to bind cultured cells (Fig. 3B), suggesting that attachment is a potential function of the sagA family.
It is of key importance in designing vaccines to understand the array of antigens presented to the host immune system. Using single-cell RT-PCR we found that multiple members of the sagA and sagB subfamilies were co-expressed in individual sporozoites and merozoites of E. tenella (Fig. 3C). Thus the parasite likely presents a complex set of antigens to the host, much like T. gondii SRSs, rather than a single one like Plasmodium falciparum PfEMP1. Analysis of stage-specific sag expression in populations of clonal E. tenella suggests that the expressed repertoire is most complex in second-generation merozoites (Fig. 3D). A small number of sagA genes peak in expression at each stage; sagB genes all peak in expression in second-generation merozoites, suggesting that they may be particularly important during the later, pathogenic, stages of infection (Fig. 3D).
The total number of sag genes varies greatly between species (Table 1). This may simply reflect the overall phylogeny (see Fig. 1A), but it is notable that species that cause the most severe pathologies have higher numbers of sags. Thus E. praecox, which causes only superficial damage and is widely regarded as the least pathogenic (Allen and Jenkins 2010), has only 19 sags, whilst E. tenella, E. necatrix, and E. brunetti, which develop deep in the mucosa causing tissue damage, inflammation, and intestinal hemorrhage (McDonald and Shirley 1987), have 89, 119, and 105 sags, respectively. However, E. mitis, with the greatest number of sags (172), does not fit this pattern; like all species it can impair bird performance and productivity but does not cause gross lesions in the intestine. It is also the case that species which induce potent immunity against reinfection after exposure to small numbers of parasites (E. maxima, E. praecox, and E. acervulina) have the lowest numbers of sags whilst those that are least immunogenic (E. necatrix and E. tenella) have high numbers; however, E. brunetti and E. mitis, which are of intermediate immunogenicity, do not fit this pattern.
We used remote homology detection to explore the evolutionary origin of the sag family. The cysteine-rich secretory protein family (CAP), found in a wide range of eukaryotes (Gibbs et al. 2008), had low but significant sequence similarity to sags (Fig. 3E). The most similar CAP-domain containing proteins were those of T. gondii, suggesting that sag genes are likely derived from CAP-domain containing genes in the common ancestor of E. tenella and T. gondi, rather than by horizontal transfer from another species such as the host. Recent protein structural evidence shows that the srs surface antigen gene family in T. gondii is related to a small group of cysteine-rich proteins in Plasmodium (Arredondo et al. 2012). These results suggest distinct evolutionary origins for the principle surface antigen gene families of the relatively closely related T. gondii and E. tenella.
Discussion
Control of pathogens such as Eimeria species has been essential for development of modern poultry production and is increasingly important for providing global food security. The availability of genomic resources for the seven Eimeria species that infect domestic chickens will underpin development and longevity of new anticoccidial drugs and vaccines.
The most striking feature of the Eimeria genomes is the disruption of more than half of all protein coding sequences with HAARs. Across every chromosome of each species we found regions where 20%–30% of the sequence comprised simple tandem repeats, interspersed with relatively repeat-free regions. Although a variety of different simple repeats were found outside of coding regions, those within coding regions were almost always based on runs of the trinucleotide CAG. We examined whether these repeats might have a particular function in the proteome but could find no association with known functional groups and showed that the repeats localized to structurally neutral regions within proteins. We hypothesize that poly-CAG is the most benign and easily evolvable coding repeat and that there is sufficient pressure on maintaining repeat banding to allow for frequent deleterious mutations. The repeat regions might function at the DNA level, perhaps in tertiary structure or gene regulation, and be selected for functional neutrality at the protein level, as we have observed. This hypothesis could be explored by chromatin immunoprecipitation and chromosome conformation capture studies. An alternative hypothesis is that rapid mutation of coding sequences provides evolutionary advantages, which would suggest selection for repeats at the protein level. Although the HAARs we examined appeared to be neutral in their effects on protein structures (presumably deleterious changes are lost in the population), occasional mutations may have been sufficiently beneficial to support the heavy burden. However, genes known to be involved in host–parasite interaction were found to be relatively protected from HAARs, and why repeats should have appeared in a banded pattern across chromosomes is not clear in this scenario.
We have characterized, for the first time, retrotransposon-like elements in an apicomplexan. Disruption or replacement of genes by transposon-mediated transgenesis holds the potential to improve our molecular arsenal for unraveling parasite biology, and also for developing attenuated vaccines. This approach has been attempted in Eimeria species using piggyBAC transposons; however, rates of insertion of these elements are low and target particular sequences (Su et al. 2012). Random, high-frequency insertions of native transposons would allow high throughput knockout screens, accelerating our understanding of Eimeria biology. The retrotransposons identified here are at best partially degraded and it is not clear whether they are actively retrotransposed. However, it opens the possibility that intact retrotransposons might be present in more distantly related Eimeria species.
To develop cheap, effective new vaccines for coccidiosis, we must understand how the parasites interact with the host immune system. Key to this may be the sag family of genes encoding surface antigens, some of which have been shown in vitro to induce pro-inflammatory cytokine responses (Chow et al. 2011). Whilst some sags are shared across all the Eimeria species studied, others are clade-specific. We found multiple sags to be co-expressed on the surface of infective parasites, suggesting that the avian immune system is presented with a diverse array of related epitopes, which could potentially aggravate inflammation. Parasites that rely on a healthy host for vector transmission are thought to use diverse arrays of related antigens to reduce pathogenicity (Spence et al. 2013) but for Eimeria, which has a simple oral-fecal life cycle, the induction of inflammation leading to diarrhea could increase parasite transmission. Eimeria sag genes have evolved from the CAP-domain superfamily of cysteine-rich secretory proteins and are unrelated to the major surface antigens of other Apicomplexa. CAP domains are also found on the surface of parasitic helminths where they are proposed to interact with host immune systems (Chalmers and Hoffmann 2012).
Methods
Parasite cultivation
The Houghton (H) strains of E. acervulina, E. brunetti, E. mitis, E. necatrix, E. praecox, and E. tenella, and the Weybridge (W) strain of E. maxima, were used for principle sequencing. The H strains were isolated at the Houghton Poultry Research Station (UK) and are the progeny of single oocyst infections. The E. maxima W strain was isolated at the Weybridge Central Veterinary Laboratory from a single oocyst infection. The Wisconsin (Wis) (McDougald and Jeffers 1976) and Nippon-2 (Nt2) E. tenella lines were used for comparative analyses. All parasites were propagated in vivo in 3- to 7-wk-old Light Sussex chickens under specific pathogen free (SPF) conditions at the Institute for Animal Health or Royal Veterinary College and purified using established methods (Long et al. 1976).
Summary of genome sequencing and assembly
Genomic DNA was prepared from purified sporulated oocysts. We used Sanger capillary sequencing data previously generated for E. tenella, described in Ling et al. (2007) and deposited in the Trace Archive (http://www.ncbi.nlm.nih.gov/Traces; CENTER_PROJECT = “EIMER”). These paired reads had a range of insert sizes to a combined coverage of ∼8×. E. tenella Illumina GAIIx sequencing libraries were prepared with insert sizes of 300 bp and 3 kbp and either 54 bp or 76 bp paired-end reads with a combined coverage of ∼160×. Capillary reads were assembled using ARACHNE v3.2 using default parameters (Batzoglou et al. 2002). IMAGE (Tsai et al. 2010) was used to fill gaps in scaffolds and extend contigs with Illumina reads, running six iterations (three with k-mer = 31 and three with k-mer = 27), mapping with BWA (Li and Durbin 2009). The consensus sequence from the ARACHNE-IMAGE assembly was corrected with Illumina reads using iCORN (Otto et al. 2010). All Illumina reads which did not map to this assembly were assembled using Velvet (Zerbino and Birney 2008) and these contigs were added to the final assembly.
For tier 2 (E. necatrix H, E. maxima W, and E. acervulina H) and tier 3 (E. mitis H, E. praecox H, and E. brunetti H) genomes, 500-bp Illumina TruSeq libraries were prepared and sequenced as 76-bp paired-end reads on an Illumina HiSeq 2000 platform to a depth of 199× theoretical genome coverage for E. acervulina, 288× for E. maxima, 559× for E. necatrix, 143× for E. brunetti, 520× for E. mitis, and 102× for E. praecox as in Kozarewa et al. (2009). Reads were assembled using Velvet (Zerbino and Birney 2008), scaffolding was performed with SSPACE (Boetzer et al. 2011), and gaps were filled using IMAGE (Tsai et al. 2010).
Full methodological details of genome sequencing, assembly, and annotation are provided in the Supplemental Material.
Whole-genome phylogeny
Each of 814 one-to-one ortholog groups shared across the seven Eimeria species with Toxoplasma gondii ME49 was aligned using MAFFT v7 (Katoh and Standley 2013). Highly variable sites were trimmed using trimal (Capella-Gutierrez et al. 2009) (“automated1” option). The alignments were concatenated using FASconCAT (Kuck and Meusemann 2010) and the resulting alignment used to construct a maximum likelihood phylogenetic tree using RAxML with the model PROTGAMMALG4XF (Stamatakis et al. 2005; Le et al. 2012), bootstrap n = 100. Bootstrap percentage support is shown along the branching nodes. The tree was rooted using T. gondii as the outgroup.
Transcriptome sequencing and analysis of Eimeria tenella
Unsporulated oocysts (two biological replicates), sporulated oocysts (single replicate), purified sporozoites (single replicate), and second-generation merozoites (single replicate) of the E. tenella H strain were selected for transcriptome analysis after harvest and purification as described previously (Novaes et al. 2012). Total RNA was extracted from oocysts using a Qiagen RNeasy kit (Qiagen) and from sporozoites and second-generation merozoites using the Qiagen RNeasy Animal Cells purification protocol as described by the manufacturer. All samples were DNase-treated using the Qiagen RNase-free DNase kit during RNA purification.
Library preparation, sequencing protocols, and data processing were the same as for Reid et al. (2012). For sag gene expression clustering in Figure 3D, mean RPKM values were taken for unsporulated oocyst samples and clustering performed using MBCluster.seq with 10 clusters (Si et al. 2014). Clusters were ordered by the stage of peak expression across each cluster. The figure was drawn with Circos (Krzywinski et al. 2009).
Synteny analysis
MCScanX (Wang et al. 2012) was used to determine regions of synteny between pairs of species based on the order of pairwise orthologs identified using OrthoMCL (Li et al. 2003). Default values were used except for the MATCH_SIZE option, which was set to 3 for comparison of E. tenella and T. gondii. For other comparisons it was set to 5.
Classification and analysis of gene families
We used several methods to identify and classify metabolic genes, protein kinases, transcription factors, and Eimeria-specific gene families. Full methodological details are available in the Supplemental Material.
Retrotransposon analysis
Approaches based on domain content and the presence of long terminal repeats were used to identify and classify retrotransposons. Full methodological details are available in the Supplemental Material.
Single-cell expression of sag genes
Single fluorescent E. tenella sporozoites and second-generation merozoites (Clark et al. 2008) were sorted into 96-well plates. RNA was reverse-transcribed for a panel of target transcripts by adding gene-specific reverse primers (Supplemental Table S9). Stage-specific multi-cell-derived cDNA or single-cell cDNA with each single-plex primer pair were included as positive controls P1 and P2. No template multiplex and no reverse transcription reactions were included for each assay as negative controls N1 and N2.
MDBK cell binding by SAGs
Confluent monolayers of MDBK cells were blocked with 1% BSA in PBS for 2 h at 4°C, washed three times in PBS, then incubated with recombinant-expressed EtSAG proteins (0.5 mg/mL) for 1 h at 40°C. Monolayers were washed four times with PBS to remove unbound proteins, then cells and bound proteins were solubilized in SDS sample buffer, separated by SDS-PAGE, transferred to nitrocellulose by electroblotting, and probed with rabbit hyperimmune sera raised to recombinant SAG proteins.
Classification of repeat regions
We used Tandem Repeat Finder (Benson 1999) to identify STRs in genomic sequences. It was run with the following parameters: 2, 1000, 1000, 80, 10, 25, 1000, as were used in Ling et al. (2007) and processed using TRAP v1.1 (Sobreira et al. 2006), run with default parameters. We defined a repeat density of ≥0.05 over a window of 1 kb as defining a repeat-rich region based on the distribution of repeat density across E. tenella. Scaffolds shorter than 5 kb were not included in the analysis.
We used SEG (Wootton and Federhen 1996) with parameters 7, 0, 0, –l, to identify HAARs of length ≥7. Repeats of “X,” the symbol used when the amino acid is unknown, and repeats which contained stop codons were removed from the SEG output file. To determine the HAAR content of protein sequences in other organisms, we ran SEG on all complete proteomes from UniProt (∼2000 species, ∼10-m sequences). We then ranked each species by the median number of repeats per sequence. We took the top 10, excluding viruses, and also T. gondii for comparison.
Full methodological details of protein structural and proteomic analyses of HAARs are available in the Supplemental Material.
Data access
Sequencing reads and assemblies for each genome have been submitted to the European Nucleotide Archive (ENA; http://www.ebi.ac.uk/ena/): E. tenella Houghton, E. maxima Weybridge, E. acervulina Houghton, E necatrix Houghton, E. mitis Houghton, E. brunetti Houghton, and E. praecox Houghton (PRJEB4918). Sequencing reads for E. tenella Nippon and E. tenella Wisconsin have been submitted to ENA (PRJEB4009). Transcriptome sequences for E. tenella Houghton have also been submitted to ENA (ERP001847) and ArrayExpress (https://www.ebi.ac.uk/arrayexpress/; E-ERAD-109).
Acknowledgments
The work was funded by BBSRC grants S17413 and S19705/6, Wellcome Trust core funding to Wellcome Trust Sanger Institute (WTSI), and faculty baseline funding from the King Abdullah University of Science and Technology (KAUST). We thank Mike Quail, Karen Mungall, Carol Churcher, and members of the core DNA pipelines for sequencing at WTSI and Bioscience core laboratories for sequencing operations at KAUST. We acknowledge Martin Aslett from WTSI for data submission and Dora Harvey and Fionnadh Carroll from the Institute for Animal Health for technical assistance. K.-L.W. would like to acknowledge funding from the Ministry of Science, Technology and Innovation, Malaysia (Project No. 07-05-16-MGI-GMB10) and the Universiti Kebangsaan Malaysia (Project No. DIP-2012-21). J.P. and S.S.H. were funded by the Canadian Institute for Health Research (CIHR #MOP84556).
Footnotes
-
↵21 Retired.
-
↵22 Deceased.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.168955.113.
Freely available online through the Genome Research Open Access option.
- Received October 29, 2013.
- Accepted July 8, 2014.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








