
High-throughput functional testing of ENCODE segmentation predictions
- Center for Genome Sciences and Systems Biology, Department of Genetics, Washington University School of Medicine, St. Louis, Missouri 63110, USA
- Corresponding author: cohen{at}genetics.wustl.edu
-
↵1 These authors contributed equally to this work.
Abstract
The histone modification state of genomic regions is hypothesized to reflect the regulatory activity of the underlying genomic DNA. Based on this hypothesis, the ENCODE Project Consortium measured the status of multiple histone modifications across the genome in several cell types and used these data to segment the genome into regions with different predicted regulatory activities. We measured the cis-regulatory activity of more than 2000 of these predictions in the K562 leukemia cell line. We tested genomic segments predicted to be Enhancers, Weak Enhancers, or Repressed elements in K562 cells, along with other sequences predicted to be Enhancers specific to the H1 human embryonic stem cell line (H1-hESC). Both Enhancer and Weak Enhancer sequences in K562 cells were more active than negative controls, although surprisingly, Weak Enhancer segmentations drove expression higher than did Enhancer segmentations. Lower levels of the covalent histone modifications H3K36me3 and H3K27ac, thought to mark active enhancers and transcribed gene bodies, associate with higher expression and partly explain the higher activity of Weak Enhancers over Enhancer predictions. While DNase I hypersensitivity (HS) is a good predictor of active sequences in our assay, transcription factor (TF) binding models need to be included in order to accurately identify highly expressed sequences. Overall, our results show that a significant fraction (∼26%) of the ENCODE enhancer predictions have regulatory activity, suggesting that histone modification states can reflect the cis-regulatory activity of sequences in the genome, but that specific sequence preferences, such as TF-binding sites, are the causal determinants of cis-regulatory activity.
It is widely reported that specific combinations of covalent histone modifications reflect the regulatory function of underlying genomic DNA sequence (Strahl and Allis 2000). As part of the ENCODE Project, the genomic locations of a variety of covalent histone modifications were determined by chromatin immunoprecipitation sequencing (ChIP-seq) in a number of cell types and cell lines. Two studies used these data to train computational models that predict different functional regions of the human genome. These unsupervised learning algorithms, Segway (Hoffman et al. 2012) and ChromHMM (Ernst and Kellis 2010, 2012), take functional genomics data as input (DNase-seq; FAIRE-seq; and ChIP-seq of histone modifications, RNA polymerase II large subunit [POLR2A], and CTCF) and return segmentation classes, which are then assigned a hypothesized function using current knowledge of histone modification function. As part of the ENCODE Project, these two sets of predictions were consolidated to create a unified annotation of the entire human genome with seven functional classes in multiple cell types. These segmentations include Transcription Start Site, Promoter Flanking, Transcribed, CTCF-bound, Enhancer, Weak Enhancer, and Repressed or Inactive segments (The ENCODE Project Consortium 2012; Hoffman et al. 2013). If histone modifications accurately reflect the regulatory activity of their associated DNA, then these segmentation classes should have measurably different cis-regulatory activities.
In this study we tested whether the segmentation classes determined by ENCODE have different effects on gene regulation in their predicted cell type. We used the accepted operational definition of enhancer activity as the ability to modulate expression of a reporter gene under control of a basal promoter. We used CRE-seq, a massively parallel reporter assay, to determine whether (1) sequences in the Enhancer, Weak Enhancer, and Repressed classes drive expression that is different from that produced by negative controls, (2) sequences in different segmentation classes drive different levels of gene expression, and (3) sequences control gene expression levels consistent with their predicted segmentation labels. We find that segmentation predictions drive distinct levels of expression. In particular, enhancer predictions drive expression that is different from the expression levels driven by negative control sequences. We find that chromatin features can distinguish highly expressed sequences with some accuracy, but transcription factor (TF)-binding preferences better identify the most highly expressed sequences.
Results
CRE-seq library and measurements
We used a high-throughput multiplexed reporter assay (Kwasnieski et al. 2012; Melnikov et al. 2012; Patwardhan et al. 2012; Sharon et al. 2012) to characterize the regulatory activity of 2100 randomly chosen sequences annotated as Enhancer, Weak Enhancer, or Repressed. Specifically, we tested sequences with the following annotations in the K562 cell line: 600 Enhancer regions, 600 Weak Enhancer regions, and 300 Repressed regions. In order to test the cell-type specificity of the segmentation predictions, we also tested 600 Enhancer predictions from the H1-hESC cell line that are not annotated as Weak Enhancers or Enhancers in K562 cells.
We sought to establish an empirical null distribution as a negative control for activity in this assay, against which to compare the activities of sequences from the different segmentation classes. We randomly selected 284 sequences from each class of predictions and scrambled the nucleotide sequence of each while maintaining dinucleotide content, in order to preserve basic sequence features of the segment such as CpG frequency and nucleosome favoring signals. We designed our experiment to compare the expression distribution for each segmentation class to the expression distributions from their corresponding scrambled negative controls. Including predicted cis-regulatory elements (CREs) and scrambled negative controls, our final experimental design included 3237 distinct reporter gene constructs (Supplemental Data 1).
We used CRE-seq, a massively parallel reporter gene assay (Kwasnieski et al. 2012), to simultaneously measure the expression of all constructs. We first synthesized 13,000 unique 200-mer DNA sequences using array-based oligonucleotide (oligo) synthesis (LeProust et al. 2010). Each predicted CRE was replicated four times on the array, and each replicate was tagged with a unique nine–base-pair (bp) barcode, providing redundancy in the expression measurements. The 200-bp limit of oligonucleotide synthesis, along with the requirement to include priming sites and restriction enzyme sites, limited our tested CREs to 130 bp of each segmentation prediction. For the Enhancer and Weak Enhancer classes, we selected the entire region of 300 short (121–130 bp) genomic segments, and the central 130 bp of 300 longer genomic segments (>130 bp). Because only a small fraction of Repressed segments are <130 bp in length, we tested only central sequences from this class. We chose the center because it is an unbiased portion that does not incorporate additional histone or sequence features beyond the algorithms’ output. This allows us to appropriately test the predictive power of the segmentations. Finally, we used the array-synthesized oligos to create a library of these CREs cloned upstream of the Hsp68 minimal promoter in which each reporter construct contains a unique sequence barcode in its 3′ UTR (Kwasnieski et al. 2012). The resulting plasmid library was then transfected into K562 cells, and RNA was isolated after 22 h.
To measure CRE activity, we quantified the level of each barcode in the transfected cells using RNA-seq, and normalized the RNA barcode counts by the abundance of each barcode in the plasmid DNA pool. The RNA/DNA ratio of barcode counts is a quantitative measure of the expression driven by each CRE in the library (Supplemental Data 2, 3; Kwasnieski et al. 2012). We performed four independent transfections in K562 cells and found that our expression measurements are precise, displaying high reproducibility between biological replicates (R2 range: 0.95–0.97) (Fig. 1A). To test the robustness of our measurements, we used a luciferase assay to measure expression driven by 12 individual CREs upstream of the minP basal promoter. Expression in the luciferase assay exhibits strong agreement with the batch CRE-seq expression measurements upstream of the Hsp68 promoter (R2 = 0.70) (Fig. 1B; Supplemental Fig. 1), demonstrating that our assay accurately measures cis-regulatory activity and that our results have little dependence on the choice of minimal promoter.
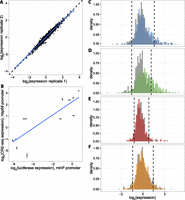
Reproducible expression measurements show differences in expression by segmentation class. (A) Representative scatterplot showing expression of each CRE in two biological replicates (R2 = 0.95, range of R2 between all replicates: 0.95–0.97). Dashed black line is line of equality and blue line is best fit. (B) Correlation between CRE-seq and luciferase assays. Expression driven by 12 CREs was measured in individual luciferase assay (upstream of minP promoter, x-axis) and batch CRE-seq assay (upstream of Hsp68 promoter, y-axis). Luciferase expression is normalized to the Renilla transfection control, and CRE-seq expression is normalized to the basal promoter alone. Error bars represent the standard error of the mean. Blue line is best fit. R2 = 0.70. (C–F) Histograms of genomic CRE expression measurements in K562 cells. Each class is compared to scrambled controls with equivalent GC and dinucleotide content (gray). Dashed lines are the fifth and 95th percentiles of the scrambled distributions. (C) K562 Enhancer class (blue), (D) K562 Weak Enhancer class (green), (E) K562 Repressed class (red), (F) H1-hESC Enhancer class (orange).
Expression of segmentation classes
We compared the activity of each class of segmentation prediction to the activity of its corresponding negative control distribution of scrambled sequences. We used two metrics to classify individual segmentations as “active” or “inactive” with respect to this negative control expression distribution (Table 1). First, we computed the fraction of CREs within a segmentation class that drives expression higher than that of the 95th percentile of the matched scrambled expression distribution. We recognized that CREs may be active even if they drive expression below the 95th percentile of the control, so we also used a second metric to capture some of these sequences. We compared the 16 replicate measurements for each CRE (four barcodes per CRE in four independent experiments) with the distribution of all of the scrambled controls (Wilcoxon rank sum test, one-tailed, P < 0.05, Bonferroni correction with N = 3236). We conducted the same test for each scrambled CRE to estimate the fraction of scrambled sequences that drive activity (Table 1, square brackets). By both of these metrics, a significant number of Enhancer and Weak Enhancer predictions are active (Fig. 1C,D; Table 1). In contrast, neither the K562 Repressed regions nor the H1-hESC Enhancer regions show activity that is significantly different from their scrambled negative controls (Fig. 1E,F; Table 1). Enhancer and Weak Enhancer regions show distinct levels of activity from both the K562 Repressed and H1-hESC Enhancer regions (Wilcoxon rank sum, P < 0.01). Moreover, segmentations from the Repressed category did not repress expression below the fifth percentile of their matched scrambled controls, suggesting that these sequences are transcriptionally inactive and not repressive (Supplemental Table 1). We get the same results regardless of whether the sequences are short segmentations included in their entirety, or longer predictions from which we included only the central 130 bp (Supplemental Fig. 2). This result indicates that our expression measurements are not biased by the method of choosing 130-bp sequences for testing. Taken together, we conclude that sequences annotated as Enhancer and Weak Enhancer segments have increased levels of activity over their corresponding null distributions, and that different segmentation classes produce distinct median levels of activity in our assay.
Percentage of active CREs by segmentation class
Our previous work (White et al. 2013) showed that CRE-seq can detect repression below basal promoter activity, particularly when the minimal promoter has detectable expression on its own. In this experiment we chose the Hsp68 promoter because it drives expression in the 48th percentile of the library of genomic sequences. Many sequences, both segmentation predictions and scrambled sequences, drove expression that was significantly lower than the scrambled distribution, indicating that we can detect repression in this assay. However, we observed no significant increase in the number of sequences with repressive activity in the segmentations as compared with the scrambled sequences, suggesting that the segmentations do not repress expression below what is expected by chance (Wilcoxon rank sum Test, P < 0.05, Bonferroni correction) (Supplemental Table 1). We conclude that Enhancer, Weak Enhancer, and Repressed segmentations do not have the ability to repress the Hsp68 promoter.
Unexpectedly, we found that sequences classified as Weak Enhancers drive a higher median level of activity than sequences classified as Enhancers (P = 3.7 × 10−4 by Wilcoxon rank sum) (Supplemental Fig. 3). The difference between the two classes is even greater when comparing the fraction of CREs we designated as “active” relative to their matched scrambled sequences (Table 1). Compared to Weak Enhancers, segmentations in the Enhancer class have higher GC content (Supplemental Fig. 4B), a sequence feature associated with higher cis-regulatory activity (Landolin et al. 2010; Lidor Nili et al. 2010; White et al. 2013). Indeed, scrambled sequences derived from the Enhancer class drive expression higher than scrambled sequences from the Weak Enhancer class (Supplemental Fig. 4A). Therefore, despite having higher GC content, a feature associated with higher expression, the Enhancer predictions drive expression lower than the Weak Enhancer predictions. This suggests that some additional determinant is responsible for the higher activation of segments labeled as Weak Enhancers.
We asked whether differences in covalent histone modifications correlate with the difference in expression between Weak Enhancers and Enhancers. We compared the levels of all histone modifications (Hoffman et al. 2013) that were measured in K562 cells between the two classes. Weak Enhancers were segmented from Enhancers by their lower levels of the histone modification H3K27ac (Fig. 2B; Creyghton et al. 2010), thought to signify active enhancers, and H3K36me3 (Fig. 2D; Barski et al. 2007), often thought to signify a transcribed gene body but recently also found in silenced genes (Chantalat et al. 2011). Surprisingly, lower levels of both of these covalent histone modifications are associated with higher expression of enhancers in our assay (Wilcoxon rank sum test, P < 10−5) (Fig. 2A,C), even within the Enhancer or Weak Enhancer classes (Supplemental Fig. 5). We did not find an association of H3K27ac signal in the larger context (up to 500 bp surrounding the selected regions). In one study, “dips” in the levels of H3K27ac correlated with enhancer activity (Kheradpour et al. 2013), which is consistent with our observation that lower levels of H3K27ac are more predictive of enhancer activity. However, in our data we did not see correlation between the H3K27ac “dip score” and cis-regulatory activity. Thus, Weak Enhancers may have more activity than Enhancers in part because they have lower enrichment of H3K27ac and H3K36me3, which associate with higher activity in our assay. These histone modifications do not fully explain the expression differences between these two classes, indicating that other sequence features must explain the higher activity of Weak Enhancers.
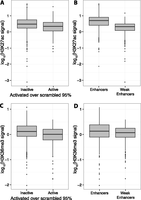
Lower H3K27ac and H3K36me3 signals are associated with higher Weak Enhancer expression. Boxplots showing that H3K27ac signal (A) and H3K36me3 signal (C) are depleted in active CREs compared to inactive CREs. H3K27ac signal (B) and H3K36me3 signal (D) are also depleted in Weak Enhancers compared to Enhancers. Active CREs are those above the 95th percentile of scrambled distribution (Table 1).
Sequence and chromatin features
We searched for sequence and chromatin features that could predict activity across all segmentation classes in our assay. Two primary sequence features (PSFs) (GC content and minor groove width as estimated by ORChID2 [Rohs et al. 2009; Bishop et al. 2011] score) and six chromatin features (The ENCODE Project Consortium 2012; Hoffman et al. 2013) (DNase I HS from Duke; DNase I HS from University of Washington [UW]; FAIRE-seq; and ChIP-seq of H3K4me1, H3K36me3, and RNA polymerase POLR2A) are significantly enriched in sequences that drive high expression in our assay (Wilcoxon rank sum test, P < 0.05 Bonferroni correction with N = 16) (Supplemental Table 2). We used these data to develop a quantitative model that distinguishes active CREs from inactive CREs. Of these eight features, DNase I HS (UW) signal best separated the active from inactive sequences (AUC = 0.685) (Fig. 3A,B), suggesting that DNA accessibility is a good indicator of the cis-regulatory potential of a sequence (Thurman et al. 2012). No other single feature performed as well as DNase I HS signal and all other single features had AUC lower than 0.6 (Supplemental Table 2). A logistic regression model with the above-mentioned six chromatin features and two PSFs improves the classification of active sequences (AUC = 0.733) (Fig. 3A), but only marginally above that of DNase I HS alone. However, even among those CREs with a high DNase I HS score (UW DNase I HS score > 5, 685/2096 CREs pass this threshold), the active CREs are enriched for seven chromatin features, suggesting that there is some additional information in the histone modifications beyond DNase I HS despite the fact that DNase I HS is by far the most predictive feature (Supplemental Table 3). As chromatin and PSFs can only classify active sequences to a moderate level, we hypothesized that additional sequence-specific binding features, such as TF-binding motifs, may better explain expression.
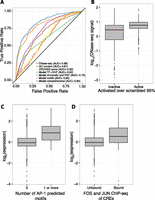
Chromatin features and sequence-specific binding identify active sequences. (A) Receiver operating characteristic (ROC) curve shows that a logistic regression model (“Model comprehensive”) incorporating sequence-specific binding motifs, chromatin features, primary sequence features (PSFs), and TF-ChIP data is best able to identify active sequences. Of logistic regression models with fewer features, one with sequence-specific binding motifs (“Model motifs”) does best, followed by a model incorporating chromatin and primary sequence features (“Model chromatin and PSF”), and a model with only significant TF-ChIP features (“Model TF-ChIP”). Minor groove width as predicted by ORChID2 score, GC content, and DNase I HS are also shown. Area under the curve (AUC) is indicated in legend. (B) Boxplot showing that active CREs are enriched in high DNase I HS signal over inactive CREs. (C) Boxplot showing that CREs with at least one predicted AP-1 motif drive expression higher than CREs with no AP-1 predicted motifs. (D) CREs overlapping with ChIP-seq peaks for a FOS (FOS or FOSL1) family member and a JUN (JUNB or JUND) family member, the constituent proteins of AP-1, drive expression higher than unbound CREs.
We investigated whether the inclusion of TF-binding specificities improved our ability to explain the expression differences we observed in our assay. Using several libraries of TF-binding models (Newburger and Bulyk 2009; Jolma et al. 2013; Mathelier et al. 2014), we searched for motifs enriched or depleted in activated CREs and found 50 significant, nonredundant motifs (Supplemental Table 4). A logistic regression model that incorporated these binding models performs better at distinguishing active sequences than the chromatin and PSF model (AIC [Akaike 1974]: 1881 vs. 1729 for model with motifs; AUC = 0.802) (Fig. 3A). We performed fivefold cross-validation on all of the models and observed little decrease in predictive power, suggesting that our model is not over-fit (Supplemental Table 5). The predicted motif for activator protein 1 (AP-1), a heterodimer of TFs in the FOS and JUN families (Hess et al. 2004), is the most significantly enriched motif in highly expressed CREs. In addition, the most significant motif found in a discriminative de novo motif analysis (Bailey 2011) was highly similar to the AP-1 motif (E = 0.0041) (Gupta et al. 2007). Among segmentations with a predicted AP-1 motif, DNase I HS (Duke) is the only chromatin feature significantly enriched in those that are active (Supplemental Table 3), suggesting that DNase I HS provides some additional information beyond the presence of the AP-1 motif. The expression driven by CREs with predicted AP-1 motifs is significantly higher than the expression driven by sequences without the motif (log2 ratio of 0.96, P < 2.2 × 10−16) (Fig. 3C). Furthermore, highly expressing CREs are significantly enriched for sequences that are bound by FOS and JUN family TFs in K562 cells (P = 8.8 × 10−10 by Fisher’s exact test, odds ratio = 4.2) (Fig. 3D; The ENCODE Project Consortium 2011). These data suggest that AP-1 is responsible for the activity of many enhancers in K562 cells, as previously reported (Muthukrishnan and Skalnik 2009; Kheradpour et al. 2013), and, as a consequence, the enhancers’ histone modification state.
Discussion
In this study we directly tested the cis-regulatory activity of segmentation predictions based on histone modification data from the ENCODE Project. We found that these predictions were cell type-specific in K562 cells and could accurately distinguish enhancer sequences from non-enhancer sequences. Our results suggest that combinations of TF-binding preferences, not histone modifications alone, are most predictive of actively expressing genomic sequences, a result supported by other attempts to define the sequence features of enhancers (Heinz et al. 2010; Lee et al. 2011; Arvey et al. 2012; Gorkin et al. 2012; Smith et al. 2013). These results support a model where TF binding and subsequent transcriptional regulation configure the immediate chromatin environment (Struhl and Segal 2013), leading to the constellation of histone modifications observed in segments with high cis-regulatory activity. However, even our model incorporating all of the available features is only moderately predictive (AUC = 0.84) and cannot quantitatively predict expression level. This suggests that more complex features determine the quantitative expression levels controlled by enhancers.
We conclude that the Repressed segmentation class consists mostly of sequences with no transcriptional activity rather than cis-regulatory sequences that actively repress transcription. We have previously shown transcriptional repression by short enhancers (White et al. 2013), indicating that the length of CREs we tested cannot explain the lack of observed repression. There are two possible explanations for why we did not see repression in this assay. First, the Repressed segmentation class contains mostly sequences with predicted low activity by either the ChromHMM or Segway algorithms, with only a small fraction of the sequences predicted to have repressive activity by these algorithms. Second, it is possible that we are unable to predict combinations of histone modifications that signal repression such that no segmentation successfully defines repressive activity. Because a large fraction of regulated gene expression works through the activity of transcriptional repressors, identifying combinations of histone modifications that reflect repression is still an important challenge.
Only a small fraction (∼26%) of predicted enhancer sequences had activity in this assay. It is therefore possible that a large fraction of the predictions in ChromHMM/Segway are false positives. Alternatively, many sequences might score as false negatives in this assay. The short length and episomal nature of the expression assay could contribute to false negatives, although we emphasize that the accepted operational definition of an enhancer is a sequence that modulates the activity of an episomal reporter gene. In addition, our comparison of segmentations to scrambled controls does not allow us to find active sequences that express at low levels. Finally, it is possible that some sequences might only be active in the context of the genome or when paired with a different minimal promoter sequence. While the relative number of active sequences between classes in our assay should be accurate, as the same experimental design was utilized for all sequences, our estimates should be taken as a lower bound of the number of active sequences.
Finally, we conclude that combinations of histone modifications often identify functional enhancers, but our interpretation of these combinations needs to be refined. In particular, high levels of the covalent histone modifications H3K27ac and H3K36me3 are thought to mark active enhancers and transcribed gene bodies or even heterochromatic regions (Barski et al. 2007; Creyghton et al. 2010; Chantalat et al. 2011). Among segments marked as Enhancers or Weak Enhancers, lower enrichment of these modifications is found at segments with high activity in this assay. This finding suggests that the precise function of these modifications needs to be explored, as it is clear that there is no simple linear relationship between the level of these modifications and expression.
Methods
CRE-seq library construction
A pool of 13,000 unique 200-mer oligos was ordered through a limited licensing agreement with Agilent Technologies. Oligos were structured as follows: 5′ priming sequence (GTAGCATCTGTCC)/NheI site/CRE/HindIII site/XhoI site/SphI site/barcode/SacI site/3′ priming sequence (CGACTACTACTACG). A more detailed diagram of array sequence is provided in Supplemental Figure 6.
The plasmid library was prepared as previously described (Kwasnieski et al. 2012), except using primers CF166 and CF167 (Supplemental Table 6) and an annealing temperature of 57°C. The amplified library product was purified on a polyacrylamide gel as previously described (White et al. 2013). The library plasmid backbone, CF10, was created from the plasmid pGL4.23, by cloning dsRed-Express2 between the Acc65I and FseI sites. Purified library amplicons were cloned into CF10 using NheI and SacI. We prepared DNA from 100,000 colonies to generate PL7_1. We then cloned the Hsp68 promoter driving DsRed into PL7_1. A cassette containing the Hsp68 promoter was amplified from pGL-hsp68 with primers CF121 and CF168 (Supplemental Table 6). pGL-hsp68 was created by amplifying the Hsp68 promoter from hsp68LacZ (kind gift of M. de Bruijn, Oxford Stem Cell Institute, Oxford, UK) using primers JKO25F and JKO25R (Supplemental Table 6). The Hsp68 DsRed amplicon was cloned into library PL7_1 by using HindIII and SphI, creating library PL7_2.
Cell culture and transfection
K562 cells were maintained in Iscove’s modified Dulbecco’s medium (IMDM) with 10% fetal bovine serum and 1% amino acids (Life Technologies). The plasmid library was purified by phenol-chloroform extraction and ethanol precipitation before transfection. The Neon transfection system (Life Technologies) was used to transfect the plasmid library. For each replicate, 1.2 million cells were pelleted by centrifugation, washed with PBS and resuspended in 100 μL of Buffer R. Twenty-seven micrograms of plasmid library DNA along with 3 μg of pMax-GFP as a positive control was transfected into the cells by using three 10-msec pulses at 1450V. The transfected cells were seeded into T-25 flasks with 5 mL of the growth medium and incubated at standard conditions. Transfection efficiency was >90% (data not shown).
Selection of segmentation predictions
Segmentation predictions (The ENCODE Project Consortium 2012; Hoffman et al. 2013) were downloaded from the Ensembl Genome Browser (Flicek et al. 2013) and converted to UCSC notation. We filtered predictions that overlapped with the ENCODE DAC Blacklisted Regions (http://moma.ki.au.dk/genome-mirror/cgi-bin/hgFileUi?db=hg19&g=wgEncodeMapability) or RepeatMasker regions (http://www.repeatmasker.org/species/homSap.html). We also removed predictions that contained restriction site sequences that we intended to use for cloning sequences into a plasmid library. To select H1-hESC Enhancer predictions, we removed H1-hESC Enhancer predictions that overlapped with K562 Enhancer or Weak Enhancer predictions. Next we sorted predictions by chromosome, and separated them by length into long (>130 bp) and short (121–130 bp). To choose the predictions to test, we selected lines of this file at regular intervals, so the tested CREs span all chromosomes of the human genome. Genomic and scrambled CRE sequences are listed in Supplemental Data 1. All genomic coordinates used are from hg19.
Preparing samples for RNA-seq
RNA was extracted from K562 cells 22 h after transfection using the PureLink RNA mini kit (Life Technologies) and then excess DNA was removed using the TURBO DNA-free kit (Applied Biosystems), following the manufacturer’s instructions. First, strand cDNA was synthesized from the RNA using SuperScript II Reverse Transcriptase (Life Technologies). Both the cDNA samples and the DNA from the original plasmid library were prepared for sequencing using a custom protocol as previously described (Kwasnieski et al. 2012). Briefly, we used PCR amplification of the sequence surrounding the barcode in the RNA transcript or plasmid using primers CF150 and CF151b (Supplemental Table 6). We then digested the PCR product using SphI and XhoI and ligated Illumina adapter sequences (MO576/582, MO577/583, MO578/584, MO579/585) (Supplemental Table 6) to these amplified sequences. Two lanes of the Illumina HiSeq machine were used to sequence this barcode region from the cDNA and DNA, and reads that perfectly matched the first 13 expected nucleotides were counted, regardless of quality score. This resulted in 77.5 million reads from the cDNA, across four biological replicates, and 34.8 million reads from the DNA. Only barcodes with ≥50 reads in the DNA pool and ≥3 reads in the cDNA pool were used for downstream analysis. The expression of each barcode was calculated as (cDNA reads)/(DNA reads) and then normalized to the expression of the basal promoter alone (Supplemental Data 2). The expression of each CRE in each biological replicate was calculated as the mean of the expression of each BC associated with it, and the overall expression of each CRE was calculated as the mean of its expression in each biological replicate. The standard error of the mean (SEM) was calculated as previously described (Kwasnieski et al. 2012) (Supplemental Data 3).
Luciferase assays
Plasmid pGL-CBR was created by inserting the click-beetle red (CBR) luciferase gene (from pCBR-Control Vector [Accession Number AY258592], Promega) into pGL4.23 (Promega) at the XbaI and NcoI sites. pGL-CBR contains the minP basal promoter from pGL4.23. Twelve individual CREs from the oligo library were amplified by PCR and inserted into pGL-CBR at the NheI and HindIII sites to form individual pGL-CBR-CRE plasmids. The 46-bp cis-regulatory element containing the HS II enhancer from Ney et al. (1990) was also cloned into pGL-CBR using annealed oligos POS1 and POS2 (Supplemental Table 6), also at the NheI and HindIII sites of pGL-CBR, to create a positive control pGL-CBR-CRE plasmid. Each pGL-CBR-CRE plasmid, along with the original pGL-CBR, was then transfected into K562 cells individually in triplicate using the Neon transfection system. Each transfection used 4 μg pGL-CBR-CRE plasmid with 0.4 μg Renilla control plasmid (pRL-CMV, Promega) and 2 × 105 cells. Transfected cells were then seeded into 12-well plates with 1 mL of growth media. Twenty-six hours later, each well was split into two wells, each in a separate 24-well plate (Krystal 24 Well Black Assay Plate, MidSci). These were then immediately imaged using I IVIS 50 (Caliper; exposure time 10–60 sec, binning 8, field of view 12, f/stop 1, open filter), with one plate imaged for CBR-luciferase using 535 μM D-luciferin (Gold Biotech), and one plate imaged for Renilla using 400 nM Coelenterazine (Biotium Inc.). The CBR-luciferase signal of each transfection sample was normalized by the corresponding Renilla signal, and the expression of each CRE was determined by the mean of the three transfections (Supplemental Data 4).
Data sources
We used the normalized chromatin ChIP-seq, FAIRE-seq, and DNase-seq data used in the integrated segmentation of the genome by Hoffman et al. (2013), which can be accessed at https://sites.google.com/site/anshulkundaje/projects/wiggler. These included (all from K562 cell line): CTCF, Duke DNase I, UW DNase I, FAIRE, H3K27ac, H3K27me3, H3K36me3, H3K4me1, H3K4me2, H3K4me3, H3K9ac, H4K20me1, RNA POLR2A, and Control. These data were produced by the ENCODE Consortium (The ENCODE Project Consortium 2012). The signal associated with each CRE we analyzed was the average signal over that segment.
The TF-binding matrices were taken from three databases: JASPAR vertebrate (146 matrices) (Mathelier et al. 2014), uniPROBE (757 matrices) (Newburger and Bulyk 2009), and high-throughput SELEX (820 matrices) (Jolma et al. 2013). FIMO (Grant et al. 2011) was used to find binding sites in the CREs used in the assay (both genomic and scrambled), using the default options with a P-value threshold of 10−4. The AP-1 binding matrix that was enriched in highly expressed sequences in our assay was from JASPAR (MA0099.2). DREME (Bailey 2011) was used for discriminative motif finding, using the sequences activated over the 90th percentile of the scrambled distribution as the positive group and all other sequences as the negative group, with the maximum motif length set at 12 bp and all other default options. The TOMTOM web module (http://meme.nbcr.net/meme/cgi-bin/tomtom.cgi) (Gupta et al. 2007) was used to find similar motifs, using default options.
TF ChIP-seq data were obtained from http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeRegTfbsClustered/. GC-content and ORChID2 (Rohs et al. 2009; Bishop et al. 2011) scores were calculated from the nucleotide sequences of the CREs.
Logistic regression models
A logistic regression model was developed to predict sequences activated over the scrambled 90th percentile. The parameters for the model were chosen from a filtered list of available genomic data and sequence features. Each of the three sets of parameters was filtered separately: histone data including PSFs (GC-content and ORChID2 scores), binding matrices, and a set of peaks from TF ChIP-seq. Those scores that had a significantly different distribution of values in the active CREs (expression >90th percentile of the matched scrambled distribution) vs. the inactive CREs passed the filter. For the parameter set with histone data and PSFs and the parameter set with binding matrices, we used the Wilcoxon rank sum test (two-tailed, P < 0.05, corrected using Bonferroni with N = 16 for histone and N = 1687 for binding matrices). For the TF ChIP-seq peak data (which is in binary form), we used Fisher’s exact test (P < 0.05, corrected using Bonferroni with N = 16). Seventy-three binding matrices, eight histone with PSF parameters (including GC-content and ORChID2 scores), and eight TF ChIP-seq parameters passed the filter. The binding matrices were further filtered to remove those that showed nearly identical binding patterns across the CREs (≥99% similar), resulting in 50 binding matrices.
A logistic regression model for predicting actively expressed CREs was created for each of the three sets of parameters separately and with all sets of parameters together (66 total parameters). Only additive terms were used. We then created receiver operating characteristic (ROC) curves attempting to correctly predict the activated CREs (>90th percentile of the matched scrambled distribution). The area under the curve (AUC) was calculated for each model as well as the best performing histone parameter (UW DNase I HS), GC-content, and ORChID2 scores. Additionally, fivefold cross validation was used to ensure our models were not over-fit. The CREs were split into five training groups, and the model was trained on the data holding out each group in turn (beginning with the filtering of the parameters) and tested on the group held out. AUC was calculated for each of these sets, and the mean AUC from the five sets was calculated (Supplemental Table 5).
Acknowledgments
We thank Shondra Miller for advice on our transfection protocol; Lynne Collins for assistance with our luciferase assay; the Kyunghee Choi laboratory for the K562 cell line; the Marella de Bruijn laboratory for the Hsp68LacZ plasmid; Ilaria Mogno, Mike White, and Brett Maricque for feedback on our project design; and members of the Cohen laboratory for their manuscript comments. This work was supported by a grant from the National Institutes of Health (R01 GM092910).
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.173518.114.
- Received February 3, 2014.
- Accepted July 17, 2014.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








