
Maize centromeres expand and adopt a uniform size in the genetic background of oat
- 1Department of Horticulture, University of Wisconsin-Madison, Madison, Wisconsin 53706, USA;
- 2Department of Plant Biology, University of Georgia, Athens, Georgia 30602, USA
-
↵3 These authors contributed equally to this work.
Abstract
Most existing centromeres may have originated as neocentromeres that activated de novo from noncentromeric regions. However, the evolutionary path from a neocentromere to a mature centromere has been elusive. Here we analyzed the centromeres of nine chromosomes that were transferred from maize into oat as the result of an inter-species cross. Centromere size and location were assayed by chromatin immunoprecipitation for the histone variant CENH3, which is a defining feature of functional centromeres. Two isolates of maize chromosome 3 proved to contain neocentromeres in the sense that they had moved from the original site, whereas the remaining seven centromeres (1, 2, 5, 6, 8, 9, and 10) were retained in the same area in both species. In all cases, the CENH3-binding domains were dramatically expanded to encompass a larger area in the oat background (∼3.6 Mb) than the average centromere size in maize (∼1.8 Mb). The expansion of maize centromeres appeared to be restricted by the transcription of genes located in regions flanking the original centromeres. These results provide evidence that (1) centromere size is regulated; (2) centromere sizes tend to be uniform within a species regardless of chromosome size or origin of the centromere; and (3) neocentromeres emerge and expand preferentially in gene-poor regions. Our results suggest that centromere size expansion may be a key factor in the survival of neocentric chromosomes in natural populations.
Centromeres can be stable for hundreds of thousands of years, but under rare circumstances have been known to change positions along the chromosomes. Examples of centromere repositioning have been documented in both plant and animal species as revealed by comparative genomics (Han et al. 2009; Rocchi et al. 2012). An early example involved the comparison of X chromosomes from human and two lemur species (Ventura et al. 2001). Gene order is strongly conserved on the three X chromosomes, yet the centromeres are in different locations, indicating that the centromeres underwent dramatic and yet poorly understood repositioning events (Ventura et al. 2001). One way to study centromere repositioning is to focus on newly established centromeres known as neocentromeres. There are many known neocentromere examples in human clinical samples (Voullaire et al. 1993; Marshall et al. 2008) as well as in different animal and plant species (Williams et al. 1998; Maggert and Karpen 2001; Nasuda et al. 2005; Ishii et al. 2008; Ketel et al. 2009; Topp et al. 2009; Fu et al. 2013). Most newly formed neocentromeres lie in moderately repetitive genomic regions interspersed with single-copy sequences (Marshall et al. 2008), whereas nearly all mature centromeres contain long arrays of satellite repeats (Henikoff et al. 2001; Jiang et al. 2003). The transition from a neocentromere to a stable mature centromere presumably involves the accumulation of repeats over long time frames (Yan et al. 2006; Kalitsis and Choo 2012).
Centromere identity is conferred epigenetically by the presence of the specialized histone H3 variant known as CENPA in humans (Earnshaw and Rothfield 1985) and CENH3 in plants (Talbert et al. 2002). The distribution of CENH3-containing nucleosomes within the boundaries of centromeres is not well understood, although it appears to be discontinuous and interspersed with canonical nucleosomes (Blower et al. 2002; Yan et al. 2008). Some human neocentromeres and several plant centromeres contain genes embedded as islands within centromeres (Saffery et al. 2003; Nagaki et al. 2004; Gong et al. 2012). While genes may closely border CENH3-containing nucleosomes, gene transcription is generally incompatible with CENH3 (Ketel et al. 2009). Centromeres in higher eukaryotes usually span hundreds of kilobases of sequence and often do not appear to have sharp edges, at least as determined by chromatin immunoprecipitation (ChIP) of CENH3 from complex plant tissues (Yan et al. 2008; Gong et al. 2012). The total number of CENH3 nucleosomes is positively correlated with genome size (Zhang and Dawe 2012), but centromere size does not necessarily correlate with chromosome size. For example, in the budding yeast Saccharomyces cerevisiae, each of the 16 centromeres contains a single nucleosome (Meluh et al. 1998; Henikoff and Henikoff 2012), although the largest chromosome (1532 kb) is six times bigger than the smallest chromosome (230 kb) (Goffeau et al. 1996). More strikingly, although the sizes of chicken (Gallus gallus) macrochromosomes and microchromosomes are vastly different (Hillier et al. 2004), all chicken chromosomes appear to have kinetochores of a similar size (Johnston et al. 2010). For instance, the Z chromosome (∼75 Mb) is 15 times bigger than chromosome 27 (∼5 Mb), but the centromeres of both chromosomes have a 30- to 40-kb CENPA-binding domain (Shang et al. 2010).
Plants are known for the capacity to tolerate very wide and even interspecies crosses. Two of the most distant plant species to ever be crossed are oat (Avena sativa, 2n = 6× = 42) and maize (Zea mays, 2n = 2× = 20), which diverged nearly 60 million years ago. Most of the maize chromosomes are stochastically lost in progeny, often retaining just one maize chromosome in the oat background (Kynast et al. 2001). Since the oat genome (11,300 Mb) is over four times bigger than the maize genome (2500 Mb), and total (summed) centromere size scales linearly with genome size (Zhang and Dawe 2012), we predicted that maize centromeres would expand in the oat background. We mapped the CENH3-binding domains of two maize neocentric chromosomes and seven normal maize chromosomes after transfer to the oat background. All nine centromeres showed a dramatic expansion of roughly twofold, principally into regions of low gene density. These results illuminate the process of centromere reorganization that follows wide species crosses. Centromere size variance may be a key factor that contributes to chromosome loss following such crosses, and centromere expansion may be an important adaptation that allows new centromeres to stabilize.
Results
Confirmation that neoM3 is an isochromosome derived from the short arm of maize chromosome 3
Several maize lines have been used to develop oat-maize chromosome addition lines (oat strains containing one maize chromosome). The first maize line used was a sweet corn hybrid known as Seneca 60. One of the Seneca 60 chromosomes identified in oat was a fragment of chromosome 3 that contained a neocentromere (Topp et al. 2009). This neocentric chromosome, neoM3, was recovered as a derivative from a full Seneca 60 chromosome 3 addition line called OMA3.01. Staining of the neoM3 chromosome with anti-CENH3 antibodies suggests that it is an isochromosome with two identical chromosome arms (the arm ratio is 1.03 ± 0.02, n = 20) (Fig. 1A,B). To confirm this, we isolated an 8.7-kb DNA segment (m3S8.7) from the distal region on the short arm of maize chromosome 3. Fluorescence in situ hybridization (FISH) using m3S8.7 as a probe produced a single hybridization signal on the short arm of maize chromosome 3 in OMA3.01 (Fig. 1C) but generated signals on both chromosomal ends of neoM3 (Fig. 1E).
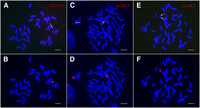
Cytological characterization of the neocentric chromosome neoM3. (A) Immunofluorescence assay of the oat-maize neoM3 addition line using anti-CENH3 antibodies. The arrow points to the CENH3 signal on the neoM3 chromosome. (B) The neoM3 chromosome is identified by sequential genomic in situ hybridization (GISH) of the same metaphase cell using maize genomic DNA as a probe. (C) The two copies of maize chromosome 3 (arrows) in the oat-maize addition line OMA 3.01 are detected by FISH using a 8.7-kb DNA probe amplified from the distal region on the short arm. (D) Identification of the maize chromosomes in the same metaphase cell as assayed by GISH. (E) FISH mapping of the 8.7-kb DNA probe on the neoM3 chromosome. Note: The probe hybridizes to both ends of the neoM3 chromosome (arrows). (F) The identification of neoM3 in the same metaphase cell is confirmed by GISH. Bars, 10 μm.
Mapping the CENH3-binding domain of neoM3 (nCenM3)
Maize centromeres contain arrays of two intermingled repetitive DNA elements, including a 156-bp satellite repeat CentC (Ananiev et al. 1998) and a centromeric retrotransposon CRM (Zhong et al. 2002). The CentC/CRM arrays span several megabases of DNA in some maize centromeres (Jin et al. 2004; Ananiev et al. 2009). The CENH3-binding domains of these heavily repetitive centromeres generally cannot be delineated by sequencing-based approaches. However, the CentC/CRM arrays account for only a portion of the CENH3-binding domains in several other maize centromeres, and in these cases, the CENH3 boundaries can be defined by mapping the sequences associated with CENH3 nucleosomes (Wolfgruber et al. 2009).
As a control for all maize chromosomes, we first conducted CENH3 ChIP, followed by Illumina sequencing (ChIP-seq) of the reference maize inbred line B73. This replicates prior ChIP experiments on B73 using lower-coverage 454 sequencing (Wolfgruber et al. 2009). We obtained a total of 84 million (M) paired sequence reads, including 12.9 M reads (one end or both ends of a paired read, 7.7% of the 168 M total ends) related to CentC or CRM repeats. We mapped 40 M read pairs to unique positions in the B73 reference genome (version 2). The CENH3-binding domain of B73 chromosome 3 (Cen3) was mapped between positions 99.78 and 100.76 Mb (chromosome 3 is 232.1 Mb long) (Fig. 2A; Table 1), which is in agreement with the Cen3 position mapped previously based on a total of 149,756 ChIP-454 sequence reads (Wolfgruber et al. 2009). We note that B73 Cen3 may, in fact, be larger than ∼1 Mb, since the assembly is not complete for this centromere.
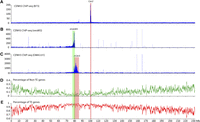
Mapping of the centromere and neocentromeres on maize chromosome 3. (A) Mapping of ChIP-seq reads from B73 on maize chromosome 3. The CENH3-binding domain of Cen3, marked by a pink box, was mapped to the region between 99.78 and 100.76 Mb. The y-axis shows the number of ChIP-seq reads in 10-kb windows along chromosome 3. (B) Mapping of ChIP-seq reads from the neoM3 line on maize chromosome 3. The CENH3-binding domain of nCenM3, marked by a green box, was mapped to the region between 78.2 and 80.3 Mb. Background reads were detected throughout 0 to 80.6 Mb. The y-axis shows the number of ChIP-seq reads in 10-kb windows along chromosome 3. Chromosome 3 sequence of maize B73 is used as the reference sequence. (C) Mapping of ChIP-seq reads from the OMA3.01 line on maize chromosome 3. The CENH3-binding domain of nCen3, marked by a pink box, was mapped in 79.3–83.9 Mb. The y-axis shows the number of ChIP-seq reads in 10-kb windows along chromosome 3. (D) Distribution of non-TE genes on maize chromosome 3. The y-axis shows the percentage of non-TE genes in 10-kb windows. (E) Distribution of TE-related genes on maize chromosome 3. The y-axis shows the percentage of TE-related genes in 10-kb windows. The vertical pink and green boxes across all panels indicate the positions of the neocentromere and original centromere on maize chromosome 3.
Sizes of maize centromeres in the native and oat backgrounds
We then conducted a ChIP-seq analysis of the neoM3 line. We generated 57 M paired reads and mapped 1.49 M reads to maize chromosome 3. Significant sequence enrichment was observed at positions 78.2–80.3 Mb of maize chromosome 3 (Fig. 2B). The read distribution on chromosome 3 showed a cliff-like drop-off around position 80.6 Mb, and very few sequence reads were mapped beyond 80.6 Mb (Fig. 2B), suggesting that neoM3 was broken in this region and the duplicated short arms fused to create an isochromosome. As the distribution of CENH3 ChIP-seq reads in all other maize chromosomes and those in several other species show a bell-shaped distribution (Yan et al. 2008; Gong et al. 2012), the complete CENH3-binding domain in nCenM3 likely includes the 78.2–80.3 Mb region on both arms. Thus, the CENH3-binding domain of nCenM3 includes a minimum of 2.4 Mb (78.2–80.6 Mb) and likely spans 4.8 Mb, which is significantly larger than the mapped size of Cen3 in B73.
Cen3 was repositioned when initially transferred to oat
A simple comparison of the location of nCenM3 to the position of B73 Cen3 would suggest that nCenM3 is far removed from the natural centromere location. However, nCenM3 is not derived from B73―it is derived from the oat line OMA3.01, which contains chromosome 3 originally derived from Seneca 60. Therefore, we also conducted a ChIP analysis of OMA3.01. We generated 212 M paired reads and mapped 1.39 M reads to maize chromosome 3. Surprisingly, we found that the CENH3-binding domain of OMA3.01 (nCen3) is also displaced relative to B73, and spans 4.6 Mb between positions 79.3 and 83.9 Mb (Fig. 2C; Table 1). The two new centromeres, nCen3 and nCenM3, partially overlap in the region of 79.3–80.6 Mb, suggesting that neoM3 was most likely derived from a centromeric misdivision event within nCen3 (Fig. 3).
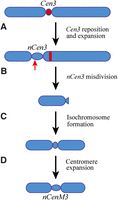
A diagrammatic illustration of reposition and expansion of maize Cen3 in the genetic background of oat. (A) Cen3 repositioned to a short-arm domain that is ∼16 Mb away from its original location, resulting in a neocentromere nCen3. nCen3 has also significantly expanded compared to Cen3. (B) A misdivision occurred in nCen3. The red arrow points to the approximate position of the misdivision. The red bar represents the centromeric DNA (Cent C and CRM repeats) associated with the original Cen3. (C) The short arm derived from the misdivision formed an isochromosome. (D) The centromere of the original isochromosome expanded, resulting in the current version of nCenM3.
We then questioned whether the maize Seneca 60 line naturally contains a centromere in a different position than B73 using an assay that involves immunofluorescence for CENH3, followed by FISH using a CentC probe. FISH mapping results showed that maize chromosome 3 retained the CentC repeats in the OMA3.1 oat line (Fig. 4A). Thus, the maize chromosome has maintained the DNA sequences from its original centromere. We analyzed chromosome 3 in 21 metaphase cells. The signals from CENH3 and CentC were completely separated on 15 chromosomes (71%) (Fig. 4A), partially overlapped on two chromosomes, and completely overlapped on four chromosomes. By comparison, among the 15 chromosomes 3 analyzed in the maize Seneca 60 line, the CENH3 and CentC signals were completely separated from each other on only three chromosomes (20%), partially overlapped on one chromosome, and completely overlapped on 11 chromosomes (Fig. 4B). These results show that centromere 3 of Seneca 60 underwent a repositioning event during the formation of OMA3.01. Both of the chromosome 3 centromeres are neocentromeres in the formal sense: nCen3 was newly formed upon introduction into oat, while nCenM3 occurred secondarily as an outcome of a misdivision event that further shifted the position toward the short arm (Figs. 2, 3).
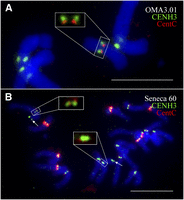
Locations of CENH3 and CentC on maize chromosome 3. (A) Locations of CENH3 (green) and CentC (red) on chromosome 3 in OMA3.01. The CENH3 and CentC signals on maize chromosome 3 are exemplified in the large square. Note: The CENH3 signals are shifted away from the CentC signals toward the short-arm direction. (B) Locations of CENH3 (green) and CentC (red) on chromosome 3 in Seneca 60. The CENH3 and CentC signals on one chromosome 3, which are completely overlapped, are shown in the large square. Arrows point to the FISH signals derived from the 8.7-kb probe associated with the short arm of chromosome 3. Bars, 10 μm.
nCen3 and nCenM3 formed in gene desert regions
We found that the position of nCen3 (79.3–83.9 Mb) represents one of the most gene-deficient regions on chromosome 3 (Fig. 2D,E). Only 21 of the 4197 nontransposable element (non-TE) genes annotated on chromosome 3 were found in this 4.6-Mb domain. The gene density in nCen3 is one gene per 219 kb, compared to one gene per 55 kb for the average of chromosome 3. A random sampling of 4.6-Mb regions from chromosome 3 suggests that there is only a 2.7% chance of selecting a 4.6-Mb region containing ≤21 non-TE genes.
Similarly, the position of nCenM3 (78.2–80.3 Mb) also represents a gene-deficient region. Only nine non-TE genes were found in this 2.1-Mb domain, representing a gene density of one gene per 233 kb of DNA.
Transcription of genes within the neocentromeres
CENH3 ChIP-seq reads were distributed unevenly within nCenM3 and nCen3, resulting in alternating subdomains enriched or depleted for CENH3 (Fig. 5). The CENH3-depleted regions most likely contain H3 nucleosomes, as has been demonstrated in rice centromeres. Active genes were detected in the CENH3-depleted regions in several rice centromeres (Yan et al. 2008).
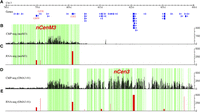
Mapping of CENH3 binding and gene expression in the neocentromeres. (A) The positions of non-TE genes annotated in nCenM3 and nCen3, which overlap in the 79.3–80.3 Mb region. (B) Distribution of ChIP-seq reads in nCenM3. Each black bar represents the number of ChIP-seq reads (y-axis) in a 1-kb window. (C) Gene expression value (FPKM, y-axis) based on RNA-seq in the neoM3 line. (D) Distribution of ChIP-seq reads in nCen3. Each black bar represents the number of ChIP-seq reads (y-axis) in a 1-kb window. (E) Gene expression value (FPKM, y-axis) based on RNA-seq in the OMA3.01 line. The green blocks indicate subdomains significantly enriched with CENH3, which are interspersed with subdomains that were not enriched with CENH3 (blocks with no color).
We conducted RNA-seq in OMA3.01 and neoM3 (two biological replicates; see Methods) to examine the transcription of 21 and nine genes annotated within the two neocentromeres. Only nine of the 21 genes in nCen3 showed transcription in OMA3.01 (FPKM > 1) (Supplemental Table S1). All nine genes were located in CENH3-depleted subdomains (Fig. 5; Supplemental Fig. S3). Similarly, we detected very low amounts of RNA-seq reads (FPKM = 0–12) for six of the nine genes in nCenM3 (Supplemental Table S1). Transcription of the remaining three genes was detected in both nCenM3 and nCen3 (Fig. 5). The first two genes, G959 and G576, were associated with CENH3-depleted subdomains. The third gene (G311) is unusually large (42.8 kb) and consists of seven small exons that together make up the 1.2-kb coding sequence (Supplemental Fig. S1; Fig. 6). Interestingly, this gene contains a small CENH3 subdomain that includes 4175 bp of the first intron, 20,110 bp of the second intron, and a 48-bp exon located in the middle of these two introns (Supplemental Fig. S1). The enrichment of CENH3 within this region was confirmed by ChIP-PCR (see Methods; Supplemental Fig. S2). In summary, only 12 expressed genes were found within the two neocentromeres, and all were located within subdomains that lack CENH3, except for a portion of gene G311 in nCenM3.
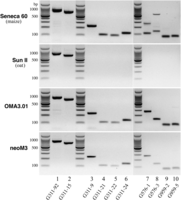
RT-PCR analysis of three active genes located in the neocentromeres. (Lane 1) Primer 311-92 amplified an 840-bp fragment from gene G311 in Seneca 60 (maize), Sun II (oat), OMA3.01 line, and neoM3 line. This fragment spans exon 1 to exon 5 of G311. (Lane 2) Primer G311-15 amplified a 707-bp fragment from gene G311 in all four lines. This fragment spans exon 2 to exon 7 of G311. (Lanes 3,4,5,6) Four different maize-specific primers were designed to amplify different parts of G311 in the two oat-maize chromosomal addition lines. (Lanes 7,8) Two different maize-specific primers were designed to amplify gene G576 in the two oat-maize chromosomal addition lines. Note: Both primers amplified two bands in Seneca 60, suggesting that G576 has a homologous gene of G576 in a different maize chromosome. Both primers, however, amplified a single band in the two addition lines. (Lanes 9,10) Two different maize-specific primers were designed to amplify gene G959 in two oat-maize chromosomal addition lines. All primer sequences are provided in Supplemental Table S3.
Alteration of gene expression due to neocentromere activation
We were interested in whether the transcription of the centromeric genes was altered due to neocentromere activation. Because both neoM3 (Fig. 1B) and OMA3.01 (Fig. 1D) lines contain two copies of the chromosomal segment spanning 0–80.6 Mb, the transcription levels of genes within this chromosomal segment can be directly compared. Three active genes (G959, G576, and G311) are associated with both lines (Fig. 5). Gene G959 is located within nCenM3 but outside of nCen3. This gene showed a similar level of transcription in the two lines (P = 0.22, 107 FPKM in neoM3 and 75.6 FPKM in OMA3.01). Gene G576 is also located within nCenM3 but is outside of nCen3, but it showed a higher level of transcription in OMA3.01 (341.4 FPKM) than in neoM3 (118.5 FPKM, P = 0). Gene G311 is located within both nCenM3 and nCen3. The amount of G311 transcript was significantly higher in neoM3 (410.8 FPKM) than in OMA3.01 (243.9 FPKM, P = 1.1 × 10−10).
We also conducted quantitative real-time PCR (qPCR) to confirm the differential expression. The maize specificity of each primer was examined using oat, maize, and oat-maize addition lines (Fig. 6). The qPCR results were consistent with the RNA-seq data: We did not detect a significant difference for the amounts of G959 transcript in the two lines. However, the amount of G576 transcript was significantly lower in neoM3 than in OMA3.01, and the amount of G311 transcript was significantly higher in neoM3 than in OMA3.01 (Supplemental Fig. S4). These data suggest that neocentromere formation can alter gene expression but that the effects are not dramatic.
Expansion of maize centromeres in the genetic background of oat
Comparison of the sizes of the CENH3-binding domains between Cen3 in B73 (0.98 Mb) and nCen3 and nCenM3 in oat (4.6 Mb and 4.8 Mb, respectively) suggests that the maize centromeres expanded significantly in the oat background. To test whether this is a general phenomenon, we conducted CENH3 ChIP-seq in seven other oat-maize chromosome addition lines developed from maize inbred B73 (maize chromosomes 1, 2, 5, 6, 8, 9, and 10) (Rines et al. 2009). Since B73 has been fully sequenced (Schnable et al. 2009), the sizes of individual B73 centromeres in the maize and oat backgrounds can be directly compared.
Cen2 is the best sequenced B73 centromere because it contains the least amount of CentC repeats (Wolfgruber et al. 2009). We mapped B73 Cen2 to the region between 92.70 and 94.73 Mb that spans 2.03 Mb, similar to prior data (Fig. 7A; Table 1; Wolfgruber et al. 2009). However, Cen2 of B73 in the oat background mapped to the region between 91.21 and 94.73 Mb, covering 3.51 Mb of the chromosome (Fig. 7B; Table 1). Interestingly, the expansion of Cen2 occurred exclusively in the short-arm direction. Mapping of all annotated non-TE genes in chromosome 2 revealed that the expanded region (91.21–92.70 Mb) represents one of the most gene-deficient regions on chromosome 2. Only nine of the 4766 genes annotated on chromosome 2 were mapped within this 1.49-Mb region (P = 0.056). We found a large gene (58.2 kb) located 23 kb away from the boundary of the CENH3 domain on the long arm (Fig. 7C). Expression of this gene has been confirmed by RNA-seq from leaf tissues (Li et al. 2010). We hypothesize that the long transcribed domain associated with this gene may prevent the expansion of Cen2 in the long-arm direction.
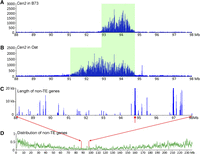
Expansion of Cen2 of B73 in the genetic background of oat. (A) The CENH3-binding domain (green box) of Cen2 in B73. The ChIP-seq read number (y-axis) was calculated in 10-kb windows per million reads. (B) The CENH3-binding domain (green box) of Cen2 in oat-maize chromosomal addition line OMA2.51. (C) The length (kb) of non-TE genes in 20-kb sliding windows (step = 10 kb). The red arrow points to a 58.2-kb transcribed gene located 23 kb away from the CENH3-binding domain. (D) Distribution of non-TE genes along maize chromosome 2. Details of gene distribution within the 88–98 Mb region are exemplified in C.
We also sequenced the ChIPed DNAs from six other oat-B73 chromosome addition lines. Significant expansion was observed in all six centromeres in the oat background (Fig. 8; Table 1). The CENH3-binding domains of Cen1 and Cen6 cannot be delineated in B73 because these two centromeres contain large amounts of CentC repeats (Albert et al. 2010); thus, the CENH3-binding domains are presumably contained entirely within the CentC repeat arrays. In the oat background, however, CENH3-binding was detected in the region between 131.68 and 134.99 Mb in Cen1, and between 47.13 and 50.70 Mb in Cen6, respectively (Fig. 8). Thus, these two centromeres expanded from the CentC repeat arrays into the flanking regions that can be delineated by ChIP-seq.
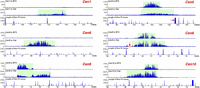
Expansion of Cen1, Cen5, Cen6, Cen8, Cen9, and Cen10 of B73 in the genetic background of oat. The top panel of each centromere shows the ChIP-seq read distribution in B73. CENH3 binding was detected in Cen5, Cen8, Cen9, and Cen10, but not in Cen1 and Cen6. The middle panel of each centromere shows ChIP-seq read distribution in the oat background. The ChIP-seq read number (y-axis) in both panels was calculated in 10-kb windows per million reads. The bottom panel of each centromere shows the length (kb) of non-TE genes in 20-kb sliding windows (step = 10 kb). The red arrow in Cen5 points to a 136.8-kb transcribed gene flanking the long-arm boundary of the CENH3-binding domain. The horizontal red bar (indicated by a red arrow) in Cen8 is a region that completely lacks ChIP-seq sequence reads. A deletion may have occurred in this region during the transfer of this maize chromosome into oat. The expansion of Cen8 surpassed this deletion.
Cen5, Cen8, Cen9, and Cen10 of B73 contain few CentC repeats (Albert et al. 2010). Their sizes in maize ranged from 1.4 Mb to 1.9 Mb, whereas in oat they were roughly two times larger, ranging from 3.3 Mb (Cen1 and Cen10) to 3.8 Mb (Cen8) (Fig. 8; Table 1). The expansion of Cen5 and Cen8 was bidirectional. In contrast, the expansion of Cen9 and Cen10 was exclusively in the long arm and short arm, respectively (Fig. 8). These results further support the assertion that the direction of expansion is not random and is likely restricted by the presence of actively transcribed genes.
We also conducted genomic in situ hybridization (GISH) using oat genomic DNA as a probe to test the possibility that oat sequences may have invaded the maize centromeres. Unambiguous hybridization signals were only detected in the telomeric regions of maize chromosome 2 in line OMA2.51 (six different oat-maize addition lines were assayed) (Supplemental Fig. S5). There was no evidence of oat sequences in any of the introduced maize centromeres.
Discussion
Centromere expansion: A requirement for survival of evolutionary new centromeres?
We demonstrate in nine cases that centromeres transferred from maize into oat increase in size, including two cases where the maize centromeres moved to different locations. Oat and maize are effectively cross-incompatible, and plants can only be recovered after embryo culture. Like many similar crosses (Laurie and Bennett 1988), when embryos survive, they are usually haploid for one of the contributing genomes (in this case, oat). Centromere incompatibility appears to be the primary cause for genome elimination in hybrids (Sanei et al. 2011). An analysis of diploidized plants from the oat-maize hybrids revealed that some progeny retain maize chromosomes that have presumably undergone a process of centromere inactivation followed by re-assembly. The process of recovering a maize chromosome in oat is roughly equivalent to a whole-chromosome transfer event followed by strong selection for stable chromosome transmission.
A prior analysis of centromere size in 10 grass species demonstrated that centromere size is correlated with genome size such that the “total centromere area” is equally distributed among the available chromosomes (Zhang and Dawe 2012). A simple tetra-ploidization event is not expected to affect centromere size because the number of centromeres increases accordingly. The oat-maize comparison is different: The oat genome is four times bigger than maize, but the number of centromeres is only doubled (42 versus 20). Therefore, we anticipated that any given oat centromere would be roughly twice as large as a maize centromere. Data shown here demonstrate that maize centromeres transferred into oat tend to stabilize at a size of ∼3.6 Mb, which is, indeed, close to twice the size of the average maize centromere, which appears to be closer to 1.8 Mb. The two unequivocal neocentromeres described here, nCen3 and nCenM3, show similar sizes to all other expanded maize centromeres. This observation, taken in the context of the general observation that centromere sizes are generally uniform within species and do not correlate with chromosome size (Johnston et al. 2010; Henikoff and Henikoff 2012; Zhang and Dawe 2012), suggests that each species maintains a centromere size equilibrium. A consistent centromere size may be favorable for chromosome alignment, as it would allow each chromosome to have an equal likelihood of being attached by a similar number of microtubules, which may be essential for random segregation and distribution of chromosomes during meiosis.
It seems highly unlikely that any newly formed centromere will spontaneously form at the optimum size. In fact, most human neocentromeres are smaller than the average native centromere (Irvine et al. 2004; Marshall et al. 2008). Therefore, we believe that the stabilization of neocentric chromosomes in natural populations will depend on whether the CENH3 domain of the neocentromere can expand into the flanking regions. While OMA3.01 was reported to be stable upon discovery (Muehlbauer et al. 2000), the original misdivision derived from it (neoM3) was mitotically unstable, and stable lines were ultimately selected and studied (Topp et al. 2009). Similarly, a highly unstable maize chromosome known as Dp3a was recently shown to have a neocentromere (Fu et al. 2013). The instability of Dp3a is likely associated with the fact that the neocentromere contains only a 350-kb CENH3-binding domain and is located in a gene dense area.
Transcription and neocentromere establishment
Centromeric chromatin is generally incompatible with gene transcription. Insertion of a marker gene in the centromeres of Schizosaccharomyces pombe chromosomes results in complete silencing of the gene (Allshire et al. 1995). Similarly, neocentromeres in multiple species generally form in gene-poor regions (Lomiento et al. 2008; Alonso et al. 2010; Shang et al. 2013); when they do form over genic areas, the affected genes are suppressed or silenced (Ishii et al. 2008; Ketel et al. 2009; Shang et al. 2013). Why does centromeric chromatin avoid actively transcribed genes? CENH3 nucleosomes cannot be modified by the histone modification pathways specific to the canonical H3 nucleosomes. CENH3 nucleosomes are also more compact and conformationally more rigid than H3 nucleosomes (Black et al. 2004). Thus, CENH3 chromatin is probably far less compatible with regulated transcription. In addition, gene transcription may actively evict CENH3 nucleosomes during periods of development when they cannot be readily replaced (Gassmann et al. 2012). Our data strongly support this extensive literature by demonstrating that 12 transcribed genes found in nCenM3 and nCen3 were all located within subdomains depleted of CENH3. The only exception was an internal domain of a long gene that contains a single 48-bp exon.
We demonstrate that both nCen3 and nCenM3 represent the most gene-deficient regions on chromosome 3 (Fig. 2). In addition, the expanded CENH3 domains on seven maize centromeres are also largely gene-deficient (Figs. 7, 8). Interestingly, a large and active gene was found to be near the boundaries of the expanded Cen2 (Fig. 7C) and Cen5 (Fig. 8). We postulate that the transcription of these large genes impedes the expansion of CENH3 domains, similar to barriers that block the spreading of heterochromatin (Noma et al. 2006; Scott et al. 2006). Some maize chromosomes were especially difficult to recover in oat-maize hybrids (Kynast et al. 2001; Rines et al. 2009), suggesting that the expansion of centromeres may have failed. For example, maize chromosome 3 was not recovered from the oat × B73 and oat × Mo 17 hybrids. In addition, the centromere of maize chromosome 3 recovered from the oat × Seneca 60 hybrid moved to a new position (Fig. 2C). Thus, active genes flanking Cen3 (Fig. 2D) may prohibit the expansion of this centromere, resulting in the loss of this chromosome or repositioning of Cen3 in the oat background.
Ishii et al. (2013) recently conducted wide crosses between oat and pearl millet (Pennisetum glaucum, 2n = 2× = 14) and developed true oat-millet hybrids that contain two complete haploid sets of all 21 oat and seven millet chromosomes (Ishii et al. 2013). The millet chromosomes appeared to adapt the oat background in these hybrids. Since the millet genome (2450 Mb) (Martel et al. 1997) is significantly smaller than the oat genome (11,300 Mb), our data suggest that the millet centromeres expanded to adapt to the overall larger genome environment. Interestingly, the centromeres of millet chromosomes contain large amounts of satellite repeats (Ishii et al. 2013) and may lack active genes, which is favorable for centromere expansion. In wide crosses between a large genome species (such as oat and wheat) and a small genome species (such as maize, pearl millet, or sorghum), chromosomes from the small genome parent were often eliminated in early embryogenesis (Laurie and Bennett 1988; Ishii et al. 2013). We propose that failure of centromere expansion of chromosomes derived from the small genome parent may be the key factor in chromosome elimination.
Methods
Plant materials
Oat-maize chromosome addition line OMA3.01 contains maize chromosome 3 derived from maize hybrid Seneca 60 (Kynast et al. 2001). The oat-maize neoM3 monosomic addition line was derived from OMA3.01 (Topp et al. 2009). Oat-maize chromosome addition lines OMA1.36, OMA2.51, OMA5.60, OMA6.34, OMA8.05, OMA9.41, and OMA10.26 contain maize chromosomes 1, 2, 5, 6, 8, 9, and 10, respectively, from maize inbred B73 (Rines et al. 2009). OMA3.01, OMA2.51, neoM3, and maize lines Seneca 60 and B73 were used in ChIP-seq and FISH experiments. Seneca 60, B73, and oat cultivar Sun II were used in PCR and qPCR experiments. All plants were grown in greenhouses. Leaf tissues and root tips were collected from the plants for experiments.
FISH, GISH, and chromosomal immunoassay
FISH, GISH, and immunoassays on chromosomes were performed according to published protocols (Jiang et al. 1995; Jin et al. 2004). In the GISH procedure, oat genomic DNA was used as a probe, and unlabeled maize genomic DNA was used as a blocker. For FISH identification of maize chromosome 3, we isolated an 8.7-kb DNA segment from the maize bacterial artificial chromosome ZMMBBb0013L21. This fragment is located in the distal region of the short arm of maize chromosome 3 and is named m3S8.7. Primers were designed based on BAC sequence and then were used in PCR (Supplemental Table S2). PCR condictions were 94°C for 3 min, followed by 35 cycles of 95°C for 30 sec, 55°C for 90 sec, and 72°C for 60 sec and ended by a 4-min extension at 72°C. PCR products were recovered by a Gel Extraction Kit (Qiagen, catalog no. 28704). The 8.7-kb DNA segment from 10 PCR products were mixed and labeled as a FISH probe.
ChIP, ChIP-seq, and RNA-seq
A CENH3 antibody developed in rice recognizes both maize and oat CENH3 (Jin et al. 2004). This antibody was used for all immunoassay and ChIP experiments. ChIP was conducted following a published protocol (Nagaki et al. 2003). Normal rabbit serum was used in a mock treatment as a negative control. ChIPed DNA was then used for ChIP-seq library construction according to the protocol provided by Illumina, including repairing the ends of DNA fragments, poly(A) tailing of the 3′ ends, ligation of paired-end adapters, fractionation of 150–300 bp adapter-ligated DNA using a 2% agarose gel, and enrichment of sized adapter-modified DNA fragments by PCR. The enriched DNA sample was sequenced using Illumina Genome Analyzer II or HiSeq platforms.
RNA-seq was performed in both neoM3 and OMA3.01 lines. Two biological replicates of young leaf tissues harvested from both neoM3 and OMA3.01 were used for RNA-seq analysis. Total RNA was extracted using an RNeasy plant kit (Qiagen, catalog no. 74904). Approximately 40 μg total RNA was converted to cDNA using the mRNA-seq kit from Illumina. RNA-seq libraries were constructed using a barcode method and sequenced using an Illumina Genome Analyzer II platform.
ChIP-seq reads were mapped to the reference genome of maize B73 version 2 using the MAQ alignment program (Li et al. 2008). We allowed 1-bp mismatch between each sequence read and the reference genome, then kept only reads that mapped to a unique position in the reference genome for further analysis. We used TopHat (Trapnell et al. 2009) to map sequence reads from RNA-seq to the same reference genome and employed Cufflinks (Trapnell et al. 2010) to measure the difference of gene expression level between OMA3.01 and neoM3.
Mapping and identification of CENH3-enriched regions followed published protocols with only minor modifications (Yan et al. 2008). We considered the genomic position of the starting nucleotide of a unique read as a uniquely mappable region and then calculated the number of unique read pairs per base pair mappable region in 1-kb windows. We used these adjusted read numbers to identify the enriched region of CENH3. We required that the enriched window be P < 1 × 10−5 and that the CENH3 region includes at least three continuous enriched windows.
RT-PCR, qPCR, and ChIP-PCR
We used RT-PCR to examine the expression of three genes, G959, G576, and G311, which are present in the neocentromere. Primers were designed to have a length between 20 and 24 bp, with the annealing temperature of 55°C–60°C (Supplemental Table S3). RNA was isolated from leaf tissues collected from Seneca 60, Sun II, OMA3.01, and neoM3. RT-PCR was conducted following a published protocol (Yan et al. 2005). We then conducted qPCR to quantify the transcript level of these three genes in the neoM3 and OMA 3.01 lines. Gene G311 is present in both maize and oat. Therefore, the RT-PCR products from Seneca 60 and Sun II of two primers, G311-92 and G311-15, were sequenced to develop maize-specific primers. Maize-specific primers were designed based on the divergence of cDNA sequences between Sun II and Seneca 60 (Supplemental Table S3). Only maize-specific primers were then used in qRT-PCR analysis. PCR reactions were carried out using the DyNAmo SYBR Green qPCR kit (Thermo Scientific) and run at 95°C for 5 min, followed by 45 cycles of 95°C for 10 sec, 60°C for 20 sec, and 72°C for 30 sec. The glyceraldehyde-3-phosphate dehydrogenase gene of oat was used as an internal reference as previously described (Jarosova and Kundu 2010). For each gene, the relative threshold cycle number was normalized over the internal control as previously described (Yan et al. 2005).
We conducted ChIP-PCR to verify the relative enrichment of three genes in the CENH3-bound fraction over the mock control (Supplemental Table S3). A primer (NEG79) designed from a region of chromosome 3, located outside of the neocentromere, was used as a negative control. We calculated the difference in the PCR threshold cycle number to determine the relative enrichment of each amplicon as described (Yan et al. 2005)
Data access
The ChIP-seq reads associated with maize chromosomes from all nine oat-maize chromosome addition lines and the RNA-seq data sets have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE47342.
Acknowledgments
We thank Drs. Ron Phillips and Howard Rines for providing the oat-maize chromosome addition lines for this research. This work was supported by grant DBI-0922703 from the National Science Foundation to R.K.D. and J.J., and grant DBI-0923640 to J.J.
Footnotes
-
↵4 Corresponding authors
E-mail kelly{at}plantbio.uga.edu
E-mail jjiang1{at}wisc.edu
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.160887.113.
- Received May 21, 2013.
- Accepted October 7, 2013.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported), as described at http://creativecommons.org/licenses/by-nc/3.0/.








