
The landscape of RNA polymerase II transcription initiation in C. elegans reveals promoter and enhancer architectures
- Ron A.-J. Chen1,5,
- Thomas A. Down1,5,
- Przemyslaw Stempor1,
- Q. Brent Chen2,
- Thea A. Egelhofer3,
- LaDeana W. Hillier4,
- Tess E. Jeffers2 and
- Julie Ahringer1,6
- 1The Gurdon Institute, and Department of Genetics, University of Cambridge, Cambridge CB3 0DH, United Kingdom;
- 2Department of Biology, Carolina Center for Genome Sciences, and Lineberger Comprehensive Cancer Center, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599-3280, USA;
- 3Department of Molecular, Cell and Developmental Biology, University of California Santa Cruz, Santa Cruz, California 95060, USA;
- 4Department of Genetics and Genome Sequencing Center, Washington University School of Medicine, St. Louis, Missouri 63108, USA
-
↵5 These authors contributed equally to this work.
Abstract
RNA polymerase transcription initiation sites are largely unknown in Caenorhabditis elegans. The initial 5′ end of most protein-coding transcripts is removed by trans-splicing, and noncoding initiation sites have not been investigated. We characterized the landscape of RNA Pol II transcription initiation, identifying 73,500 distinct clusters of initiation. Bidirectional transcription is frequent, with a peak of transcriptional pairing at 120 bp. We assign transcription initiation sites to 7691 protein-coding genes and find that they display features typical of eukaryotic promoters. Strikingly, the majority of initiation events occur in regions with enhancer-like chromatin signatures. Based on the overlap of transcription initiation clusters with mapped transcription factor binding sites, we define 2361 transcribed intergenic enhancers. Remarkably, productive transcription elongation across these enhancers is predominantly in the same orientation as that of the nearest downstream gene. Directed elongation from an upstream enhancer toward a downstream gene could potentially deliver RNA polymerase II to a proximal promoter, or alternatively might function directly as a distal promoter. Our results provide a new resource to investigate transcription regulation in metazoans.
Transcription is a fundamental process that plays a central role in development and cellular responses. The regulation of transcription is complex, often involving interplay between promoters and regulatory elements such as enhancer regions. A prerequisite for understanding the mechanisms and principles of this process requires identification of these sites and their relationships.
Caenorhabditis elegans is a widely used and powerful model organism for functional genomic studies. The genome of 100 MB is well-annotated and 30× smaller than that of humans, making genomic studies highly accessible (The C. elegans Sequencing Consortium 1998). However, despite these advantages, study of transcription regulation has been hampered because the transcription initiation sites of most C. elegans genes are unknown. More than 70% of protein-coding genes are trans-spliced to a 22-nt leader RNA encoded from another region of the genome (Allen et al. 2011; Blumenthal 2012). The region from the initial 5′ end to the trans-splice site, termed the “outron,” is spliced off and degraded (Bektesh and Hirsh 1988; Blumenthal 1995). Consequently, the annotated start sites for most C. elegans genes mark trans-splice sites rather than transcription initiation sites because cDNAs and RNA-seq data derived from total RNA indicate the structure of the mature trans-spliced transcript. Using these positions or translational start sites as transcription start sites (TSSs), some promoter features have been described (Ooi et al. 2006, 2010; Gerstein et al. 2010; Liu et al. 2011b). However, the true sites and features of RNA polymerase II transcription initiation and the chromatin landscape at these sites remain to be discovered.
In eukaryotes, regulatory and chromatin features of protein-coding TSSs have been extensively documented (de Hoon and Hayashizaki 2008; Jiang and Pugh 2009; Venters and Pugh 2009). For example, transcription initiation sites often possess an initiator element (Inr), and a subset of genes have a TATA-box 25 to 32 nt upstream of the TSS which has a role in focusing start-site selection (Weis and Reinberg 1992; Smale 1997; Juven-Gershon et al. 2008; Lenhard et al. 2012). An Sp1 motif has also been reported to play a role in transcript initiation (Segal et al. 1999; Wierstra 2008). Active promoters usually contain a nucleosome-depleted region (NDR) flanked by well-positioned nucleosomes (Bernstein et al. 2004; Lee et al. 2004; Nishida et al. 2006; Henikoff 2007; Jiang and Pugh 2009; Rhee and Pugh 2012). In addition, nucleosomes near active protein-coding TSSs are enriched for trimethylation of H3K4 and acetylation of H3K27 (Turner 2000; Barrera and Ren 2006). Furthermore, histone variants H3.3 and H2A.Z, associated with unstable nucleosomes, are also enriched in promoter regions (Zhang et al. 2005; Millar et al. 2006; Raisner and Madhani 2006; Whittle et al. 2008; Weber et al. 2010).
Recent high-throughput sequencing studies in other organisms have uncovered novel transcriptional phenomena at promoters and enhancers. For example, in yeast and mammalian cells, it has been observed that transcription at protein-coding promoters often initiates bidirectionally, whereas bidirectional transcription is infrequent in Drosophila (Core et al. 2008; Neil et al. 2009; Seila et al. 2009; Xu et al. 2009; Nechaev et al. 2010; Flynn et al. 2011; Kharchenko et al. 2011; Wei et al. 2011). How bidirectional transcription initiation drives productive transcription elongation primarily in the coding direction is not well understood (Seila et al. 2008, 2009; Flynn et al. 2011), but a recent report has suggested that gene loops can play a role (Tan-Wong et al. 2012). In addition, it has been shown that mammalian enhancer regions are transcribed, but transcription initiation within enhancer regions has not been well characterized (De Santa et al. 2010; Kim et al. 2010; Melgar et al. 2011; Ong and Corces 2011; Preker et al. 2011; Wang et al. 2011). The mechanism and functions of transcription at enhancers are unclear. Proposed models suggest regulatory functions or, alternatively, that the process of transcription could act to generate regions of open chromatin in order to enhance the accessibility of downstream coding promoters (Travers 1999; Ong and Corces 2011; Natoli and Andrau 2012).
Here, we globally identify sites of RNA polymerase II transcription initiation and elongation in C. elegans. We assign transcription start sites to protein-coding and noncoding genes and find that bidirectional transcription is widespread. Furthermore, we discover 2361 mapped transcription factor binding sites that overlap transcription initiation clusters, defining a set of transcribed enhancers. We show that productive transcription elongation across these regions is usually oriented toward and in the same orientation as that of the nearest downstream gene. Our characterization of this regulatory architecture and provision of transcription initiation and elongation resources will facilitate studies of transcription regulation in metazoans.
Results
Identification of C. elegans RNA polymerase II transcription initiation sites
To globally capture RNA polymerase II initiation sites, including nascent transcripts before they have been trans-spliced, we isolated short (20–100 nt) nuclear RNAs with a 5′ cap from mixed-stage embryos, and prepared and sequenced RNA-seq libraries (hereafter called short cap RNAs). We also made strand-specific RNA-seq libraries from capped nuclear RNAs over 200 nt long to assess nuclear expression of elongated transcripts (hereafter called long cap RNAs). In total, we obtained ∼50 million uniquely mappable reads for the short capped RNA libraries and ∼55 million uniquely mappable reads for the long capped RNA libraries. As expected for RNA polymerase II transcription initiation sites, there is strong enrichment for Ser5 phosphorylated RNA Pol II (the initiating form) near the 5′ ends of short cap RNA reads (Fig. 1A).
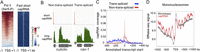
Transcription initiation regions (TICs) are occupied by RNA Pol II and enriched near the 5′ end of coding genes. (A) Heat map displays of forward strand TICs (n = 36,662) ranked by enrichment of Pol II Ser5 ChIP-chip signal. All panels are plotted for the enrichment of signal in 2-kb windows centered at cap RNA TSSs. (B) Genome browser views of short cap RNA (orange) and long cap RNA (green) signals at a non-trans-spliced and a trans-spliced gene. (*) The outron region. (C) The fraction sites where at least two cap RNA starts were analyzed relative to WormBase transcript start sites of trans-spliced (blue) and non-trans-spliced (red) genes are plotted in an 800-bp window. (D) Mononucleosome signal (adult) anchored at either cap RNA TSSs (red) or WormBase transcript starts (gray).
We observed that short cap RNA reads often mapped near the 5′ ends of genes. For non-trans-spliced genes, reads initiated at or near the annotated starts of WormBase transcripts (Fig. 1B,C). In contrast, for trans-spliced genes, where the WormBase transcript starts usually mark the trans-splice site, short cap RNA reads initiated upstream, and we observed long cap RNA signals covering the outron region between the trans-splice site and the short cap RNA tags (Fig. 1B,C). The outron signals are not observed in RNA-seq libraries prepared from whole-cell (mainly cytoplasmic) poly(A)+ RNAs (Supplemental Fig. S1). Initiation sites are broadly distributed upstream of trans-spliced transcripts (Fig. 1C), indicating that transcription does not initiate at a fixed position relative to trans-splice sites.
As in other organisms, we observed that the initiation sites of short cap RNAs are usually closely clustered (Supplemental Fig. S1). We grouped the 5′ tags into clusters, which defined 73,500 RNA polymerase II transcription initiation clusters (TICs) (see Methods; Supplemental Tables S1, S2). To link TICs to annotated WormBase genes, TICs mapping from −200 bp to +100 bp of an annotated WormBase transcript start, or where long cap RNA signal was present between an annotated transcript start and a TIC, were assigned as potential transcription start sites for protein-coding genes (n = 10,106 TICs assigned to 7691 genes) (Supplemental Tables S1, S2). A further 14,810 TICs mapped within the body of protein-coding genes and might mark alternative 5′ ends or have regulatory functions, and 3637 TICs were assigned to ncRNA genes. The remaining 44,947 TICs were unassigned. Unassigned TICs map on the anti-sense strand in gene bodies or not within −200 bp to +100 bp of an annotated transcript start (from −200 bp to +100 bp).
Chromatin features of transcription initiation sites
To characterize the identified TICs, we analyzed the distributions of chromatin features that have well-characterized properties at promoters, transcribed regions, and enhancers using heat map plots. We selected the mode of each TIC as a representative TSS for analysis and ranked the TSSs based on the ratio of H3K4me3 to H3K4me1 ChIP signals to more easily visualize promoter and enhancer characteristics (Guenther et al. 2007; Heintzman and Ren 2007; Gerstein et al. 2010; Pekowska et al. 2011; Rada-Iglesias et al. 2011).
TSSs assigned to protein-coding genes display chromatin features typical of promoters in other organisms. Near the TSSs, we observed generally higher H3K4me3 than H3K4me1 and high levels of H3K27ac (Supplemental Fig. S2), a signature of active protein-coding promoters (Ruthenburg et al. 2007; Lenhard et al. 2012). Productive elongation detected by long cap coverage is observed downstream from the assigned coding TSSs, and H3K36me3 signal, a mark of transcriptional elongation of protein-coding genes (Guenther et al. 2007; Sati et al. 2012), is usually also present (Supplemental Fig. S2). Regions with the highest levels of H3K4me3 appear to be divergent promoters, as these additionally display H3K36me3 and reverse strand long cap RNA signals upstream of the TSSs.
To further validate the protein-coding gene TSS assignments, we compared the patterns of chromatin signals anchored at the starts of WormBase transcripts to the same set of genes anchored at the newly assigned cap RNA TSSs (Fig. 1D; Supplemental Fig. S3). We observed that promoter-associated chromatin signals are sharper at short cap TSSs (Supplemental Fig. S3). Additionally, plotting nucleosome signal at short cap RNA TSSs reveals a wave-like pattern, indicating that nucleosomes are well-phased at C. elegans promoters (Fig. 1D; Supplemental Fig. S3), as observed in other eukaryotic cells (Bernstein et al. 2004; Lee et al. 2004; Nishida et al. 2006; Henikoff 2007; Jiang and Pugh 2009; Rhee and Pugh 2012). Such positioning is not clear using WormBase TSSs.
In contrast to the cap RNA TSSs assigned to protein-coding genes, unassigned TSSs and those assigned to noncoding RNA (ncRNA) genes are enriched for chromatin features typical of enhancers, with higher H3K4me1 relative to H3K4me3 and low levels of H3K36me3 (Supplemental Fig. S2). ncRNA and unassigned TSSs are frequently embedded within long cap RNA signal, indicating they lie in regions with productive transcriptional elongation (Supplemental Fig. S2). To summarize, TSSs assigned to protein-coding genes show conventional promoter chromatin signatures, whereas ncRNA and unassigned TSSs are enriched for enhancer-like chromatin signatures.
Core promoter elements are correlated with different initiation types
We observed that the TICs have a range of patterns, from a single strong peak to a broad distribution of initiation events (Supplemental Fig. S4), as has been seen in humans and Drosophila (Carninci et al. 2006; Ni et al. 2010). We developed a shape score, which we defined as the fraction of reads associated with the highest point in the TIC, to categorize TICs containing at least 10 tags (see Methods). A sharp TIC where all tags map to the same position has a score of 1, whereas a broad TIC containing multiple starts of similar heights will have a low score. We defined sharp promoters as TICs with scores of 0.7–1.0 and broad ones as those between 0 and 0.2 (Supplemental Fig. S4). The remaining TICs have intermediate characteristics.
To look for fixed sequence features in the protein-coding promoters, we aligned TSSs of sharp and broad promoters using the WebLogo sequence analysis tool (Fig. 2A; Crooks et al. 2004). As in other organisms (Zhang and Dietrich 2005; Carninci et al. 2006), we found that transcription preferentially initiated at the adenine of a TCA sequence, with a stronger preference in sharp compared to broad promoters. In addition, in sharp promoters, there is a weak enrichment for T/A-rich sequence between −25 to −30, corresponding to the region where TATA-boxes reside (Juven-Gershon et al. 2008).
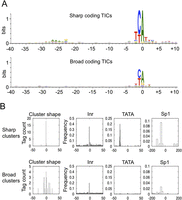
Protein-coding promoter sequence features. (A) WebLogo analysis of sequence aligned at sharp and broad TSSs. (B) Inr, TATA, and Sp1 motif occurrence at sharp and broad protein-coding TSSs.
To further investigate core promoter sequence features, we examined the occurrence of Inr and TATA motifs as well as that of Sp1, a widely active transcription factor, using published position weight matrices (Lenhard et al. 2012). We found that the Inr motif was present in both types of protein-coding promoters, with slightly higher frequency in sharp promoters (Fig. 2B). In contrast, the TATA motif was only associated with sharp promoters (Fig. 2B); a bias for TATA occurrence in sharp promoters was also reported in human and Drosophila (Ni et al. 2010; Rach et al. 2011). Similar to the TATA motif, we found that the Sp1 motif was associated primarily with sharp promoters (Fig. 2B). Of note, sharp TSSs with a TATA box were substantially less likely to harbor an Inr motif at the TSS than those without a TATA box, suggesting some degree of redundancy between TATA and Inr (P = 5 × 10−15; χ2 test).
Characterization of bidirectional transcription
In the heat map analyses, we observed that reverse strand TSSs were often located a short distance upstream of forward strand TSSs, indicative of bidirectional transcription initiation (Supplemental Fig. S2). To investigate the occurrence of bidirectional transcription, we plotted the density of forward and reverse strand short cap initiation sites across the three classes of TSSs. This analysis showed a strong peak of reverse strand signal ∼120 bp upstream of the initiation sites of all TSS classes (protein-coding, noncoding, and unassigned) (Fig. 3); 39% of all TICs have an upstream reverse strand initiation within 200 bp. The bidirectional signal observed for protein-coding genes is not simply due to divergent protein-coding transcription (head to head configuration, where the upstream gene is transcribed in the opposite orientation) because it is also clearly evident at tandemly transcribed protein-coding genes (tail to head, where the upstream gene is in the same orientation) (Supplemental Fig. S5). We conclude that transcription initiation in C. elegans is often bidirectional.
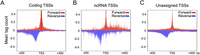
Bidirectional transcription is widespread in C. elegans. The frequency of forward (red) and reverse (blue) transcription initiations were analyzed at (A) coding, (B) ncRNA, and (C) unassigned TSSs. The distribution of signal is plotted in 1-kb windows at mode TSSs of the TICs.
Most transcription factor binding sites initiate transcription
Recent studies in mammalian cells identified transcripts and engaged RNA polymerase II within enhancer regions, indicating that these regions are transcriptionally active (De Santa et al. 2010; Kim et al. 2010; Melgar et al. 2011; Wang et al. 2011; Natoli and Andrau 2012). However, where transcription initiates within enhancer regions and the relationship between transcription factor binding sites and enhancer transcription initiation are unclear.
To investigate these questions, we used a set of C. elegans transcription factor (TF)-binding regions defined from ChIP mapping of 22 transcription factors (Gerstein et al. 2010). We focused on “factor-specific regions” bound by 1–4 TFs (n = 13,156, 79% of binding sites), as these are enriched for containing expected sequence motifs and being located near genes known to be under their regulation. As expected, we observed that most factor-specific TF regions have a classical enhancer signature of high H3K4me1 and low H3K4me3 (Fig. 4B), in contrast to protein-coding promoters, which have the opposite pattern (Fig. 4A).

TF-binding regions initiate bidirectional transcription. Comparison of the enrichment of Pol II (Ser5), H3K4me3, and H3K4me1 at TSSs of ubiquitously expressed protein-coding genes (n = 4282) (A), and factor-specific TF-binding regions (n = 13,516) (B). Heat map rows are ranked by H3K4me3/H3K4me1 ratio and all signals plotted in 2-kb windows centered at (A) TIC mode TSSs, or (B) the midpoint of TF-binding regions. (C) Forward (red) and reverse (blue) TIC coverage plotted in a 2-kb region centered at the midpoints of factor-specific TF-binding regions; 60% of sites have both forward and reverse strand signals.
Using our short and long cap RNA data, we next examined the relationship between transcription factor binding, transcription initiation, and productive transcription elongation. We first asked whether transcription initiates near TF-binding sites. Remarkably, we observed that 65% of factor-specific TF-binding regions overlap a TIC. Therefore, TF-binding sites frequently initiate transcription. To further explore the transcription initiation activity of TF-binding sites, we plotted the frequency and positions of transcription initiation events relative to TF-binding sites. We observed peaks of transcription initiation closely flanking the TF-binding sites, with 60% of transcribed binding sites having both forward and reverse strand short cap RNA signals (Fig. 4B,C). RNA Pol II is also clearly enriched at TF-binding regions (Fig. 4B). These results demonstrate that bidirectional transcription is initiated at TF-binding sites.
Characterization of a novel widespread regulatory architecture
We used the criterion of overlap between transcription factor binding regions and transcription initiation sites to define active enhancers (Melgar et al. 2011). We first selected intergenic TF-binding sites that were at least 500 bp from an annotated transcript start and that overlapped a TIC, then removed regions that appeared to be unannotated promoters based on having a signature of high H3K4me3/low H3K4me1 (Fig. 5). These intergenic TF-binding regions that initiate transcription define 2361 transcribed enhancers. Supporting the view that these sites identify active enhancers (Supplemental Fig. S6), they overlap previously published functionally validated embryonic enhancer regions (Landmann et al. 2004; Lei et al. 2009).
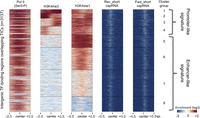
Intergenic TF-binding regions overlapping TICs are enriched for enhancer-like chromatin signature. Heat map analysis for the indicated enrichment signals displayed as 8 k-means clustered groups based on H3K4me3 and H3K4me1 (n = 3137). All signals were plotted in 2-kb windows centered at the midpoint of TF-binding regions. A promoter-like signature of high H3K4me3/low H3K4me1 is found in clusters 1– 4 (n = 776). In contrast, clusters 5–8 show an enhancer-like chromatin signature, high H3K4me1 and low/under-enriched H3K4me3 (n = 2361).
We find that features of enhancer transcription initiation sites differ from those of protein-coding promoter TSSs. First, although enhancer TSSs are enriched for the Inr initiation element, TATA and Sp1 elements are essentially absent (Supplemental Fig. S7). Second, chromatin features also differ at enhancer and promoter TSSs. In addition to differences in relative levels of H3K4me3 and H3K4me1 described above, HTZ-1 is strongly enriched at protein-coding TSSs, but nearly absent at enhancer TSSs (Supplemental Fig. S7C). Further, whereas histone H3 and mononucleosomes are depleted at protein-coding TSSs, these show weak enrichment at enhancer TSSs (Supplemental Fig. S7C). In summary, promoter sequence elements and the chromatin signature of enhancer TSSs differ from those of protein-coding genes.
We next examined whether transcription initiation at enhancers was productively elongated or, alternatively, whether initiation events generally produced short transcripts. To answer this question, we analyzed the abundance of nuclear long cap RNA signals (generated from nuclear capped RNAs > 200 nt) on each strand of intergenic enhancers. We find that enhancers generally produce elongated transcripts and that there is a striking bias for transcription to be predominantly on one strand (Fig. 6A,B). Therefore, although most enhancers initiate bidirectional transcription, productive elongation is primarily in one direction.
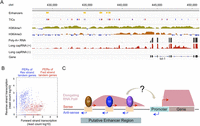
Definition of a novel regulatory architecture. (A) Genome browser image of signals indicating enhancers, TICs (red: forward strand; blue: reverse strand), H3K4me3, H3K4me1, whole-cell (mainly cytoplasmic) RNA, and nuclear long cap RNA from the plus and minus strands, with gene annotation below. (B) RNA Pol II elongation across enhancers is in the same orientation as that of the nearest downstream gene. Plots show forward strand long cap RNA-seq read count (log10 scale) relative to reverse strand signal over enhancers upstream of forward (red) or reverse (blue) strand tandem genes. The red dots showing enhancers upstream of the tol-1 gene (forward strand) shown in A were circled. (C) Proposed model illustrating the architecture and potential regulatory roles of transcribed enhancers.
Since enhancers are often located upstream of their target genes, we investigated the relationship between enhancer and downstream gene transcription. We assigned intergenic enhancers to the nearest downstream protein-coding gene, and asked whether the direction of transcription across the enhancer was related to that of the downstream gene. To be confident in the assignment of the downstream gene, we limited this analysis to intergenic enhancer regions upstream of tandemly transcribed genes, excluding divergently transcribed genes. Remarkably, for 90% of enhancer regions, the majority of elongated transcription is in the same orientation as that of the downstream gene (Fig. 6B). We also found that the level of transcription across enhancers is positively correlated with that of the downstream gene (ρ = 0.45, Spearman's P < 10−7). Together, these results support the view that transcribed enhancers are active regulatory regions for downstream gene expression (Fig. 6C).
Discussion
Here, we characterized the global landscape of RNA polymerase II transcription initiation in C. elegans. As in other eukaryotic cells (Lenhard et al. 2012), we find that transcription initiation in C. elegans is often bidirectional. In addition to identifying transcription initiation sites at protein-coding genes, we also documented abundant intergenic and intronic transcription initiation sites and defined thousands of active enhancers based on the transcriptional activity of mapped transcription factor-binding sites. The global identification of transcription start sites will facilitate future studies of gene expression regulation in C. elegans and allow comparative analyses with other eukaryotes.
Mammalian enhancer regions have also been shown to be transcribed, but this phenomenon is not well-characterized, and its function is not yet known (De Santa et al. 2010; Kim et al. 2010; Koch et al. 2011; Melgar et al. 2011; Wang et al. 2011; Natoli and Andrau 2012). In addition, both unidirectional and bidirectional transcription has been observed. The relationships between the different reports of mammalian enhancer transcription are unclear because the studies used different cell lines and methods and assayed different types of sites: DNAse I hypersensitive sites, CBP-binding sites, TF-binding sites, or regions with enhancer-like chromatin signatures. In C. elegans, we observed that although transcriptional initiation at enhancers is usually bidirectional, productive elongation across them is predominantly in one direction, usually oriented toward the downstream gene. These enhancer transcripts are either unstable or not exported from the nucleus, as they are primarily apparent in libraries made from nuclear RNA and usually undetectable in libraries generated from whole-cell poly(A)+ (mainly cytoplasmic) RNA. We note that since the binding sites for only a small percentage of C. elegans transcription factors are available, the set of enhancers identified here is unlikely to be complete.
Oriented transcription across enhancers may seem to be at odds with the classical view that enhancers act in an orientation-independent manner. However, oriented enhancer function has been documented (Swamynathan and Piatigorsky 2002; Hozumi et al. 2013). Furthermore, although transcription across C. elegans enhancer regions is oriented, we do not know whether the enhancer function of these binding sites is oriented as well. For example, orientation could be determined through interactions with other regulatory elements associated with the downstream gene (e.g., the promoter).
Transcription from enhancer regions toward proximal promoters might create an open chromatin environment (Hirota et al. 2008) or could potentially deliver RNA polymerase II to the promoter (Vernimmen et al. 2011). Another possibility is that upstream enhancers could function as alternative promoters for protein-coding genes. Such a function would be in line with a recent report in mouse showing that active upstream intergenic enhancers can function as alternative tissue-specific promoters (Kowalczyk et al. 2012). C. elegans genes associated with upstream transcribed enhancers are enriched for having developmental, signaling, or neuronal functions, types of genes that usually show tissue or temporally restricted expression, and underenriched for genes with general cellular functions (Supplemental Table S4). Direct tests will be necessary to address whether enhancers have promoter activity because trans-splicing would remove upstream transcribed regions from the mature transcript. However, if upstream enhancers do function as promoters, they appear to have different characteristics than proximal promoters (Supplemental Fig. S7). In the future, it will be of interest to functionally analyze enhancer transcription and explore the similarities and differences in regulatory architecture between different organisms. Our finding of a novel widespread regulatory architecture in C. elegans and generation of new resources will facilitate future studies of gene expression regulation in metazoans.
Methods
Preparation of nuclear capped RNA-seq libraries
Worms were grown in liquid culture and two million adults bleached to obtain mixed staged embryos as previously described (Stiernagle 2006). Nuclei were isolated from embryos using the method of Ooi et al. (2010). Nuclei were further purified by centrifugation through a 1.8 M sucrose cushion in 10 mM Tris pH 7.5, 10 mM MgCl2. RNA was extracted from the purified nuclei using Tripure (Roche).
To prepare short cap RNA-seq libraries, we followed the published method by Nechaev et al. (2010) with minor modification. In brief, total nuclear RNA (20 to 25 μg) was run on a polyacrylamide gel and a size range of 20 to 100 nt extracted. To enrich for capped RNA, the purified short nuclear RNA was incubated with 5′ to 3′ RNA nuclease, Terminator (Epicentre), to deplete noncapped RNA. The enriched short capped RNA was cloned and PCRed using Illumina short RNA-seq adapters according to the manual instruction. To construct long cap RNA-seq libraries, total nuclear RNA was incubated with DNAse I (Ambion) to remove genomic DNA contamination, followed by Qiagen Mini-Elute columns (cut-off size > 200 nt RNAs). These cleaned-up “long” nuclear RNAs were treated with Terminator nuclease to enrich for capped RNA. Strand-specific long cap RNA libraries were made using the dUTP replacement method (Levin et al. 2010) and Illumina paired end adapters.
Short cap RNA and long cap RNA-seq libraries were sequenced using on Illumina GIIA instruments (SE36 and PE36, respectively). Two biological replicates were prepared for each library type.
Mapping, clustering of 5′ end tags, and shape score
Short cap reads were mapped to the WS220 reference sequence using BWA 0.5.9 (Li and Durbin 2009). The 36-bp reads were first mapped in their entirety, and those that did not align were then trimmed of any 3′ adaptor match and then retried. Mappings with quality < 10 were discarded at this point. Mapped reads were split by strand, with forward and reverse strand mappings analyzed independently, and mappings with the same strand and 5′ end position (cap-stacks) were combined. To define cap clusters, all cap-stacks containing two or more tags were clustered using a single-linkage approach, merging two or more stacks if they lie within 50 bp of one another. Singleton cap-stacks (not considered in the initial clustering step) were added to a cluster if they lay within the clustered region or 25 bp on either side. The final boundary coordinates of each cluster were then defined to include any singleton stacks. Finally, clusters overlapping rRNA, tRNA, miRNA, snRNA, snoRNA, or snlRNA genes (from Ensembl release 61/WS220) were excluded from the set. Using pooled cap RNA-seq data sets (two biological and one technical replica; 55,021,362 mapped reads), we obtained 73,500 clusters defining transcription initiation regions (TICs). For analyses where a single TSS position was required, we considered the distribution of cap 5′ ends within the TIC, and selected the position with the most tags (the mode). In the case of a tie (two or more positions with the same number of tags), we selected the mode closest to the median of the TIC. TIC shape score was calculated as the fraction of total tags in the cluster present at the mode position (the “mode weight”). Only TICs with at least 10 tags were used for the shape analysis. To monitor elongation, long cap RNA reads were mapped as read pairs using BWA 0.5.9 and filled in. For browser analyses of RNA-seq data, 36-bp long cap RNA sequence reads were mapped using BWA 0.5.9 in single-end mode.
TSS assignments
TICs were assigned to WormBase TSSs (via the Ensembl release 61/WS220 gene set) using a stepwise approach. In all cases, assignments were strand-specific (i.e., forward-strand TICs could only be assigned to forward-strand genes). First, any TIC overlapping a window from −199:+100 relative to a WormBase TSS was assigned (type “wormbase_tss”) to the gene associated with that TSS. Second, the remaining TICs were assigned (type “transcript_body”) to a gene if they lie anywhere within its body. Third, long-cap RNA mappings were assembled into continuous “rafts” of overlapping tags from the same strand. Rafts that overlap the TSS of exactly one gene (on the correct strand) were associated with that gene. The remaining (intergenetic) TICs were assigned (type “raft_to_wormbase_tss”) if they overlap a window from −199:+100 relative to the 5′ end of a transcript raft on the appropriate strand. All remaining TICs were left unassigned. Supplemental Table S2 gives the list of TICs and associated information.
Motif plots
Motif plots were performed on a subset of TICs made nonredundant by randomly selecting one representative from any set of TICs lying within 250 bp (taking both strands into account). The following position weight matrices (PWMs) were used: Inr (Chalkley and Verrijzer 1999), TATA (V$TATA_01 from TRANSFAC [Wingender 2008]), and Sp1 (V$SP1_Q6 from TRANSFAC [Wingender 2008]). We plotted the fraction of sequences with a match for the motif at base-pair resolution (for TATA and Inr) and at 20-bp resolution for the much-more-sparsely distributed Sp1. For TATA and Sp1, we consider the first position in the PWM to be the position of the motif match. For Inr, we follow convention in taking the strongly constrained A (third position in the PWM) as the location of the motif match.
Defining enhancers
Intergenic regions bound by one to four transcription factors were selected from Niu et al. (2011), discarding those lying within 500 bp of the TSS of a protein-coding gene (n = 4914). Regions overlapping at least one TIC (excluding TICs assigned to the TSSs of protein-coding genes) were identified (n = 3137). The Cistrome k-means clustering tool (Liu et al. 2011a) was used to generate eight groups of H3K4me3 and H3K4me1 histone modification signals to identify and discard likely unannotated promoters (clusters 1–4 in Fig 5). The remaining 2361 regions were defined as enhancers (Supplemental Table S3; clusters 5–8 in Fig 5).
Each enhancer was associated with its two neighboring protein-coding genes. If both these genes are in the same orientation (tandem genes), the enhancer was assigned to the downstream gene. Enhancers lying between transcribed genes were associated with the nearest downstream gene. Enhancers lying between convergently transcribed genes were left unassigned. Assignments are given in Supplemental Table S3.
To assess the directionality of transcription across enhancers assigned to tandem genes, we counted the number of long-cap fragments mapping in each direction over each enhancer, then calculated the moderated log ratio of forward vs. reverse reads, i.e., log10(f+1/r+1), where f is the number of forward-strand reads and r is the number of reverse-strand reads. Moderated log-ratios were used because there is a substantial number of enhancers with zero reads on one or the other strand. We then plotted forward/reverse ratios for enhancers between tandem forward-strand genes and enhancers between tandem reverse-strand genes.
Data sets, processing, and visualization
MNase-digested mononucleosome data for embryos (GSM514735) (Ooi et al. 2010) and adults (GSM777719) (Steiner et al. 2012) were downloaded from Gene Expression Omnibus. Transcription factor ChIP binding region data sets are from Niu et al. (2011). The following embryo ChIP-seq data were obtained from modENCODE (http://www.modencode.org/), with the following accession numbers: H3K4me3 (modENCODE_5166), H3K4me1 (modENCODE_5158), H3K36me3 (modENCODE_5165), H3K27ac (modENCODE_5159), H3K27me3 (modENCODE_5163), and HTZ-1 (modENCODE_6218). ChIP-chip RNA Pol II 4H8 data is from modENCODE_4148. Data are available from http://submit.modencode.org/submit/public/download/accession_number (e.g., for H3K4me3 data: submit.modencode.org/submit/public/download/5166).
Raw ChIP-seq data sets obtained from modENCODE were aligned using BWA with default settings (Li and Durbin 2009), normalized using BEADS (Cheung et al. 2011), then converted to log2 ratios of BEADS scores (enrichment relative to input), standardized so the mean of the autosomal signal was 0 and the standard deviation 1, and then z-scored. For RNA Pol II 4H8 ChIP-chip data, log2 ratios of IP/Input were obtained and standardized so the signal had mean 0 and standard deviation 1.
The heat map analyses were performed using the heat map analysis function in Cistrome (Liu et al. 2011a). The IGV Genome Browser was used for visualization (Robinson et al. 2011).
Nuclear protein-coding gene expression levels were calculated using the method of Hiller et al. (2009), calculating depth of coverage per million reads (dcpm) on the appropriate strand from the long cap RNA sequence data.
The GO term analysis was performed on tandem genes associated with enhancers at http://http://gostat.wehi.edu.au/) (Beissbarth and Speed 2004) and GO terms summarized using REVIGO (http://revigo.irb.hr/) (Supek et al. 2011).
To generate graphical representations of multiple sequence alignments at TSSs, extracted sequence data were uploaded to WebLogo (Crooks et al. 2004), a web-based application for sequence logos (http://weblogo.threeplusone.com/). The plots were generated using the default settings with adjustment of 36% GC composition for the C. elegans genome.
Data access
The RNA-seq data from this study have been deposited in the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE42819.
Acknowledgments
We thank R. Durbin for helpful comments on the manuscript and C. Bradshaw for bioinformatics support. R.A.-J.C., P.S., and J.A. were supported by a Wellcome Trust Senior Research Fellowship to J.A. (054523), T.A.D. by a Wellcome Trust Research Career Development Fellowship, and Q.B.C., T.A.E., and T.E.J. by National Human Genome Research Institute modENCODE grant 1-U01-HG004270-01. J.A. and T.A.D. also thank the Wellcome Trust and Cancer Research UK for core funding support.
Footnotes
-
↵6 Corresponding author
Email ja219{at}cam.ac.uk
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.153668.112.
Freely available online through the Genome Research Open Access option.
- Received December 13, 2012.
- Accepted March 29, 2013.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported), as described at http://creativecommons.org/licenses/by-nc/3.0/.








