
Widespread plasticity in CTCF occupancy linked to DNA methylation
- Hao Wang1,5,
- Matthew T. Maurano1,5,
- Hongzhu Qu1,2,5,
- Katherine E. Varley3,
- Jason Gertz3,
- Florencia Pauli3,
- Kristen Lee1,
- Theresa Canfield1,
- Molly Weaver1,
- Richard Sandstrom1,
- Robert E. Thurman1,
- Rajinder Kaul1,
- Richard M. Myers3 and
- John A. Stamatoyannopoulos1,4,6
- 1Department of Genome Sciences, University of Washington, Seattle, Washington 98195, USA;
- 2Laboratory of Disease Genomics and Individualized Medicine, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing 100029, China;
- 3HudsonAlpha Institute for Biotechnology, Huntsville, Alabama 35806, USA;
- 4Department of Medicine, University of Washington, Seattle, Washington 98195, USA
-
↵5 These authors contributed equally to this work.
Abstract
CTCF is a ubiquitously expressed regulator of fundamental genomic processes including transcription, intra- and interchromosomal interactions, and chromatin structure. Because of its critical role in genome function, CTCF binding patterns have long been assumed to be largely invariant across different cellular environments. Here we analyze genome-wide occupancy patterns of CTCF by ChIP-seq in 19 diverse human cell types, including normal primary cells and immortal lines. We observed highly reproducible yet surprisingly plastic genomic binding landscapes, indicative of strong cell-selective regulation of CTCF occupancy. Comparison with massively parallel bisulfite sequencing data indicates that 41% of variable CTCF binding is linked to differential DNA methylation, concentrated at two critical positions within the CTCF recognition sequence. Unexpectedly, CTCF binding patterns were markedly different in normal versus immortal cells, with the latter showing widespread disruption of CTCF binding associated with increased methylation. Strikingly, this disruption is accompanied by up-regulation of CTCF expression, with the result that both normal and immortal cells maintain the same average number of CTCF occupancy sites genome-wide. These results reveal a tight linkage between DNA methylation and the global occupancy patterns of a major sequence-specific regulatory factor.
The polyfunctional regulator CTCF plays a central role in multiple complex genomic processes, including transcription (Baniahmad et al. 1990; Filippova et al. 1996; Vostrov and Quitschke 1997), imprinting (Bell and Felsenfeld 2000; Hark et al. 2000), and long-range chromatin interactions and subnuclear localization (Yusufzai et al. 2004; Splinter 2006; Hou et al. 2008). Cohesin, a major mediator of chromosomal contacts during mitosis (Seitan et al. 2011), is tightly co-localized with CTCF, indicating a key function for CTCF in chromosome pairing (Parelho et al. 2008; Rubio et al. 2008; Wendt et al. 2008). CTCF has also been connected with multiple malignancies, including by the association of mutations in its gene locus (Filippova et al. 1998), through its anti-proliferative effect (Rasko et al. 2001), and through regulatory interactions with tumor suppressor genes (Butcher et al. 2004; Witcher and Emerson 2009; Soto-Reyes and Recillas-Targa 2010; Dávalos-Salas et al. 2011).
CTCF is ubiquitously expressed, and it is widely believed that CTCF binding patterns are largely invariant between cell types (Kim et al. 2007; Cuddapah et al. 2008; Heintzman et al. 2009), though diverse regulatory mechanisms at individual loci have been described (Lefevre et al. 2008; Sekimata et al. 2009; Witcher and Emerson 2009; Lai et al. 2010; Shukla et al. 2011). In addition, at a small number of loci, variable CTCF occupancy has been linked with DNA methylation in vivo (Kanduri et al. 2000; Pant et al. 2003), and in vitro studies suggest that methylation may hinder CTCF binding at certain sequence elements (Bell and Felsenfeld 2000; Hark et al. 2000; Filippova et al. 2001; Renda et al. 2007). However, neither the degree to which CTCF binding patterns vary between different cell types nor the relationship of such variability with DNA methylation is currently known.
We therefore sought to establish the cellular selectivity of CTCF binding and to define its relationship with methylation on a global scale. By using genome-wide occupancy profiling and reduced representation bisulfite sequencing (RRBS), we establish that a majority of CTCF sites are cell-selective, and link 41% of this variable CTCF occupancy to differential DNA methylation. We further observe markedly different CTCF binding patterns distinguishing normal and immortal cells, which are associated with increased methylation and up-regulation of CTCF expression. These results indicate a global linkage between DNA methylation and the occupancy patterns of an important genome regulator.
Results
Widespread plasticity of CTCF occupancy patterns
To assess CTCF binding variation genome-wide, we localized and quantified CTCF occupancy by ChIP-seq in 19 diverse cell types, including seven immortal cell lines and 12 normal cell types. We generated two biological replicates for each cell type. Both replicates were of high enrichment and exhibited high concordance (average correlation of 0.93) (Supplemental Fig. S1). We found that CTCF binds an average of about 55,000 sites in each tested cell type (Supplemental Fig. S1A). In total, we identified 77,811 distinct binding sites across all 19 cell types.
To survey binding variability genome-wide, we conservatively assessed how many cell types demonstrated binding at each site using a dual-threshold strategy to prevent bias toward variable sites (see Methods). In all 19 cell types, 27,662 binding sites were present. However, 50,149 binding sites were found to be unbound in at least one cell type (Supplemental Table S2). Thus, 64% of CTCF sites are found to vary in at least one cell type, demonstrating the existence of a widespread variability in CTCF occupancy. These variable sites exhibited clear occupancy differences between bound and unbound cell types, including at the well-known H19/IGF2 imprinted locus (Fig. 1A–C). Variable binding sites were occupied in an average of 10 of 19 cell lines, implying a high degree of shared regulation between cell types (Fig. 1D). Indeed, between any two cell types, an average of 72% of bound sites were in common (Supplemental Fig. S3). Variable sites had a similar genomic localization (Fig. 1E) compared with constitutive sites.
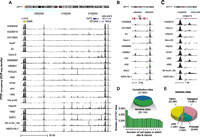
CTCF in vivo binding exhibits widespread plasticity. (A–C) Constitutive and variable CTCF sites. (A) The H19/IGF2 imprinted locus in multiple human cell types. Note the total silencing in two cell lines of the seven CTCF sites in the differentially methylated region (DMR; yellow box at left), and the complex pattern of cell-selective CTCF binding flanked by constitutive sites. Location (hg19), chr11:2,015,000–2,184,000. (B,C) Additional examples of variable sites. (D) Genome-wide analysis of CTCF binding in 19 cell types reveals 77,811 distinct binding sites; 27,662 sites are constitutively present in all cell types; 50,149 variable sites exhibiting a wide range of selectivity are present in a subset of one to 18 cell types (below). (E) Genomic distribution of variable sites is similar to constitutive sites (Supplemental Fig. S2A).
Distinct CTCF binding landscapes in normal vs. immortal cells
To understand whether binding variability follows a similar pattern in related cell types, we performed an unsupervised hierarchical clustering of variable CTCF binding sites (see Methods). We found that the variable CTCF binding landscape distinguished three groups (Fig. 2A). The first group of immortal cells consists of malignancy-derived and EBV-immortalized cell lines, including several carcinomas (colorectal, Caco-2; cervical, HeLa-S3; hepatocellular, HepG2), neuroblastoma (SK-N-SH_RA), and retinoblastoma (WERI-RB-1) and EBV-transformed lymphoplastoid (GM06990). The remaining two groups consist of normal cell types of limited proliferative potential: The second group consists of three epithelial cell types, including renal cortical (HRE), small airway (SAEC), and esophageal (HEEpiC) mucosal epithelia, and the third group consists of fibroblasts, including abdominal (AG10803), toe skin (AG09309), gum (AG09319), aortic adventitial (AoAF), foreskin (BJ), mammary (HMF), pulmonary artery (HPAF), and pulmonary (HPF) and brain microvascular endothelium (HBMEC). Principal component analysis and bootstrap assessment of the uncertainty in the hierarchical clustering confirmed a separation between the normal cell types and remaining cell lines, although the epithelial line HRE was less clearly distinguished (Supplemental Fig. S4). We then sought to identify the specific binding differences characterizing these three groups. We identified 4146 specific binding sites whose occupancy was significantly different between these groups at a false-discovery rate (FDR) of 1% (Methods) (Fig. 2B; Supplemental Table S3). These results suggest that CTCF occupancy exhibits major regulatory differences distinguishing immortal cell lines from normal epithelium, endothelium, and fibroblasts.
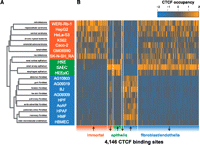
CTCF occupancy distinguishes similar cell types. (A) Unsupervised hierarchical clustering of binding at all CTCF sites. (B) CTCF occupancy at 4146 variable binding sites that distinguish immortal cell lines, epithelia, fibroblasts and endothelia (Methods). x-axis, CTCF binding sites in chromosomal order, separated into sites that are up-regulated and down-regulated (arrows) in each of the three groups (immortal, epithelial, fibroblast, and endothelial). Color corresponds to Z-score of normalized ChIP-seq density.
Variable CTCF occupancy linked to CpG methylation
Pre-existing methylation can antagonize CTCF binding in vitro (Bell and Felsenfeld 2000; Hark et al. 2000; Kanduri et al. 2000). Therefore we asked whether differential methylation was associated with variable sites in vivo. To study this, we compared CTCF occupancy and RRBS data (Fig. 3A). We studied a subset of CTCF sites in 13 cell types (n = 6,707) for which RRBS data were available from the ENCODE project (KE Varley, J Gertz, KM Bowling, SL Parker, TE Reddy, F Pauli, MK Cross, BA Williams, JA Stamatoyannopoulos, GE Crawford, et al., in prep.). We obtained methylation status of 44,048 CpGs dinucleotides in the region centered on these sites (see Methods), with each CpG monitored in an average of 12 out of 13 cell types (Supplemental Fig. S6).
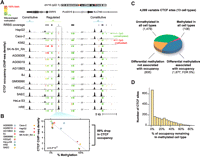
Impact of DNA methylation on cell-selective CTCF binding. (A) Example CTCF binding sites, where occupancy (above) quantitatively increases as local CpG methylation decreases (below). Green indicates CpG is 0% methylated; yellow, 50%; and red, 100%. (B) Quantitative analysis of methylation at the boxed CTCF binding site in A. (C) Global impact of methylation at variable CTCF sites monitored by RRBS. Sixty-five percent of sites with cell-type selective patterns of methylation also exhibited differences in occupancy. (D) At methylated binding sites, occupancy was reduced on average by 87% compared with cell lines without methylation at the same site. Shown are sites where increased methylation was associated with decreased occupancy (98% of all significant sites).
First, we assessed the overall methylation status at the 6707 CTCF sites with RRBS data. We found that methylation was substantially more variable at variable CTCF sites than at constitutive ones (Supplemental Fig. S5). Only 10% of these sites tested showed intermediate methylation status (between 25% and 75% methylation) (Supplemental Fig. S6). Overall, 98% of CTCF sites were unmethylated (defined as <50% methylation) in at least one of the cell types tested, confirming an inverse relationship between methylation and CTCF occupancy. However, 47% of CTCF sites were methylated (>50% methylation) in at least one cell type, suggesting a widespread potential link between methylation and CTCF occupancy.
To quantify the global association of differential methylation status with variable CTCF occupancy, we performed a linear regression analysis at the 6707 sites for which we had RRBS data (Fig. 3B; see Methods). Four thousand ninety-nine (61%) of these sites exhibited variable CTCF binding in the 13 cell types tested. Of the 4099 variable sites with RRBS data, 1677 (41%) showed a significant association (5% FDR) between methylation and occupancy (Fig. 3C). At significant sites, increased methylation was negatively associated with occupancy in 98% of cases. The magnitude of the association between methylation and occupancy was strong: Occupancy was on average 87% lower at significant sites in the methylated cell types relative to the unmethylated cell types (Fig. 3D). Further supporting a strong link to methylation, 67% of variable methylation was associated with a concomitant affect on occupancy. The remaining 36% of sites with variable methylation that was not associated with occupancy nevertheless demonstrated an aggregate reduction in occupancy in methylated cell types (Supplemental Fig. S7), confirming the overall inverse association of methylation with CTCF occupancy but suggesting that this relationship may be complicated by additional factors at this subset of sites.
Next we asked if the inverse relationship between methylation and CTCF occupancy is characterized by regional hypermethylation or if instead methylation is concentrated specifically at the region of protein–DNA interaction. We examined the location of all CpG dinucleotides relative to the CTCF motif at sites with variable methylation. Indeed, sites of differential methylation associated with occupancy differences showed an enrichment of CpG dinucleotides at two positions in the CTCF recognition sequence (Fig. 4). This finding is consistent with previous reports showing methylation outside the recognition sequence does not affect CTCF binding in vitro (Engel et al. 2004; Chadwick 2008). Within the recognition sequence, methylation at one of these CpGs (position 1) has been shown to inhibit binding of CTCF in vitro (Renda et al. 2007). The second (position 11) is the predominant CpG in the motif, which has been shown to have a higher rate of C–T transitions at vertebrate-conserved binding sites (Kim et al. 2007), consistent with germline methylation. Interestingly, constitutively unmethylated CTCF sites also showed an enrichment of CpGs at these two positions compared with differentially methylated sites without an association to occupancy (Supplemental Fig. S8). Given that the latter sites nevertheless exhibit substantial methylation variability, this suggests that the absence of CpGs at these positions may decouple CTCF occupancy from differential methylation at these sites. Overall, 29% of CTCF recognition sequences genome-wide contain a CpG at positions 1 and/or 11, and 52% of recognition sequences contain a CpG anywhere in the sequence. The genome-wide prevalence of “susceptible” CTCF sites suggests a widespread potential for interaction between CTCF and methylation.
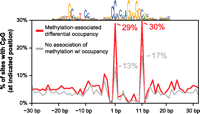
Sites significantly affected by methylation are enriched for CpGs at two positions. Frequency of a CpG (y-axis) at positions relative to the CTCF motif (x-axis) is shown for sites with variable methylation that is associated (red) and is not associated (gray) with occupancy changes. Note that at positions 1 and 11, there is a 2.2- and 1.8-fold enrichment, respectively, for the presence of a CpG at sites where the variable methylation was not associated with occupancy. Twenty-nine percent of CTCF motifs genome-wide contain a CpG at one or both of these positions.
Methylated-associated remodeling of CTCF binding in immortal cell lines
Paralleling prior reports of widespread hypermethylation in cancer (Jones and Baylin 2007; KE Varley, J Gertz, KM Bowling, SL Parker, TE Reddy, F Pauli, MK Cross, BA Williams, JA Stamatoyannopoulos, GE Crawford, et al., in prep.), we observed a bimodal pattern of methylation at CTCF sites distinguishing normal and immortal cell types (Fig. 5A). At 31% of the sites where differential methylation was associated with CTCF occupancy, methylation was observed throughout the 13 normal and immortal cell types (average number of methylated cell types, 7.3). In contrast, the remaining 69% of sites were characterized by cell-specific hypermethylation constrained to the six immortal lines (average number of methylated cell lines, 2.1) (Fig. 5A, strip at right). Notably, although the neuroblastoma line SK-N-SH_RA clusters with epithelial cell types based purely on CTCF binding (Fig. 2A), it exhibits the hypermethylation characteristic of the other immortal lines. Surprisingly, the increased methylation in immortal lines does not correspond to a decrease in the total number of bound CTCF sites (Fig. 5B). Strikingly, we also observed that CTCF transcript levels are significantly higher in the immortal cell lines (Fig. 5C). This disruption of CTCF binding in immortal cell lines is further distinguished by a unique association between CTCF occupancy and methylation at promoter sites. Of the promoter CTCF sites where methylation was significantly associated with occupancy, 98% (281 of 288) of these sites were characterized by hypermethylation in the immortal lines (Fig. 5D). These results suggest a widespread methylation-associated remodeling of the CTCF binding landscape in immortal cell lines.
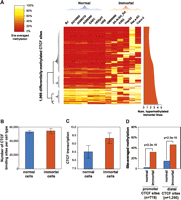
Cell-selective patterns of methylation associated with occupancy differences. (A) Methylation status at 1969 CTCF sites where differential methylation is significantly associated with occupancy differences. Color corresponds to the percentage of bisulfite sequencing tags at each site overlapping methylated CpG positions. Dendrogram (left) highlights pattern of hypermethylation in immortal cell lines. (Right) Smoothed plot of number of immortal lines exhibiting hypermethylation at each site. (B) Immortal lines show no significant difference in number of occupied CTCF sites (y-axis, mean). Error bars, SD. (C) immortal lines demonstrate increased CTCF transcript levels (y-axis, mean). Error bars, SD. (D) Immortal lines exhibit increased methylation relative to the other cell types, though significant promoter methylation is rarely observed in normal lines. y-axis, genome-wide median of per-site methylation. P-values, Wilcoxon. Promoter, ±2.5 kb of RefSeq transcription start site.
Discussion
Surprising plasticity of the CTCF occupancy landscape
This study exposes a previously unappreciated degree of plasticity within the binding landscape of the master genomic regulator CTCF. Previous studies in a small number of cell types had uncovered only limited cell-type specificity (Kim et al. 2007; Cuddapah et al. 2008; Heintzman et al. 2009). We further associate differential methylation with 41% of this variable binding at a subset of sites overlapping existing RRBS data in 13 cell types. We specifically linked this variable methylation to the presence of a CpG at two key positions relative to the consensus motif. Finally, we observe the maintenance of a stable total amount of CTCF genomic binding sites in immortal cell lines despite their altered localization associated with increased methylation. Our results show that methylation is indeed a global feature of the regulatory diversity of CTCF, and our approach is readily extensible to the repertoire of vertebrate transcription factors.
Methylation-associated disruption of CTCF binding in immortal lines
Although CTCF binding varied across all 13 cell types, we observed unique patterns of CTCF occupancy specific to the immortal cell lines. Interestingly, CTCF overexpression has previously been associated with resistance to apoptosis in breast cancer cell lines (Docquier et al. 2005) and with DNMT3B overexpression (Butcher et al. 2004). Further, the unique occurrence of hypermethylation-associated abrogation of CTCF occupancy at promoters in immortal lines is notable, given the involvement of CTCF in the methylation-associated silencing of known tumor suppressors and oncogenes (Witcher and Emerson 2009; Lai et al. 2010; Soto-Reyes and Recillas-Targa 2010). We found that the immortal cell lines we profiled have the same overall amount of genomic CTCF binding sites despite a redistribution of CTCF occupancy from binding sites subject to hypermethylation. The concomitant up-regulation of CTCF expression may therefore represent a cancer-associated compensatory mechanism. This inverse correlation is compatible with the existence of a stabilizing mechanism acting through increased CTCF expression to maintain a constant level of genomic binding despite increased methylation at its target sites, although further study in an expanded set of cell types will be necessary.
The role of DNA methylation in regulation of transcription factor occupancy
Although DNA methylation is widely invoked as a causal mechanism for transcriptional repression, surprisingly little in vivo evidence is available. While experimentally directed methylation can prevent binding of CTCF and other factors in vitro (Tate and Bird 1993; Renda et al. 2007), the mechanisms establishing methylation patterns in vivo remain unknown, and its precise relationship with gene expression remains unclear (Enver et al. 1988; Selker 1990; Walsh and Bestor 1999). Likewise, our results do not distinguish whether demethylation facilitates subsequent CTCF binding or whether bound CTCF maintains an unmethylated domain.
An alternative model has DNA methylation deposited passively in the wake of independent abrogation of transcription factor binding. This model is equally consistent with evidence that transcription factor binding sites appear to be generally depleted for DNA methylation (Mukhopadhyay et al. 2004; Lister et al. 2009; Thurman et al. 2012) and that binding sites recognized by certain sequence-specific factors have been associated with lack of methylation (Straussman et al. 2009; Dickson et al. 2010; Gebhard et al. 2010; Lienert et al. 2011). Indeed, there is evidence that the binding of some transcription factors, including CTCF, is sufficient to effect a local demethylated state (Matsuo et al. 1998; Lin et al. 2000; Stadler et al. 2011). But if in vivo methylation was deposited generally at unoccupied binding sites, then how would this process interact with the in vitro methylation sensitivity of common transcription factors?
The well-investigated H19/Igf2 imprinted locus offers an appropriate example: CTCF binding there has been shown necessary to maintain an existing unmethylated state (Schoenherr et al. 2002; Pant et al. 2004). However, CTCF is not the originator of the unmethylated state (Matsuzaki et al. 2010), implying a limited capacity to directly affect methylation. Perhaps methylation instead acts as a cooperative switch to prevent the return of CTCF after a reprogramming event. In this model, rather than guiding binding localization, methylation is a general amplifier of perturbations to transcription factor occupancy.
Other sources of variable CTCF binding
Although we have shown that 41% of overall CTCF occupancy variation is significantly linked to methylation at tested sites, 36% of variable CTCF sites overlap no variable methylation at all. It is unlikely that much of this variability is associated with genetic variability in CTCF recognition sequences (Maurano et al. 2012), though some sites may associate with modified forms of CTCF (Klenova et al. 2001; Yu et al. 2004; MacPherson et al. 2009). One likely possibility is that the constantly unmethylated variable CTCF sites may represent instances of cooperative regulation that complicate a direct relationship between methylation and CTCF occupancy. Accordingly, CTCF has been known to interact with a number of cofactors that could potentially govern its selectivity at these sites or, alternatively, maintain demethylation in the absence of CTCF binding (Chernukhin et al. 2007; Donohoe et al. 2007, 2009; Parelho et al. 2008; Rubio et al. 2008; Wendt et al. 2008; Ohlsson et al. 2010; Liu et al. 2011). Interestingly, we found that of the 36% of variable sites despite constant methylation, 76% were within 2.5 kb of a RefSeq transcription start site, compared with 38% of the variable sites associated with methylation differences. Recent work has further observed an enrichment of tethered CTCF peaks at promoters (Neph et al. 2012b), suggesting that the remaining variation in CTCF occupancy may derive from complex regulation of co-factors or variation in its specific interaction partners. Given the breadth of CTCF's regulatory functionality, our observation of global binding variation implies a widespread potential role in the translation of epigenetic marks to genome organization at thousands of sites.
Methods
Cell culture
Cells were cultured in an appropriate growth medium, with the addition of growth factors and supplements according to the suppliers' instructions (Supplemental Table S1). Cell lines were maintained in a humidified incubator at 37°C in the presence of 5% CO2.
ChIP-seq
Suspension cells were cross-linked with formaldehyde (Sigma) at a final concentration of 1% for 10 min at room temperature. Adherent cells were first detached from the plates by 0.05% Trypsin-EDTA and Trypsin neutralizer solution (Invitrogen) and then cross-linked by 1% formaldehyde. Glycine was added to a final concentration of 0.125 M for 5 min. Cells were rinsed twice with phosphate buffered saline, lysed in lysis buffer (50 mM Tris-HCl at pH 8.0, 10 mM EDTA, 1% SDS) containing protease inhibitor cocktail (Roche), and sheared by Bioruptor (Diagenode). The chromatin was incubated with Dynabeads (M-280, sheep anti-rabbit IgG, Invitrogen)-conjugated anti-CTCF polyclonal antibody (Cell Signaling no. 2899).
The CTCF–DNA complexes were washed, eluted, and reverse cross-linked. The DNA was RNase A–, Proteinase K–treated, and purified by phenol-chloroform-isoamyl alcohol extraction and ethanol precipitation. DNA was end-repaired (End-it DNA End-repair kit, Epicentre), followed by the addition of adenine to the 3′ ends (Taq DNA polymerase, NEB), and ligated to an adapter (Illumina). Purified ligation product was PCR amplified and run on a 2% agarose gel. The size-selected libraries were sequenced on an Illumina Genome Analyzer (Illumina) by the High-Throughput Genomics Center (University of Washington) according to a standard protocol.
For each cell type, experiments were conducted on two independent biological replicates.
Identification and quantification of CTCF binding sites
We obtained Uniform Element Calls from the ENCODE project for each cell line. Briefly, peaks were called using SPP (Kharchenko et al. 2008). The set of peaks reproducible in both replicates were identified based on an irreproducible discovery rate (IDR) of 0.25% (Li et al. 2011). We then combined peak calls from 19 cell types to generate a master list of all distinct CTCF binding sites. We adjusted the peak locations to center on matches to the nearest CTCF motif (P < 10−5, fimo) if the motif was within 50 bp.
To distinguish between variable and constitutive binding sites, for each site we examined the presence of a peak in each of 19 cell types. We used the peak calling program Hotspot (John et al. 2011) to enable a conservative procedure for the identification of variable binding sites. To reduce the misclassification of sites near the peak-calling threshold as variable, we employed separate cutoffs for calling peak presence and absence. First, for each CTCF binding site called above, we additionally required that it overlap a 0.5% FDR hotspot in both replicates of at least one cell line. Then, a binding site was counted as occupied in subsequent cell lines if a looser 1% FDR hotspot was present in one or both replicates for that cell line. Employing this looser criteria for binding in subsequent cell types results in conservative identification of variable sites. We confirmed that binding sites in cell types considered absent were substantially closer to background that sites in cell types considered active (Supplemental Fig. S2B).
ChIP-seq data were mapped to the human genome (GRCh37/hg19) using bowtie (Langmead et al. 2009) with the options “bowtie–mm -n 3 -v 3 -k 2–phred64-quals,” allowing up to three mismatches. Reads mapping to multiple locations were then excluded, and reads with identical 5′ ends and strand were presumed to be PCR duplicates and were excluded. Smoothed density tracks were generated using bedmap (http://code.google.com/p/bedops/) to count the number of tags overlapping a sliding 150-bp window, with a step width of 20 bp (Neph et al. 2012a). Density tracks were normalized for sequencing depth by a global linear scaling to 10 million tags. We measured occupancy by the maximum normalized ChIP-seq tag density over the 134-bp region.
Reproducibility of ChIP-seq experiments was tested using Pearson correlation on normalized density tracks of chromosome 19 between each replicate.
Clustering of cell-selective CTCF binding sites
We converted the presence and absence of a given peak to 1 and 0, respectively, in 19 cell lines. We then performed hierarchical clustering with the hclust function in R, using the “average” method and Euclidean distance metric. We cut the dendrogram (Fig. 2A) into three groups, of immortal cell lines, epithelia, and fibroblasts. To assess the significance of these three groups, we used the R package pvclust (Suzuki and Shimodaira 2006) and principal components analysis (Supplemental Fig. S4). We then used the package DESeq (Anders and Huber 2010) on the tag count at each peak to identify differentially occupied sites between each of these three groups (FDR 1%).
RRBS genome-wide methylation profiling
We downloaded RRBS methylation data for 13 cell lines from the “HAIB Methyl RRBS” track (KE Varley, J Gertz, KM Bowling, SL Parker, TE Reddy, F Pauli, MK Cross, BA Williams, JA Stamatoyannopoulos, GE Crawford, et al., in prep.) of the UCSC Genome Browser. To measure methylation in each cell line, we combined counts for both strands in both replicates and removed data for samples with less than 8× coverage. We retained only CpGs monitored in at least six samples (Supplemental Fig. S6B).
We applied a linear regression to measure whether methylation status is associated with occupancy. We normalized CTCF occupancies using the getVarianceStabilizedData function of DESeq and then averaged replicate signals. We regressed CTCF occupancy onto the average proportion methylated of all monitored CpGs in a 134-bp region centered around the CTCF peak. We excluded 1806 sites missing RRBS data and ChIP-seq data for seven or more cell types or having too great a difference in the number of CpGs monitored between any two cell types (more than six CpGs monitored). We averaged the methylation level of all CpGs within a 134-bp window to increase sensitivity and reliability. We excluded sites where the number of monitored CpGs differed by more than four among any two cell lines. We used the R package qvalue to estimate an FDR (Storey and Tibshirani 2003).
RNA expression analysis
For each cell line, total RNA was extracted in two replicates from 5 × 106 cells using Ribopure (Ambion) according to the manufacturer's instructions. RNA quality was ascertained using RNA 6000 Nano Chips on a bioanalyzer (Agilent). Approximately 3 μg of total RNA for each sample was used for labeling and hybridization (University of Washington Center for Array Technology) to Affymetrix Human Exon 1.0 ST arrays (Affymetrix) using a standard protocol. Exon expression data were analyzed through Affymetrix Expression Console using gene-level RMA summarization and sketch-quantile normalization method. Measurements from both replicates were then averaged.
Data access
CTCF ChIP-seq data have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession no. GSE30263. Affymetrix exon array data are available under accession no. GSE19090. RRBS methylation data are under accession no. GSE27584. All three sets are available for viewing in the UCSC Genome Browser (http://genome.ucsc.edu/).
Acknowledgments
We thank Jeff Vierstra, Andrew Stergachis, and Sam John for critical reading of the manuscript and many helpful suggestions. We also thank Daniel Bates, Morgan Diegel, and Doug Dunn at the University of Washington High-Throughput Genomics Center for technical assistance. This work was supported by National Institutes of Health grants U54HG004592 (J.A.S.) and U54HG004576 (R.M.M.).
Author contributions: H.W., M.T.M., and J.A.S. conceived the study. H.W. and T.C. cultured cells. H.W. and K.L. produced ChIP-seq data. H.W. and M.W. generated Illumina libraries. K.E.V., J.G., and F.P. generated RRBS data under the supervision of R.M.M. M.T.M., R.S., and R.E.T. processed data. M.T.M. and H.Q. analyzed data. R.K. oversaw production data collection and aspects of primary analysis. H.W. and M.T.M. wrote the manuscript, with contributions from J.A.S.
Footnotes
-
↵6 Corresponding author
E-mail jstam{at}uw.edu
-
[Supplemental material is available for this article.]
-
Article and supplemental material are at http://www.genome.org/cgi/doi/10.1101/gr.136101.111.
Freely available online through the Genome Research Open Access option.
- Received December 9, 2011.
- Accepted April 30, 2012.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported License), as described at http://creativecommons.org/licenses/by-nc/3.0/.








