
Human postmeiotic sex chromatin and its impact on sex chromosome evolution
- 1Division of Reproductive Sciences, Perinatal Institute, Cincinnati Children's Hospital Medical Center, Cincinnati, Ohio 45229, USA;
- 2Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, Ohio 45267, USA;
- 3Department of Integrative Cancer Therapy and Urology, Andrology Unit, Kanazawa University Graduate School of Medical Science, Kanazawa, Japan
Abstract
Sex chromosome inactivation is essential epigenetic programming in male germ cells. However, it remains largely unclear how epigenetic silencing of sex chromosomes impacts the evolution of the mammalian genome. Here we demonstrate that male sex chromosome inactivation is highly conserved between humans and mice and has an impact on the genetic evolution of human sex chromosomes. We show that, in humans, sex chromosome inactivation established during meiosis is maintained into spermatids with the silent compartment postmeiotic sex chromatin (PMSC). Human PMSC is illuminated with epigenetic modifications such as trimethylated lysine 9 of histone H3 and heterochromatin proteins CBX1 and CBX3, which implicate a conserved mechanism underlying the maintenance of sex chromosome inactivation in mammals. Furthermore, our analyses suggest that male sex chromosome inactivation has impacted multiple aspects of the evolutionary history of mammalian sex chromosomes: amplification of copy number, retrotranspositions, acquisition of de novo genes, and acquisition of different expression profiles. Most strikingly, profiles of escape genes from postmeiotic silencing diverge significantly between humans and mice. Escape genes exhibit higher rates of amino acid changes compared with non-escape genes, suggesting that they are beneficial for reproductive fitness and may allow mammals to cope with conserved postmeiotic silencing during the evolutionary past. Taken together, we propose that the epigenetic silencing mechanism impacts the genetic evolution of sex chromosomes and contributed to speciation and reproductive diversity in mammals.
The germline is the only heritable lineage across generations and drives evolution. In the male germline, X and Y chromosomes exist in hemizygosity and are vulnerable to genetic exchanges (Vicoso and Charlesworth 2006; Ellegren 2011). Therefore, the genetic contents of sex chromosomes are significantly diverged from those of autosomes. Sex chromosomes are thought to have evolved from ordinary autosome pairs during the evolutionary past (Ohno 1967). While the Y has degenerated, the sex chromosomes have accumulated unique genomic features and retain evolutionary traces of ancestral autosomes (Lahn and Page 1999; Skaletsky et al. 2003).
In the male germline, a hallmark event on the sex chromosomes is meiotic sex chromosome inactivation (MSCI) (Turner 2007; Inagaki et al. 2010; Payer et al. 2011). Sex chromosomes are largely unsynapsed during meiosis and are thereby treated differently from synapsed autosomes and specifically inactivated. MSCI presumably has impacted genomic evolution, such as through extensive retrotransposition of X-linked genes to autosomes (Emerson et al. 2004; Wang 2004; Potrzebowski et al. 2008), and the chromosomal distribution of male-biased genes. While male-biased genes that express during early spermatogenesis are enriched on the X-chromosome, male-biased genes that express during late spermatogenesis are underrepresented on the X chromosome due to MSCI (Khil et al. 2004). On the other hand, a recent study identified newly evolved male-biased genes in mammalian lineages that are enriched on the X chromosome and tend to escape sex chromosome inactivation in the round spermatid of mice, while a large number of older genes, which are expressed in the round spermatids, are enriched on autosomes (Zhang et al. 2010).
An intriguing feature of MSCI is the epigenetic maintenance of silencing. Once it is established during meiosis, silencing is maintained through meiotic cell divisions into spermiogenesis in mice (Greaves et al. 2006; Namekawa et al. 2006; Turner et al. 2006), and sex chromosomes occupy a silent compartment termed “postmeiotic sex chromatin” (PMSC) in postmeiotic spermatids (Namekawa et al. 2006). However, it remains largely unclear whether postmeiotic silencing is conserved in humans and how this epigenetic maintenance process of sex chromosome inactivation has impacted the evolution of mammalian sex chromosomes. To understand the evolutionary impact of epigenetic silencing of sex chromosomes, we investigated sex chromosome inactivation in humans and compared the evolutionary past of sex chromosomes between humans and mice.
Mammals have evolved distinct strategies between males and females to achieve epigenetic silencing of sex chromosomes. A hypothesis predicted that epigenetic memories of MSCI persisted into female early embryos and established selective inactivation of the paternal X chromosome (i.e., imprinted X-inactivation) in mice (Huynh and Lee 2003; Namekawa et al. 2006, 2010) and in marsupials (Namekawa et al. 2007). However, recent evidence suggests that the dynamics of X-chromosome inactivation in females diverges in each species of placental mammals, and that imprinted X-inactivation is not observed in preimplantation embryos of humans and rabbits (Okamoto et al. 2011), raising the question as to whether epigenetic regulation of sex chromosomes is evolutionarily conserved in the human germline. In contrast with the divergent dynamics of female X-inactivation, here we identify human PMSC and establish that there are conserved epigenetic mechanisms underlying male sex chromosome inactivation in humans and mice. Furthermore, our comprehensive analyses suggest the evolutionary impact of the epigenetic maintenance process of sex chromosome inactivation on the mammalian genome.
Results
Initiation of MSCI is mechanistically conserved in humans and mice
To elucidate the potential mechanism underlying human sex chromosome inactivation, we performed cytological analyses using paraffin sections of human testis. Paraffin sections allowed us to detect the intact chromatin structure of human germ cells. Using our optimized protocols (Methods) (Namekawa and Lee 2011), we have obtained very specific signals of immunostaining in combination with DNA fluorescence in situ hybridization (FISH) (Supplemental Fig. S1). MSCI occurs in the pachytene stage of meiosis, followed by two rounds of meiotic divisions, and leads to round spermatids (Fig. 1A). In mice, MSCI is initiated by action of the DNA damage response pathway centered on the mediator of DNA damage checkpoint 1 (MDC1) (Ichijima et al. 2011). MDC1 is a binding partner of the phosphorylated form of histone variant H2AX (γH2AX) and amplifies the γH2AX signals via a feedback loop with an ATR activator, TOPBP1, to recognize the chromosome-wide domain of sex chromosomes at the onset of MSCI. To determine whether human MSCI is conserved in mice, we examined the localization of factors associated with the DNA damage response pathway, which is a master regulator of mouse MSCI (Ichijima et al. 2011). We found that γH2AX, MDC1, and TOPBP1 consistently localize on the XY body of human spermatocytes (Fig. 1B,C), suggesting that the DNA damage response pathway centered on MDC1 is the potential mechanism for the initiation of MSCI in humans, as is the case with mice.
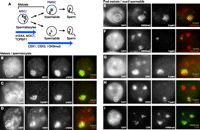
Identification of human PMSC and conserved mechanisms of sex chromosome inactivation in mammalian males. (A) Schematic of sex chromosome inactivation in males, and summary of modifications on the sex chromosomes. (B–D) Identification of modifications on the XY body in human primary spermatocytes by immunostaining. Locations of the XY body are highlighted with dotted circles. (E–H) Identification of human PMSC and its modifications in human round spermatids. Immunostaining is combined with DNA FISH to detect modifications on the region of the sex chromosomes. Locations of PMSC are highlighted with dotted circles. (I) Organization of the chromocenter detected by immunostaining with anti-centromere antibody in human round spermatids. All images are wide-field images.
Identification of human postmeiotic sex chromatin: Conserved maintenance mechanisms underlying male sex chromosome inactivation
Next, to determine whether epigenetic silencing of the sex chromosomes is maintained after meiosis in human round spermatids, we sought to identify human PMSC. Previously, we showed that heterochromatin proteins CBX1 (also known as HP1β) and CBX3 (also known as HP1γ) and trimethylation of histone H3 lysine 9 (H3K9me3) were conserved markers of PMSC both in mice and opossums (Namekawa et al. 2007). In humans, we found that CBX1, CBX3, and H3K9me3 start to accumulate on the sex chromosomes in the pachytene-to-diplotene transition during meiosis (Fig. 1D; Supplemental Fig. S1; Metzler-Guillemain et al. 2003). In human round spermatids, both the X and Y chromosomes are illuminated with H3K9me3 (Fig. 1E,F; 100% overlap with either the X or Y chromosome paints, respectively, n = 50 each). Also, CBX1 and CBX3 accumulate on the sex chromosomes (Fig. 1G,H; 82% of overlap with CBX1 and X paints, n = 50; 94% of overlap with CBX3 and Y paints, n = 50). The regions of sex chromosomes are slightly DAPI-enriched (Fig. 1E–H), although it is not as clear as mouse PMSC (Namekawa et al. 2006). We conclude that humans have PMSC and that CBX1, CBX3, and H3K9me3 are conserved modifications of PMSC in mammals. Notably, we found that the structure of the chromocenter, which is a cluster of pericentromeric heterochromatin, is largely distinct between humans and mice. As illuminated with anti-centromere staining, the human chromocenter is segregated in multiple spots in the nucleus (Fig. 1I), while the mouse chromocenter is usually a single spot and occasionally two spots in the nucleus (Namekawa et al. 2006). Despite this morphological difference, PMSC shares modifications with pericentromeric heterochromatin and is also largely associated with the chromocenter, suggesting that sex chromosomes maintain silent epigenetic modifications in the mammalian germline using the same type of mechanism as pericentromeric heterochromatin.
The above results suggest that human PMSC is a transcriptionally inert compartment. To examine the chromosome-wide silencing of the sex chromosome on human PMSC, we performed immunostaining of RNA polymerase II (Pol II). Pol II is largely excluded from the XY body in the pachytene stage (Fig. 2A), and also is continuously excluded from PMSC in round spermatids (Fig. 2B,C). On the other hand, autosomes occupy relatively large chromosome territories and spread over the Pol II–positive regions (Fig. 2D), indicating that chromosome-wide silencing is specifically on sex chromosomes with a silent compartment, PMSC. Taken together, we establish that sex chromosome inactivation in males is highly conserved between humans and mice. Our results suggest that chromosome-wide silencing in MSCI is triggered by a DNA damage response pathway and epigenetically maintained on PMSC with silent epigenetic modifications among mammals (Fig. 1A).
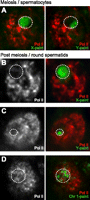
Transcriptional silencing on human PMSC. Immunostaining of RNA polymerase II (Pol II) and combined DNA FISH. (A–C) Transcriptional silencing of the human XY body and human PMSC. Locations of the XY body and PMSC are highlighted with dotted circles. (D) The chromosome territory of chromosome 1 (dotted circle) extends over the Pol II–positive regions. All images are deconvolved single Z-sections.
Comprehensive expression profiles of human sex chromosomes during spermatogenesis
Next, to understand the profiles of human sex chromosome inactivation on a gene-by-gene basis, we establish comprehensive expression profiles of MSCI and postmeiotic silencing in the human germline. Previously, to determine the profile of MSCI in mice, we examined gene expression changes during the mitosis-to-meiosis transition by comparing microarray data between spermatogonia and pachytene spermatocytes (Namekawa et al. 2006). To determine the profiles of human sex chromosome inactivation, we analyzed previously published microarray data of human spermatogonia (SG), pachytene spermatocytes (PS), and round spermatids (RS) (see Methods) (Chalmel et al. 2007; Wu et al. 2009).
In the chromosome-specific average expression, we observed a significant reduction of X-linked gene expression in the mitosis-to-meiosis
transition and modest derepression in postmeiotic RS (Fig. 3A). To analyze the X-linked gene expression on a gene-by-gene basis and determine the depth of chromosome-wide silencing in
the human germline, we sorted X-linked genes into the same representative groups of expression patterns we observed in the
mouse data set (Fig. 3B; Namekawa et al. 2006; see Methods). The Affymetrix Human U133 Plus 2.0 Array contains 1675 arrays that match with X-linked loci. Among these,
916 arrays representing distinct X-linked genes were selected. We specifically analyzed 351 expressed genes and eliminated
565 low-expression genes (which do not express above an expression value of 100 at any developmental stage), because low-expression
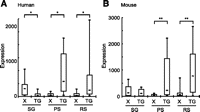
genes tend to exhibit nonspecific fluctuation. Next, we determined the depth of silencing in PS by this criterion: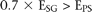 (E represents the expression of the cell type denoted in the subscript).
(E represents the expression of the cell type denoted in the subscript).
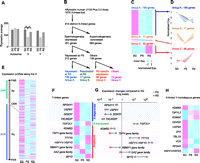
Microarray analysis of human MSCI and postmeiotic silencing. (A) Expression average of spermatogonia (SG), pachytene spermatocytes (PS), and round spermatids (RS). (**) p < 10−4, Tukey's multiple comparison test. (B) Classification of X-linked genes based on the expression profiles. (C) Expression heatmaps of normalized gene expression profiles for each group. (D) Normalized expression patterns of each group. (E) Expression heatmap along the location of the X chromosome. (F) Expression heatmap of Y-linked genes; genomic features are indicated. (G) Expression changes of Y-linked genes compared with SG. (H) Expression heatmap of X-linked Y-homologous genes.
This criterion was based on our previous study in mice (Namekawa et al. 2006) and illuminates a consistent profile of MSCI between mice and humans. By this criterion, 212 out of 351 expressed genes
are identified as repressed in PS (Fig. 3B), indicating that a large portion of X-linked genes are repressed by MSCI, as was estimated in a precedent study (Mulugeta Achame et al. 2010). To determine whether these repressed genes are reactivated in RS, we calculated the “recovery rate” as we had previously
for the mice data sets (Supplemental Fig. S2):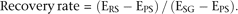
If the recovery was >0.5 (i.e., ERS recovered >50% of the repressed value between SG and PS), we defined that gene as reactivated. By this formula, we found that the vast majority of X-linked genes stay silent among the repressed genes at PS due to MSCI (195 out of 212 genes) (Fig 3B–D; Supplemental Fig. S2). We classified these genes as Group A, in which a large portion of X-linked spermatogenesis-expressed genes belong (Fig. 3C,D; Supplemental Fig. S2). In contrast, only a small portion of genes subjected to MSCI are reactivated in RS (Group B). We conclude that the majority of genes subject to MSCI are continuously repressed in RS by postmeiotic silencing, consistent with our cytological identification of human PMSC (Figs. 1, 2). Intriguingly, a group of genes highly expressed in RS exhibit specific expression only in RS (Group C in Fig. 3B–D). The other unclassified genes were categorized as Group D. The distribution of Groups A, B, and C is very similar to that in mice (Fig. 3B–D; Supplemental Table S1; Namekawa et al. 2006), suggesting the existence of conserved, chromosome-wide regulation between humans and mice.
To examine the expression profiles along human sex chromosomes, we arranged the microarray data in the order of gene locations along the X chromosome. We confirmed that MSCI and postmeiotic silencing occur in a chromosome-wide manner (Fig. 3E). Although silencing is maintained into round spermatids, as judged by the recovery rate (Supplemental Fig. S2), chromosome-wide silencing can be incomplete at this stage, as shown in mice (Namekawa et al. 2006). The human X chromosome is divided into the XAR (X-chromosome adding region) and the XCR (X-chromosome conserved region) (Ross et al. 2005). In human female X-inactivation, escape genes tend to locate in the XAR (Carrel and Willard 2005). In contrast, we did not observe any biased distribution of escape genes to either the XCR or the XAR in the male germline. This difference reflects distinct properties of escape genes between human female X-inactivation and male sex chromosome inactivation.
Notably, the human Y chromosome exhibits distinct gene expression patterns depending on the gene's properties. Genes on the ampliconic region of the Y tend to be highly expressed in both PS and RS, while X-degenerated genes on the Y are generally repressed in PS and RS (Fig. 3F,G; Supplemental Table S2). X-degenerated genes on the Y have homologs on X, presumably due to homologous recombination between ancient X and Y chromosomes (Ellegren 2011). X-linked genes that have Y-chromosome homologs are not highly expressed in RS (with exceptions, such as TSPYL2, KDM5C, and HSFX1/HSFX2) (Fig. 3H; Supplemental Table S3). Therefore, it is unlikely that X-Y homologs compensate each other's dosage during spermatogenesis.
A subset of multicopy, sex-linked genes escapes sex chromosome inactivation
A previous study showed that X-linked multicopy genes tend to be expressed in RS and that they exhibit higher mean expression values than X-linked single-copy genes (Mueller et al. 2008). This study suggested that X-linked multicopy genes tend to be repressed by MSCI but reactivated in RS. However, we do not observe any specific human multicopy X-linked genes exhibiting temporal suppression by MSCI and subsequent reactivation in RS (Fig. 4A; Supplemental Table S4). Furthermore, most of the genes in Groups B and C that are highly expressed in RS are single-copy genes in humans and mice (Supplemental Tables S1, S8). To investigate this disparity further, we have reanalyzed the gene-expression profile of mouse X-linked multicopy genes during spermatogenesis using the published microarray data (Namekawa et al. 2006). We found that mouse multicopy X-linked genes are mainly classified into two major groups. The first group is repressed by MSCI and remains repressed in RS; the other group is expressed in an RS-specific manner and contains genes that are already silent in the mitotic cells, regardless of MSCI (Fig. 4B; Supplemental Table S5). It should be noted that the data in the previous study (Mueller et al. 2008) match the average expression profile we found for these two major groups (Fig. 4C). Therefore, our analysis is in line with the previous conclusion (Mueller et al. 2008), but further reveals different classes of multicopy X-linked genes.

Expression profiles of X-linked multicopy genes. (A) Expression heat map of human X-linked multicopy genes in this study (see Methods) (Chalmel et al. 2007; Wu et al. 2009). (B) Expression heatmap of mouse X-linked multicopy genes. Published microarray data were reanalyzed (Namekawa et al. 2006). SG in mice data sets represents the average expression levels between spermatogonia A and B. Mean expression patterns are shown in right panels. A previous study proposed that most multicopy X-linked genes are repressed by MSCI and reactivated in RS in mice (Mueller et al. 2008). However, we found that mouse multicopy X-linked genes are mainly classified into two major groups. The first group is repressed by MSCI, and these genes also stay repressed in RS (repressed at PS and RS); the other group is expressed in an RS-specific manner, and these genes are already silent in the mitotic cells, regardless of MSCI (RS-specific). We found that only two genes (Plemel3 and Ott in panel B) showed repression by MSCI and reactivation in RS in mice. (C) Of note, when we calculate the average expression of the two major groups, “Repressed at PS and RS” and “RS-specific,” our data mirror the expression pattern from the previous study (Mueller et al. 2008).
The previous study (Mueller et al. 2008) and our current data suggest that amplification of copy number is the potential mechanism for escape from MSCI and postmeiotic silencing. Sex-linked multicopy genes have reproductive functions; for instance, the DAZ gene family promotes later stages of meiosis and the development of haploid gametes (Kee et al. 2009). The copy number of the DAZ gene family varies across primate genomes, and there are no DAZ gene family orthologs on the mouse Y chromosome, suggesting that the DAZ gene family is a recently evolved gamete protein and amplification of copy number that occurred after the emergence of the primate lineage. We propose that MSCI and postmeiotic silencing are the underlying evolutionary forces that amplify the copy number to ensure sufficient expression of reproductive genes against silencing.
Autosomal X-retrotransposed genes compensate for postmeiotic silencing
Another evolutionary trace of MSCI is extensive retrotransposition of X-linked housekeeping genes to autosomes; this process drove the differentiation of the X chromosome (Emerson et al. 2004). To investigate the gene expression profiles of retrotransposed genes, we identified 20 X-retrotransposed genes in humans and 27 genes in mice (Supplemental Tables S6, S7; see Methods). In humans, X-retrotransposed genes are highly expressed in PS and RS (Fig. 5A), supporting the notion that X-retrotransposed genes compensate for the expression of X-linked homologs silenced by MSCI. Expression profiles of X-retrotransposed genes are also consistent in mice (Fig. 5B). Notably, in both humans and mice, X-retrotransposed genes are continuously expressed in RS, and the X-linked homologs stay silent in RS. Therefore, X-retrotransposed genes compensate not only MSCI but also postmeiotic silencing in mammalian evolution, in agreement with a study in mice (Potrzebowski et al. 2008). Taken together, retrotransposition of X-linked housekeeping genes to autosomes could have been an alternative evolutional strategy to cope with MSCI and postmeiotic silencing in mammals.
Autosomal X-retrotransposed genes compensate postmeiotic silencing. (A,B) Comparison of gene expression levels of X-linked parental genes (X) and X-retrotransposed genes (TG) in humans and mice, respectively. The central dot is the median, the boxes encompass 50% of the data points, and the error bars indicate 90% of data points. (*) p < 0.05; (**) p < 0.001, unpaired t-test.
Postmeiotic silencing impacts sex chromosome evolution
Although the above results suggest that amplification of copy number and retrotranspositions are potential evolutionary strategies to cope with MSCI and postmeiotic silencing, we also identified a subset of X-linked genes that exhibit a unique expression pattern acting against postmeiotic silencing (Group C) (Fig. 3B–D). In this group, we identified 66 genes that are specifically expressed in RS in humans. These 66 genes are composed of six multicopy genes and 60 single-copy genes (Supplemental Tables S1, S8), suggesting that copy number is not the sole determinant of RS-specific expression. In mice, 55 genes showed RS-specific expression and were classified as mouse Group C genes (Supplemental Table S8; Namekawa et al. 2006). Remarkably, only 12 out of 66 human Group C genes consistently show RS-specific expression in both humans and mice (Fig. 6A, List A; Supplemental Table S8), while expression of the rest of the human Group C genes significantly diverged in mice. Also, out of the 55 genes in the mouse Group C, 20 genes (Fig. 6A, List B; Supplemental Table S8) are uniquely found in the rodent lineage, and nine genes (Fig. 6A, List C; Supplemental Table S8) only exist in the mouse (see Methods for identification of de novo genes in mouse and human Group C genes). These rodent-specific genes emerged after the divergence of rodents and other mammalian organisms ∼80 million years ago. After the divergence of rodents and other mammalian lineages, several Group C genes appeared only in primates (Fig. 6A, List D; Supplemental Table S8), and some Group C genes exclusively emerged in Great Apes (Fig. 6A, List E; Supplemental Table S8). These newly evolved Group C genes are often determined as multicopy genes, reflecting that the X chromosome underwent recent genomic rearrangements during primate evolution. A precedent analysis revealed that newly evolved genes tend to escape male sex chromosome inactivation in mice, especially in the round spermatids (Zhang et al. 2010). Consistent with this notion, we demonstrate that escape genes are often de novo genes during mammalian evolution. In addition to the de novo acquisition of Group C genes, we also found that a total of 43 human and mouse Group C genes show distinct expression patterns between humans and mice (Fig. 6A, List F). In sum, these results suggest that mammals have acquired escape genes to cope with postmeiotic silencing by either acquiring de novo genes or changing expression patterns.
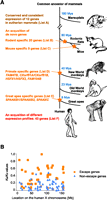
Evolutionary impacts of postmeiotic silencing. (A) Evolutionary history of escape genes from postmeiotic silencing in the course of mammalian evolution. Genes in lists A–F are displayed in Supplemental Table S8. Stars denote the acquisition timing of de novo genes. (Mya) Million years ago. (B) Distribution of Ka/Ks values of X-linked escape genes and non-escape genes (listed in Supplemental Table S9) along the length of the X chromosome. Ka/Ks values are calculated between humans and mice.
Divergent acquisition of escape genes from postmeiotic silencing is likely to have impacted speciation, because genes specifically expressed in gametes may impinge on the success of fertilization and early embryonic development of the next generation. Therefore, we hypothesized if the acquisition of escape genes from postmeiotic silencing is beneficial to reproductive fitness, this would be reflected by rapid changes in the amino acid sequences of escape genes and their subsequent adaptation during mammalian evolution. To test this hypothesis, we measured synonymous and nonsynonymous nucleotide changes between escape genes and non-escape genes during postmeiotic silencing. Synonymous substitution does not alter the amino acid sequences, whereas nonsynonymous change has an effect on encoded protein. If the rate of nonsynonymous substitution occurs frequently in genes, these genes are generally thought to be beneficial to fitness and amino acid changes and are rapidly fixed. We calculated the number of nonsynonymous substitutions per nonsynonymous site (Ka) and the number of synonymous substitutions per synonymous site (Ks) between human and mouse orthologous genes (see Methods). Remarkably, the average Ka/Ks value of X-linked escape genes from postmeiotic silencing in humans is 0.36, which is significantly higher than that of X-linked non-escape genes, which are consistently repressed by postmeiotic silencing in both humans and mice (i.e., genes subject to postmeiotic silencing: Ka/Ks = 0.14; Unpaired T-test, P < 1 × 10−4) (Supplemental Table S9). Also, higher Ka/Ks values of X-linked escape genes are distributed in a chromosome-wide manner along the human X chromosome (Fig. 6B). Notably, high Ka/Ks values were observed for X-linked escape genes that evolved specifically in the rodent lineage (the average Ka/Ks value of List B genes in Fig. 6A between mouse and rat homologous genes is 0.60). These results suggest that the chromosome-wide acquisition of escape genes along the X chromosome is beneficial during mammalian evolution. Thus, we propose that chromosome-wide silencing with PMSC is one of the evolutionary driving forces behind the acquisition of beneficial escape genes for reproductive fitness and that epigenetic mechanisms underlying PMSC impact the genetic evolution of sex chromosomes.
Discussion
Epigenetic silencing impacts sex chromosome evolution
In this study, we identified human PMSC, the silent compartment of sex chromosomes in the round spermatids, and demonstrated that conserved components illuminate MSCI and postmeiotic silencing between humans and mice. Therefore, it is likely that conserved mechanisms underlie both MSCI and postmeiotic silencing, in which a DNA damage response pathway initiates MSCI, and epigenetic modifications such as CBX1, CBX3, and H3K9me3 maintain postmeiotic silencing (Fig. 1A). Furthermore, our gene expression analysis revealed that the mammalian genome experienced dynamic changes (amplification of copy number, retrotransposition, acquisition of de novo genes, and acquisition of different expression profiles) to cope with stable sex chromosome inactivation during the evolutionary past. Previous studies suggested that MSCI is the evolutionary driving force behind sex chromosome evolution (Potrzebowski et al. 2008; Zhang et al. 2010; Ellegren 2011). This study extends the knowledge of these precedent studies and demonstrates that not only MSCI, but also PMSC have impacted these distinct features of sex chromosome evolution, and, therefore, the epigenetic maintenance process of sex chromosome inactivation acts as an evolutionary driving force.
Evolutionary driving forces behind sex chromosome evolution
Remarkably, despite the conserved feature of chromosome-wide silencing, profiles of escape genes from postmeiotic silencing are significantly diverged between humans and mice. We identified 66 human Group C genes that specifically escape from postmeiotic silencing (Fig. 3); however, only 12 genes are common between human and mice Group C genes (Fig. 6A). Given the conservation of >90% of X-linked genes between mice and humans (Ross et al. 2005), this is a significant divergence between humans and mice.
Why did our genome need to go through such dynamic changes in the course of evolution despite the conserved sex chromosome inactivation? Dynamic changes on the X chromosome could be interpreted as an outcome of the battle between sex chromosome inactivation and another unique evolutionary force, called “sexual antagonism,” underlying the X chromosome. Sexual antagonism is considered to be the genetic change that is beneficial for the reproductive fitness of one sex, but deleterious for the other sex. Rice's hypothesis predicted that sexual antagonism is the unique driving force acting on the X chromosome to accumulate male-biased genes due to its hemizygosity in males (Rice 1984). Specifically, male-biased alleles such as male reproductive genes are predicted to accumulate preferentially on the X chromosome because the recessive mutation is readily available in the male hemizygosity, but masked in the female by the homozygous allele.
Indeed, male-biased genes expressed before entering meiosis are enriched on the X chromosome (Khil et al. 2004). However, X-linked genes expressed in late spermatogenesis are considered to be underrepresented due to MSCI (Khil et al. 2004). One potential interpretation of the present study is that the X chromosome is a preferred place for the male reproductive genes, even under the pressure of sex chromosome inactivation. Indeed, escape genes from postmeiotic silencing mainly have male reproductive functions (Namekawa et al. 2006). Thus, escape genes would be a manifestation of two opposing evolutionary driving forces: sex chromosome inactivation and sexual antagonisms.
Consistent with this notion, male-biased genes that are newly evolved during the recent evolution of mammals accumulate on the X chromosome and tend to escape postmeiotic silencing in mice (Zhang et al. 2010). On the other hand, male-biased genes that evolved prior to the split between marsupials and placental mammals ∼180 million years ago (Mya) are enriched on autosomes (Zhang et al. 2010). MSCI is thought to have evolved independently in different sex chromosome systems, and the mammalian type of MSCI co-emerged with the mammalian sex chromosomes around the time of the split between marsupials and placental mammals (Potrzebowski et al. 2008). Therefore, emergence of both sex chromosome inactivation and sexual antagonisms appear to have been concomitant, and to have manifested as the emergence of X-linked male-biased genes that escape from postmeiotic silencing.
How has our genome solved the battle of two opposing evolutionary driving forces? One strategy to cope with stable sex chromosome inactivation would be amplification of the copy number of the genes. Interestingly, multicopy X-linked genes tend to escape postmeiotic silencing in mice (Mueller et al. 2008) and in humans (Fig. 4). These genes frequently locate on the inverted repeats (IRs) (Mueller et al. 2008), and enrichment of IRs is the signature of the X chromosome (Warburton et al. 2004). On the other hand, the Y chromosome is enriched with newly evolved palindrome structures in ampliconic regions (Skaletsky et al. 2003), and these regions tend to escape postmeiotic silencing (Fig. 3F). Therefore, two opposing evolutionary driving forces may have impacted the evolution of sex chromosomes and conferred their unique genomic structure.
Another consequence of sex chromosome inactivation is the retrotransposition of X-linked genes to autosomes (Fig. 5). Compensation of gene expression by autosomal X-retrotransposed genes was also found in a variety of organisms including Drosophila (Vibranovski et al. 2009), suggesting that compensation by the retrotransposed copies is a common strategy among species. Of note, these classes of genes are housekeeping genes on the X chromosome (Wang 2004); retrotransposed genes on the autosomes continuously compensate their X-linked parental genes when MSCI and postmeiotic silencing occur (Fig. 5). Therefore, consequences of sex chromosome inactivation could be attributed to their gene functions in two different ways: X-linked housekeeping genes are transposed to autosomes to compensate for their parental genes, or male-biased genes accumulate on the X chromosome due to the benefit there for reproductive fitness in male hemizygosity, and temporally express in the round spermatids as escape genes.
Divergent acquisition of escape genes: Implications in the reproductive diversity of mammals
Why are escape genes significantly diverged between mice and humans? One theory, termed the “faster-X effect,” predicted that the evolution of X-linked genes is faster than autosomes if these alleles are recessive and beneficial to the reproductive fitness of males, because the mutation could readily fix to the male germline (Charlesworth et al. 1987). The data in our study are in accordance with the prediction of this theory and demonstrate that escape genes from postmeiotic silencing in humans and/or mice exhibit rapid amino acid changes (i.e., higher Ka/Ks values) (Fig. 6B). This accordance further supports the idea that the acquisition of escape genes may have been beneficial to reproductive fitness, as discussed in the potential effects of sexual antagonisms above. In turn, X-linked genes subjected to MSCI and postmeiotic silencing exhibit lower rates of amino acid changes compared with escape genes (Fig. 6B); therefore, sex chromosome inactivation could have buffered the faster-X effects.
Another striking feature of escape genes is the de novo acquisition of new escape genes such as rodent-specific genes and primate/Great Ape–specific genes (Fig. 6A). Acquisition of these de novo genes further corroborates the prediction of faster-X effects because hemizygosity of the X chromosomes could have allowed these rapid genomic changes and the acquisition of new alleles. Accumulating evidence on the genomics of sex chromosomes is in line with this notion. For example, IRs are rapidly evolved on the X chromosomes (Warburton et al. 2004), and cancer testis (CT) genes located on these amplicons, including IRs and simple duplications, were also expanded dramatically in recent evolution (Simpson et al. 2005). In the case of the Y chromosome, the palindrome structure of the Y-ampliconic regions shows massive sequence discrepancies between species that were generated after the divergence between human and chimpanzee, ∼6–8 Mya (Hughes et al. 2010). Taken together, we suggest that the evolutionary force underlying sex chromosome inactivation is tightly associated with the divergence of the mammalian genome.
A recent study indicated that hybrid sterility is associated with the MSCI defect (Good et al. 2010), suggesting that the integrity of MSCI needs to be maintained for proper fertility. It would be intriguing to consider how the acquisition or expression changes of escape genes might have been beneficial to increasing reproductive fitness during mammalian evolution. Indeed, recent evidence indicates that the profile of an X-linked gene changed even within the subspecies of mice (Homolka et al. 2011), implicating a continuous rapid change of escape genes in recent times. These dynamic changes on the sex chromosomes might have ultimately contributed to reproductive diversity of mammals.
Methods
Human subject
Subjects underwent either open testicular biopsy or retrieval of sperm from testicular tissues while attempting to extract testicular sperm for intracytoplasmic sperm injection. To confirm normal spermatogenesis, we conducted histological, semen, and hormone analysis according to conventional methods (Johnsen 1970; WHO 1999). In addition, karyotype abnormalities were checked by sequence-tagged sites (STS) analysis as previously described (Sin et al. 2010). The Committee of Kanazawa University Hospital approved the study, and informed consent was obtained from the participants (approved no. 172 H20.8.8).
Human spermatogenesis slide preparation
To prepare paraffin blocks, testes were fixed with 4% paraformaldehyde in PBS, ethanol-dehydrated, and embedded in paraffin. Six-micrometer paraffin sections were prepared with a microtome (Leica) and deparaffinized prior to immunostaining.
FISH and immunofluorescence
Details of immunostaining and DNA FISH are described elsewhere (Namekawa and Lee 2011). For immunofluorescence, slides were incubated in PBT (0.15% BSA, 0.1% Tween 20 in PBS) for 60 min prior to overnight incubation at room temperature with the following antibodies: γH2AX (Millipore), 1:3000; MDC1 (AbD Serotec), 1:1000; TOPBP1 (gift from Junjie Chen), 1:500; CBX1 (Abcam), 1:100; CBX3 (Millipore), 1:500; H3K9me3 (Millipore), 1:100; RNA polymerase II CTD 8WG16 (Millipore), 1:200. Thereafter, slides were washed three times for 5 min in PBS plus 0.1% Tween 20, incubated with secondary antibodies (Invitrogen or Jackson ImmunoResearch) at 1:500 for 60 min in PBT, washed in PBS plus 0.1% Tween 20, and mounted in Vectashield with DAPI. For combined immunostaining and DNA FISH, we performed immunofluorescence detection of proteins first, followed by DNA FISH. After immunostaining, slides were fixed with 4% paraformaldehyde in PBS for 10 min to preserve the immunofluorescent signals. Probes for DNA FISH (human chromosome X, Y, and 1) were purchased from ID Labs Biotechnology, Inc. For the competitor DNA for hybridization, human Cot-1 DNA (Invitrogen) was used.
Image acquisition and analysis
All images were acquired with a TE2000-E microscope (Nikon) and a CoolSNAPHQ camera (Photometrics). All image acquisition, including z-sections and deconvolution, were performed using Phylum software (Improvision). For immunostaining of RNA polymerase II, z-sections were captured and deconvolved with Phylum software to eliminate any secondary signals from other z-sections. A representative z-section was shown. Adobe Photoshop was used for composing figures.
Microarray analysis
For microarray analysis in human spermatogenesis, we analyzed published data sets of the Affymetrix Human U133 Plus 2.0 Array deposited in the European Bioinformatics Institute (EBI) ArrayExpress database (http://www.ebi.ac.uk/arrayexpress; accession no. E-TABM-130) (Chalmel et al. 2007), and the National Center for Biotechnology Information Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo; accession no. GSE18914) (Wu et al. 2009). For microarray analysis in mouse spermatogenesis, we analyzed a published data set of the Affymetrix MG-430 2.0 Array deposited in the GEO database (http://www.ncbi.nlm.nih.gov/geo/; accession no. GSE4193) (Namekawa et al. 2006).
All data sets were analyzed together using Affymetrix Expression Console Software for the calculation of expression levels, with each array normalized with the RMA algorithm. Temporal expression patterns observed in genes expressed at low levels are not reliable, so low expression probes or those that do not show expression level 100 in at least one developmental time point were eliminated for the profiling of the X chromosome and all heatmap analyses (Figs. 3, 4) as we previously described (Namekawa et al. 2006). However, for the sake of references, we have included all low-expression probes for the Supplemental Tables for expression profiles of Y-linked genes and their homologous X-linked genes (Supplemental Tables S2, S3), multicopy genes (Supplemental Tables S4, S5), and retrotransposed genes (Supplemental Tables S6, S7). For the expression analysis, probes that do not specifically annotate the genome were eliminated from the analysis. R and Excel software were used to generate the figures. We used current and approved nomenclature for each gene symbol. However, for many multicopy genes, each homologous copy is almost identical, and we cannot distinguish the copies in our microarray analysis. If there are two nomenclatures for homologous copies, then we show both nomenclatures (e.g., HSFX1/HSFX2). If there are more than three nomenclatures for homologous copies, then we show these cases as “gene family” (e.g., DAZ gene family instead of DAZ1, DAZ2, DAZ3, and DAZ4).
Bioinformatic screening of X-retrotransposed genes in humans and mice
To capture X-retrotransposed genes that are retrotransposed from the X chromosome to autosomes, human and mouse whole peptide and DNA information were downloaded from Ensembl release 61 (February 2011). All of the sequences were screened and aligned with 50% of amino acid homology at the minimum threshold that showed at least 70% sequence coverage when compared with X-linked genes using ClustalW (Thompson et al. 1994). Following widely accepted criteria of retrotransposed genes (Emerson et al. 2004), we calculated nonsynonymous (Ka/Ks) versus synonymous (Ks) changes within each pair of X-linked and X-retrotransposed genes. If the Ka/Ks value was below 0.5, we defined the pair as functionally conserved retrotransposed genes. To discard false-positive data, we confirmed all of the candidates by a manual process that led us to remove intron-containing sequences and sequences on the same chromosomal location. Finally, we collected 20 human X-retrotransposed genes and 27 mouse X-retrotransposed genes.
Determination of emergence timing of escape genes from postmeiotic silencing during mammalian evolution
We estimated emergence timing of de novo escape genes from postmeiotic silencing during the evolution of eutherian mammals. To determine a presence and an absence of the gene ortholog of interest during mammalian evolution, whole mammalian expressed sequence tag (EST), RefSeq mRNA, and nonredundant databases were screened with nucleotide sequences of escaping genes using BLAST, version 2.2.25 with the “-q -e -e 0.000001 -F T” option (Altschul and Lipman 1990; Altschul et al. 1990; Betran et al. 2002). Subsequently, we searched peptide orthologs of escaping genes using BLASTP. The orthologs were determined when at least 30% sequence coverage showed >80% similarities. De novo genes were defined when both nucleotide and peptide alignment did not present any annotation information or did not satisfy our thresholds.
Calculation of Ka and Ks
Ka (synonymous substitutions per synonymous site) and Ks (nonsynonymous substitutions per nonsynonymous site) values were calculated using only coding sequences. First, we aligned sequences and then manually removed insertion and deletion sequences from the alignment data. Ka and Ks values were calculated by the Wisconsin Package (Version 11.1, Genetics Computer Group).
Acknowledgments
We thank Misako Ichijima for technical assistance and artwork in Figure 6; S.K. Dey, Jeannie Lee, Yuya Ogawa, Norishige Yamada, and Fumika Hamada for comments on the manuscript; and Erin L. Adams and Serenity Curtis for editing the manuscript. Y.I. is a research fellow of the Japan Society for the Promotion of Science. This work was supported by the Basil O'Connor Starter Scholar Award from the March of Dimes Foundation, the Developmental Fund and Trustee Grant at Cincinnati Children's Hospital Medical Center, and NIH grant GM 098605 to S.H.N.
Footnotes
-
↵4 Corresponding author.
E-mail satoshi.namekawa{at}cchmc.org.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.135046.111.
- Received November 22, 2011.
- Accepted February 27, 2012.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported License), as described at http://creativecommons.org/licenses/by-nc/3.0/.








