
Abstract
Genetic testing for disease risk is an increasingly important component of medical care. However, testing can be expensive, which can lead to patients and physicians having limited access to the genetic information needed for medical decisions. To simplify DNA sample preparation and lower costs, we have developed a system in which any gene can be captured and sequenced directly from human genomic DNA without amplification, using no proteins or enzymes prior to sequencing. Extracted whole-genome DNA is acoustically sheared and loaded in a flow cell channel for single-molecule sequencing. Gene isolation, amplification, or ligation is not necessary. Accurate and low-cost detection of DNA sequence variants is demonstrated for the BRCA1 gene. Disease-causing mutations as well as common variants from well-characterized samples are identified. Single-molecule sequencing generates very reproducible coverage patterns, and these can be used to detect any size insertion or deletion directly, unlike PCR-based methods, which require additional assays. Because no gene isolation or amplification is required for sequencing, the exceptionally low costs of sample preparation and analysis could make genetic tests more accessible to those who wish to know their own disease susceptibility. Additionally, this approach has applications for sequencing integration sites for gene therapy vectors, transposons, retroviruses, and other mobile DNA elements in a more facile manner than possible with other methods.
Genetic testing has become increasingly common for diagnosing or determining risks for a variety of conditions (Andermann and Blancquaert 2010). For example, mutations in BRCA1 and BRCA2 play a major role in familial breast and ovarian cancer (Miki et al. 1994; King et al. 2003) and account for a large fraction of the incidence of such cancers in women less than 50 yr old. Affected and potentially affected women have a high level of interest in having these genes sequenced to help them make appropriate medical decisions (Lerman et al. 1995). New technologies such as next-generation sequencing have the potential to further accelerate knowledge in this area and provide prodigious amounts of data from a wide range of samples (Kahvejian et al. 2008). Although sequencing costs have been dramatically lowered, these technologies have not quickly transitioned to the molecular diagnostics arena, partially because the complicated sample preparation and complex informatic analyses makes logistics and per-sample costs relatively high (ten Bosch and Grody 2008; Milos 2009; Tucker et al. 2009). Targeted resequencing is often carried out for determination of genetic risk. However, the sample preparation burden and cost are escalated by the need for hybrid enrichment and/or PCR for gene targeting prior to sequencing. Gene targeting is currently carried out using multiple approaches for selective capture of specific DNA sequences, but all involve complex sample manipulations and require many steps including at least one and sometimes multiple rounds of amplification (Hedges et al. 2011).
One of the most common tests for determination of genetic risk, detection of mutations in BRCA1 and BRCA2, uses PCR amplification prior to classical Sanger sequencing. Other methods of sequencing, such as oligonucleotide arrays or next-generation sequencing technologies, have also been used (Morgan et al. 2010; Schroeder et al. 2010; Walsh et al. 2010), but there is extensive sample preparation required, raising the costs and increasing the opportunity for sample-handling errors in a clinical setting. Additionally, many tests have become mired in intellectual property issues due to gene patents (Cho 2010).
We have used a single-molecule sequencing technology (Harris et al. 2008; Bowers et al. 2009) with a custom flow cell that allows natural DNA to be sequenced directly after being extracted and sheared with no amplification at any point. Instead of a flow cell with oligo(dT)50 primers typically used in a Helicos Genetic Analysis System, a gene-specific flow cell was constructed with capture primers for defined genomic regions. These generate reads on both strands that allow sequence variant calling. The entire gene can thus be sequenced without copying the DNA or using any enzymes prior to sequencing.
Results
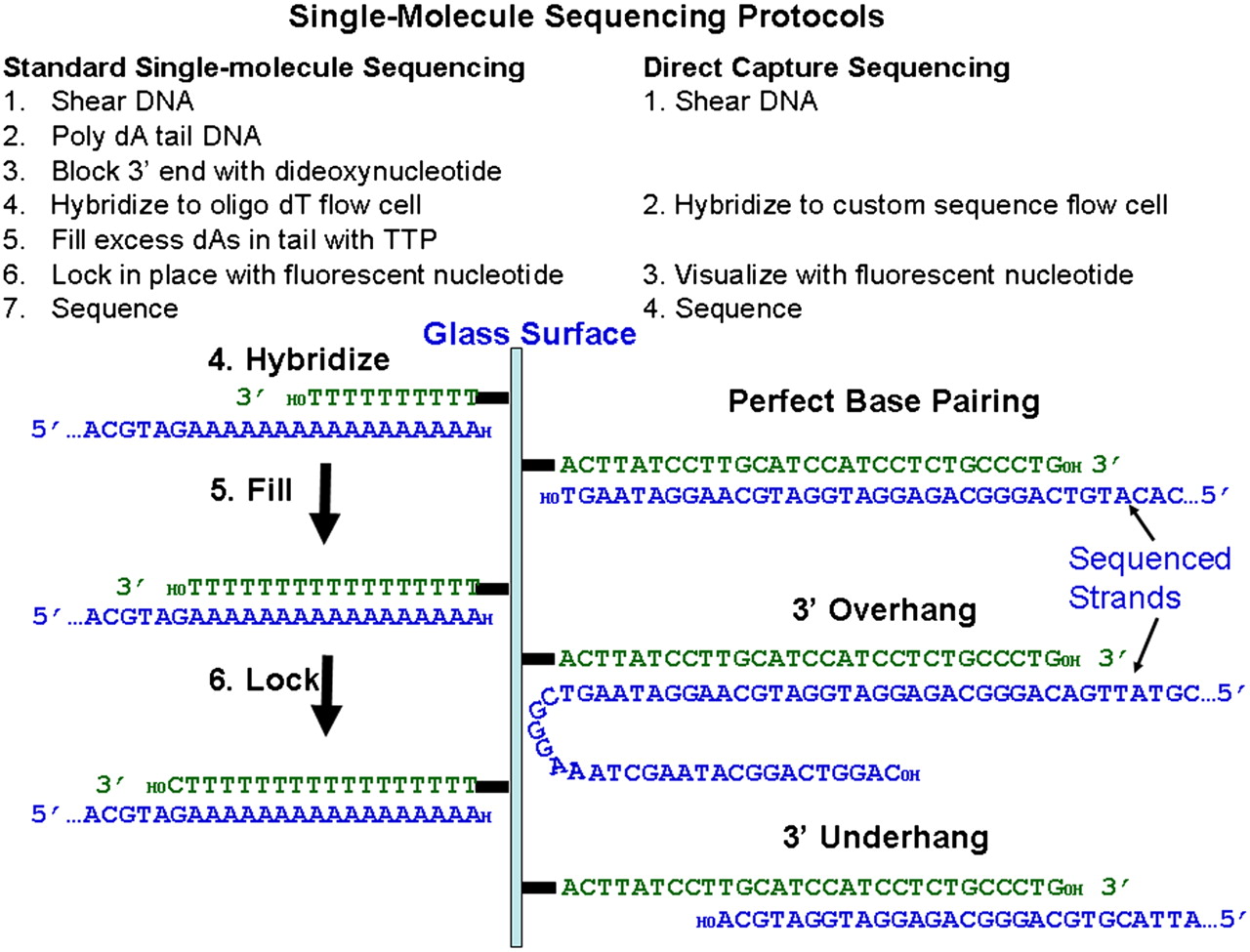
Single-molecule sequencing requires neither gene isolation nor amplification during sample preparation or during the sequencing reaction. The standard Helicos single-molecule sequencing protocol, described previously for genomic (Pushkarev et al. 2009), ChIP (Goren et al. 2010), and cDNA (Lipson et al. 2009), includes tailing DNA with terminal transferase and dATP followed by hybridizing to a flow cell with surface-attached dT50 oligonucleotides. This allows all nucleic acids in a sample to be sequenced. However, if only a subset of DNA regions is of interest, the standard dT50 oligonucleotides can be substituted with specific primers to capture and sequence the desired regions. This has the added benefit of simplifying sample preparation by eliminating the need for poly(A) tailing. Additionally, tailed samples require a blocking step in which the 3′ end of the DNA is inactivated. This is unnecessary when using flow cells with a defined sequence because most of the randomly sheared fragments hybridizing to capture primers are longer than the primer and thus have single-stranded 3′ overhangs (Fig. 1; Supplemental Fig. 1). Because there is no template across from the 3′ end of overhanging genomic DNA, the polymerase does not add extra bases to that end (Supplemental Fig. 2). Incoming DNA with ≥20-nt overhangs hybridizes to the surface as well as molecules with no overhangs. The upper limit for allowable overhangs has not been determined and is likely to be sequence-dependent (data not shown). Secondary structures that involve the targeted region can also affect hybridization efficiency. Although some sources of DNA require shearing, DNA that is already fragmented requires absolutely no sample preparation and can truly be sequenced directly.
Single-molecule sequencing protocols. The standard single-molecule sequencing protocol using tailed DNA and oligo(dT) flow cells (left) and the novel protocol using Direct Capture (right). The diagram on the left shows three steps in the standard protocol, while the diagram on the right shows the hybridization step only for the Direct Capture protocol with three different possible types of hybridizing molecules: a DNA that is only partially complementary to the capture primer (underhang), a DNA that is perfectly complementary to the capture primer, and a DNA that has a 3′ overhang. All of these molecules are sequenceable.
Flow cells were made that included 564 oligonucleotides (24–41 nt) for BRCA1 (Supplemental Table 1). The target sequences were chosen based on the exons and adjacent intronic sequences known to harbor clinically significant variants as listed in the Breast Cancer Information Core database (BIC; http://research.nhgri.nih.gov/projects/bic/), an international collaborative effort maintained by NHGRI. This targeted region included all coding sequences including exons 2, 3, and 5–24 as well as 20 bp of intronic sequence at each acceptor splice site and 10 bp of intronic sequence at each donor splice site. 5′-C12-amine-modified oligonucleotides were covalently attached to an epoxide-containing surface. Additional oligonucleotides can be added to the same flow cells for sequencing of BRCA2 and other genes.
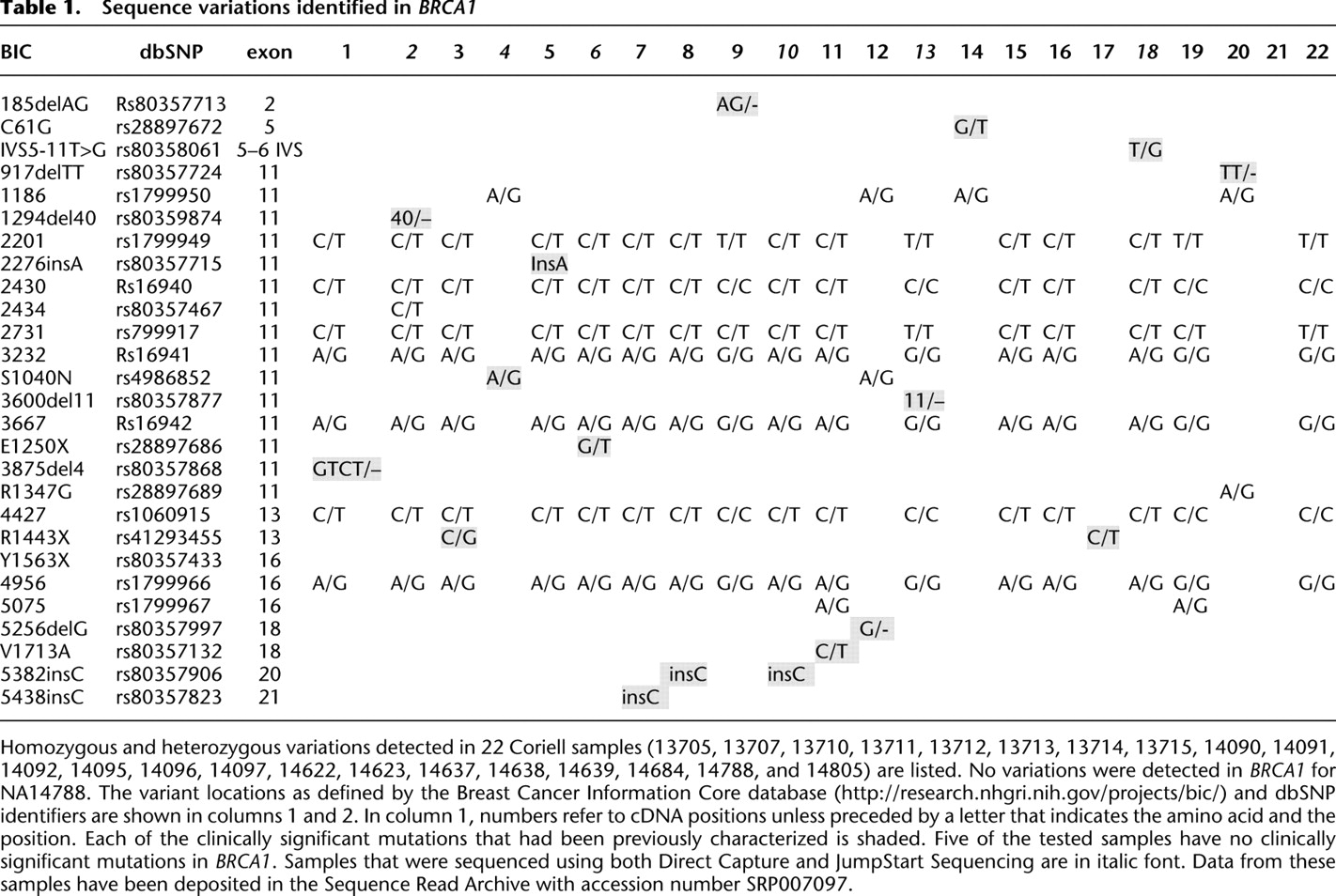
To demonstrate that extracted DNA could be sequenced directly with no prior amplification, selection, or purification, DNAs with known mutations (shaded in Table 1) were obtained from the Coriell Institute for Medical Research biorepository, sheared to 200 bp, and used with no additional sample preparation. Typically, 1 μg of genomic DNA was hybridized overnight in each channel of a 25-channel flow cell, but ranges of 0.1–20 μg of genomic DNA and shorter hybridization times have also been used successfully. Higher DNA amounts yield more sequence reads from BRCA1, although it is possible to use substantially less DNA, if necessary. DNAs were sequenced for 120 cycles of nucleotide addition. The sequence reads were filtered for artifacts caused by DNA molecules spaced too closely to be resolved by the camera and for length, keeping all reads of ≥25 nt. Reads were then aligned (Giladi et al. 2010) to the BRCA1 reference sequence. With the highest DNA amounts, >20% of the resulting sequences aligned to the BRCA1 gene. Since BRCA1 exons represent 0.0002% of the human genome, this is an enrichment of >100,000-fold. Of the reads that do not align to BRCA1, >90% align sporadically to the remainder of the genome. A typical alignment of sequence reads to the reference is shown in Figure 2. Variants detected in 22 samples are shown in Table 1.
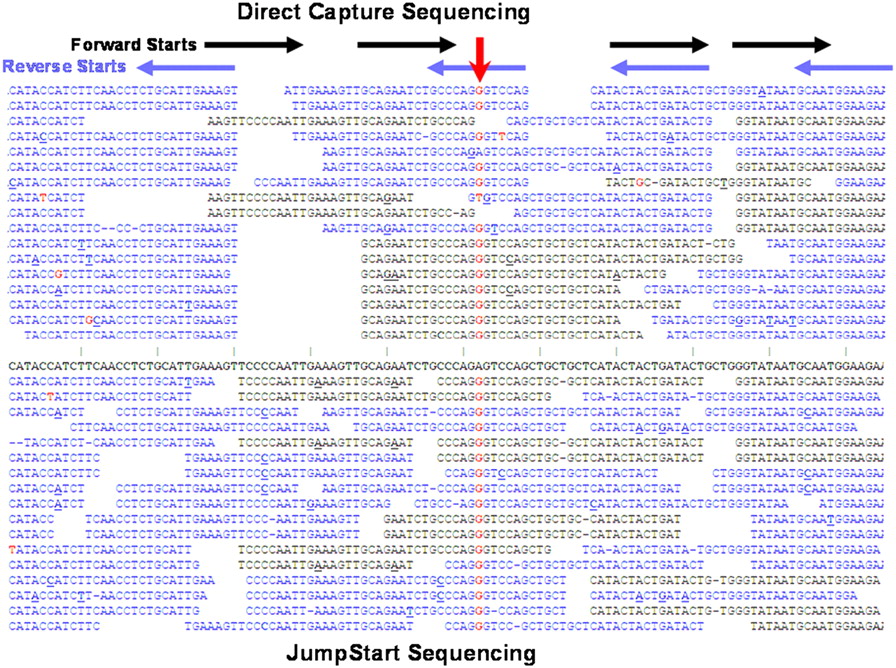
Sequence reads from representative region using Direct Capture and JumpStart Sequencing. Representative sequence reads from one region of BRCA1 exon 16 in the forward (black) and reverse (blue) directions are shown for the NA14096 sample. In the top half of the figure with Direct Capture Sequencing, horizontal black and blue arrows show where sequencing begins after each capture primer. In the lower half of the figure with JumpStart Sequencing, start sites were randomized by extension with polymerase prior to sequencing. A histogram for start sites is shown in Supplemental Figure S4. Sequence reads are aligned to the reference in the center (bold) and differences noted in red for substitutions, with dashes for deletions, and with bolded, underlined bases for insertions. Gaps are included between sequences so that individual reads can be distinguished. Random errors occur throughout the reads, but these are easily distinguished from real variants in which differences from the reference occur much more often (downward red arrow). This position corresponds to dbSNP variant rs1799966, a common substitution SNP. The reads shown are a sampling of those obtained for this region.
To call substitutions and insertion/deletions of <5 bp, the Helicos open-source SNP caller was used with modifications specific for the Direct Capture method. In addition to the standard methods for calling variants, coverage of all regions was compared among samples. Because the hybridization and sequence yield is very consistent for a given set of hybridization conditions, variations in coverage are diagnostic for larger insertions and deletions. Thus, sequencing can be used to directly detect large rearrangements, in contrast to Sanger methods, which do not distinguish large insertions/deletions from normal DNA. Since large indels are estimated to occur in 12% of BRCA high-risk patients (Walsh et al. 2006), this approach has a significant advantage in detecting a wider range of clinically significant variants without additional assays.
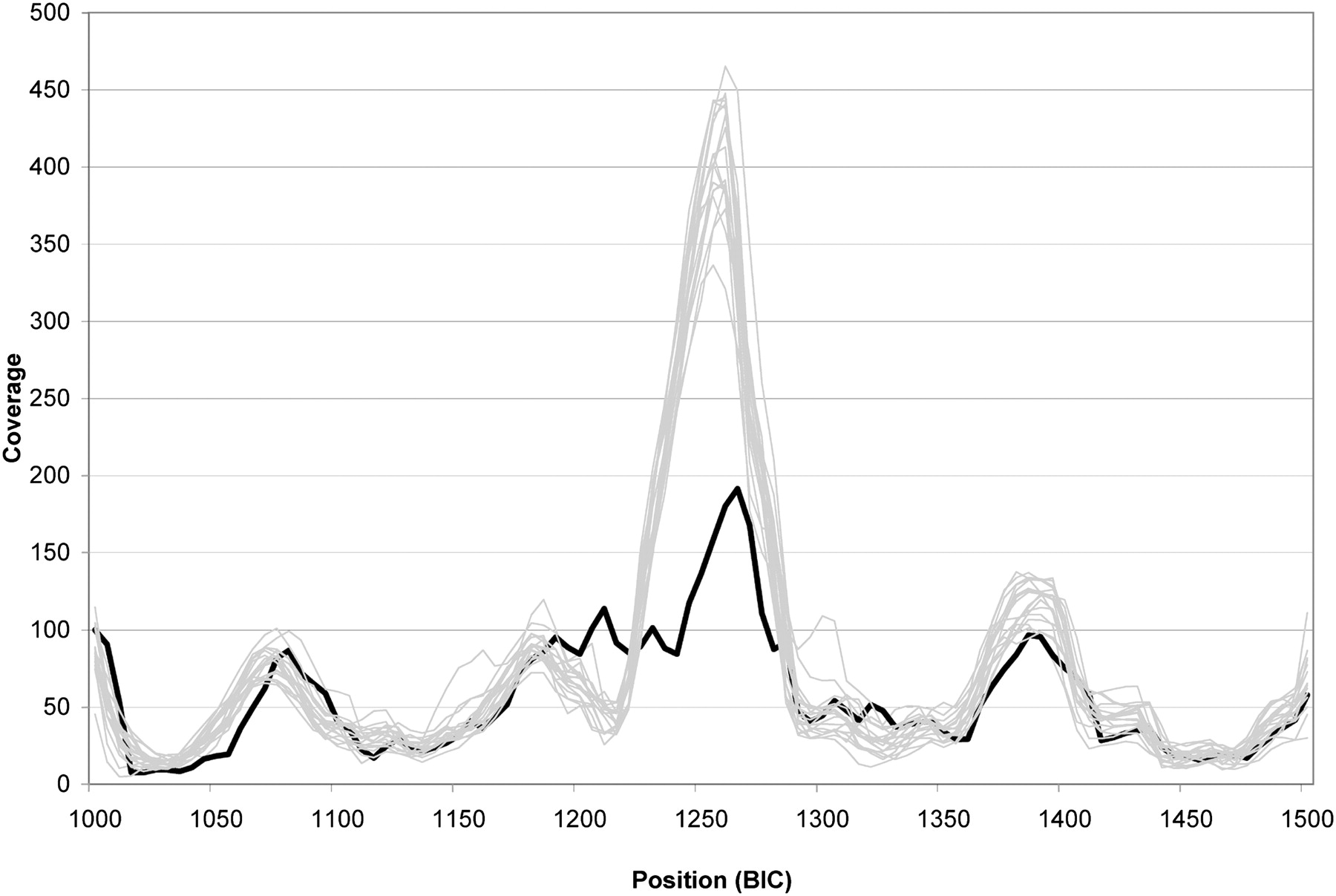
The coverage profile for the BRCA1 region for 19 samples run simultaneously on the same flow cell is shown in Supplemental Figure 3. The median coverage at each position for this sample set was 47×. Although there are coverage differences across this region caused by differing hybridization efficiencies of each oligonucleotide, the reproducibility at a given location is very high from sample to sample, making it possible to assess variations in coverage caused by large insertions and deletions. For example, there is a drop in coverage in exon 11 of NA13707, which contains a 40-bp deletion. The coverage for the 500 bp surrounding the deletion is provided for 19 DNAs with the lower coverage of the highlighted NA13707 DNA standing out (Fig. 3). Furthermore, the exact boundaries of the deletion can be determined by examining reads that align to two separate segments of the reference (Fig. 4). In this region, reads corresponding to both the reference allele and the deleted allele are found. The novel sequence junction arising from the deletion can be easily observed.
Sequence coverage. Coverage for 19 of the DNAs listed in Table 1 that were hybridized and sequenced on the same flow cell is shown for 500 bp of exon 11. Coverage bins of 5 bp were normalized to achieve the same amount of total coverage for each sample by multiplying by the appropriate factor (range = 0.70 to 2.7). DNA sample NA13707, which contains a heterozygous 40-bp deletion in this region (heavy black line); all others (thin gray lines). Numbering for the region is as found in the BIC database. The lowered coverage over a larger region than 40 bp and not centered on the deletion is caused by the presence of primers located entirely within or overlapping the deletion that generate no reads from those primers for the deletion allele.
Deletion sequences. The exact coordinates of the 40-bp deletion in NA13707 can be determined by examining reads that map to the reference sequence in the region of interest in an interrupted manner. The reference sequence is shown in the center with the known 40-bp deletion region underlined. From 928 reads aligning to this region, 28 selected reads with different end points are shown. Forward reads (left to right) are in black, and reverse reads (right to left) are in blue. Deletions relative to the reference are shown by dashes and insertions by bolding and underlining the adjacent base.

While this method can yield complete coverage of BRCA1, problems can arise if coverage is not complete due to the presence of repetitive regions or regions that do not hybridize well. Unless primers are placed close together, there are regions of low coverage because reads from one primer may end before the next starts. Also, some repetitive regions are inaccessible due to the inability to place unique primers within those regions. To circumvent both problems, we have carried out a short, on-surface extension of the capture primer after hybridization but prior to sequencing. By including low concentrations of natural deoxynucleotides and polymerase, primers on the surface are extended by variable amounts into the captured DNA, allowing a randomization of the sequencing start sites and enabling reads within repetitive regions. Such random starting, which we refer to as “JumpStart Sequencing,” improves evenness of coverage (Fig. 2; Supplemental Fig. S4) relative to standard Direct Capture Sequencing (with no extension). Flow cells using this approach can also be generated with fewer, more broadly spaced primers.
Discussion
The new method of capturing and sequencing natural DNA in a single step provides a much simpler approach to targeted sequencing and, as shown in Table 1, allows detection of all the previously identified variants in the targeted regions in the tested samples. Although the current flow cell is directed at the entire coding sequence and 20 nt of adjacent intronic sequence, it is straightforward to incorporate any other sequences that might be found to be medically relevant. Repetitive sequences that are more than twice as long as the read length and hence inaccessible to standard Direct Capture Sequencing are observable using JumpStart Sequencing. Indeed, by generating very long extensions to primers prior to sequencing, repetitive sequences could be analyzed by capturing DNA with unique sequences just outside the repetitive regions and then extending into the repeat sequences prior to starting the sequence process. All reported clinically associated variants in BRCA1 and BRCA2 are within sequences that are amenable to this technique, although there will be repetitive regions of the genome that will be difficult to cover.
A significant advantage of this approach relative to Sanger and amplification-based approaches is the ability to more readily detect large insertions and deletions because of the quantitative nature of the sequencing. Primers differ in their hybridization efficiency relative to each other due to sequence context, but each primer is consistent across multiple samples, allowing coverage at any given position to be compared across many DNAs. The lack of amplification allows heterozygous changes in copy number caused by insertions or deletions to be readily detected. Deletion of whole exons or multiple exons will show extended blocks of reduced coverage. Smaller, intra-exonic deletions will show shorter regions of reduced coverage, but these will be further supported by sequence reads that span the new junction sequences caused by the deletion (Fig. 4). Because insertions have the potential to introduce novel sequence, the fixed set of surface primers will only allow full sequencing of short inserts. However, the presence of a long insertion will make it likely that a pathogenic variation has been introduced, and this could be followed up for a more complete sequence determination using extended JumpStart Sequencing conditions.
Although DNA extracted from cell lines and blood has been used thus far, other DNA sources should be suitable substrates as well. For example, many clinical samples are stored as formalin-fixed paraffin-embedded blocks, and the DNA from such sources is frequently degraded during processing. In such cases, no sample preparation would be necessary for the extracted DNA. The DNA could be loaded directly on the flow cell and sequenced. Similarly, many buccal samples produce fragmented DNA and thus are sequenceable with no sample processing.
The novel approach described here holds broad potential not only for BRCA1 but also for any other gene for which accurate, quantitative sequencing is medically important. The minimal sample handling and lack of amplification translate directly to lower sample-preparation costs and reduced sample-handling errors critical for molecular diagnostic laboratories. Additionally, informatic analysis is simplified due to the high enrichment for the region of interest. Beyond focusing on particular genes of interest, this selective capture approach can also be used for other purposes. For example, whole exome studies typically yield sequence on only ∼80% of the originally targeted sequences (Hedges et al. 2011). Regions difficult to target or amplify using other selective capture methods are reproducible and could be addressed with this technology. Also, sequences present at low quantities in complex mixtures such as from a bacterial or viral infection or a tumor sample could be applied to custom flow cells in order to detect DNA present at low quantities. Additionally, it is frequently useful to be able to determine integration sites for gene therapy vectors, transposons, retroviruses, and other mobile elements in genomic DNA (Gawronski et al. 2009; Hackett et al. 2010). This method of single-step capture and sequencing makes such studies much more facile even if the sequence of only a single end of the mobile element is known (data not shown). Thus, direct sequencing technology offers multiple possibilities for expanding the use of next-generation sequencing as well as for making it feasible for much larger numbers of interested people to determine their genetic risk for a variety of diseases.
Methods
Flow cell design
Surface capture oligonucleotides were designed complementary to both strands of BRCA1. Attempts were made to keep GC content within 20%–80%, make all Tm’s >65°C, and minimize 3′ and total self-complementarity within a capture primer through the use of the program BatchPrimer3. When possible, oligonucleotides were spaced 20–30 nt apart so that adjacent sequence reads would overlap. While these criteria could be met for most of the sequence, there were regions in which it was not possible, so some criteria were relaxed to ensure that there was no gap of >35 nt. Oligonucleotides were compared to all human genome sequences to ensure that the 3′-terminal 20 nt were not perfect matches to any other location in the genome. Where high levels of homology were present, efforts were made to place mismatches as close to the 3′ end as possible while retaining reasonable spacing. For example, the distal end of exon 2 and the targeted intronic sequence have 182 of 195 nt that are identical with a pseudogene, so sequences had to be carefully selected to take advantage of the BRCA1-specific differences to ensure that sequence was read from exon 2 and not from the pseudogene. In addition, the gene-specific oligonucleotides were spiked with a 29-nt oligonucleotide (5′-ACTTATCCTTGCATCCATCCTCTGCCCTG-3′). This is included so that quality-control oligonucleotides could be included without interfering with the BRCA1 hybridizations and to provide a sufficient number of actively sequencing DNA molecules to maintain image registration during sequencing cycles. HPLC-purified, 5′-amine-C12 oligonucleotides were ordered from either IDT or Operon and deposited on epoxide-modified glass at 37°C at individual concentrations of 10 pM for 1 h. Glass was assembled into 25 channel flow cells for use on the HeliScope Genetic Analysis System. Flow cells with oligonucleotides for other specific DNA sequences of interest can be ordered on a custom basis from Helicos.
DNA preparation and hybridization
HapMap genomic DNA was ordered from the Coriell Institute for Medical Research (Camden, NJ) and K562 DNA from Biochain. The DNA was sheared to various sizes, generally to an average of 200 bp, using the Covaris Acoustic Shearing method as described by Covaris. Up to 25 μg of DNA was sheared in a single 120-μL shearing reaction. After shearing, 0.1–20 μg of genomic DNA was denatured for 5 min at 95°C and snap-cooled on ice. This was then added to 10 μL of 2× hybridization buffer. A variety of standard buffers (Hutton 1977; Tsai et al. 2005) was tried, and most provided similar results. Most data presented were hybridized with 5× SSC and varying concentrations of formamide with either 0.02% Triton X-100 or 0.1% SDS present. Hybridization temperatures between 37°C and 60°C were assessed with optimal results varying depending on the formamide concentration (1%–25%). Hybridizations were carried out for 1–24 h in a humidified incubation oven set to the desired temperature. Flow cells were then placed in the HeliScope Genetic Analysis System and sequenced as described previously (Bowers et al. 2009). Most runs were 550 fields of view rather than the standard 1100 fields of view to allow faster data collection. Further reductions in fields of view are possible because significantly more sequence was being generated than was necessary for variant calling.
Read filtering and alignment
Prior to alignment, reads were filtered to remove sequences that would not be useful. Reads shorter than a specified minimum length, generally 25 nt, were removed. Also, artifactual sequences arise when DNA molecules are too close on the surface or if nucleotides tend to stick to a particular spot on the flow cell. Both of these situations lead to an apparent over-incorporation of nucleotides, so reads that closely resemble the base addition order are removed. Filtered reads were aligned to a set of sequences corresponding to the expected target sequences of the capture probes in a left-justified setting (i.e., alignments were penalized for starting away from the first base of the reference) for standard sequencing and to the whole gene reference for JumpStart Sequencing as well as the sequences corresponding to the spiked oligonucleotides. In parallel, reads were also aligned to the entire human genome, and any read that had a higher-scoring alignment to the untargeted genome than the target sequence was discarded. Aligned reads were then optimally positioned with respect to the target BRCA1 gene sequence.
Sequence variant detection
For detection of sequence variants in the aligned read data, we used SnpSniffer, a Helicos-developed algorithm. SnpSniffer calls SNPs and short indels using a multi-stage process. At first, the nucleotide counts at each position are tallied, based on aligned reads. The nucleotide counts are compared to those expected by chance due to machine error using a binomial model, and any positions that differ from the reference in a statistically significant fashion are flagged. The next stage identifies the most likely alleles at each candidate location and realigns the reads to the candidate alleles. This step reduces alignment errors substantially in locations with true sequence variants and avoids spurious allele calls. Following the realignment stage, mutations are called again using the realigned reads.
Homopolymer length mutations were determined by comparing the distribution of homopolymer lengths indicated by reads that span a homopolymer to the expected distributions. The expected distribution is computed using the machine deletion and insertion rates. The models to which the empirical data are compared correspond to both homozygous and heterozygous length mutations. Detected sequence variants were then filtered by minimal frequency, statistical significance, and average read length of sequence reads. Variants that did not pass these filters were considered artifacts and were ignored.
Insertions and deletions of 5 nt or longer were detected by comparing the relative coverage of each individual sample to the median of all samples on the same run. Regions that had significantly different coverage profiles were flagged, and then all possible deletions were examined for consistency with the observed sequences. Exact sequences for deletions can be determined in this manner. Insertions can be detected in this way as well, but, if the insertion was much longer than the read length, determination of their exact sequence would require a longer JumpStart extension in order to span the sequence. Knowledge that a long insertion had occurred at a particular site would generally be sufficient to implicate that variant as clinically significant.
Data access
Sequencing data from the 22 Coriell samples listed in Table 1 have been submitted to the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi) under accession no. SRP007097.
Acknowledgments
This publication was made possible by grant RC2 HG005598 from the National Human Genetics Research Institute (NHGRI) at the National Institutes of Health. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NHGRI.
References
- ↵A Andermann, I Blancquaert. 2010. Genetic screening: A primer for primary care. Can Fam Physician 56: 333–339.
- ↵J Bowers, J Mitchell, E Beer, PR Buzby, M Causey, JW Efcavitch, M Jarosz, E Krzymanska-Olejnik, L Kung, D Lipson, . 2009. Virtual terminator nucleotides for next-generation DNA sequencing. Nat Methods 6: 593–595.
- ↵M Cho. 2010. Patently unpatentable: implications of the Myriad court decision on genetic diagnostics. Trends Biotechnol 28: 548–551.
- ↵JD Gawronski, SM Wong, G Giannoukos, DV Ward, BJ Akerley. 2009. Tracking insertion mutants within libraries by deep sequencing and a genome-wide screen for Haemophilus genes required in the lung. Proc Natl Acad Sci 106: 16422–16427.
- ↵E Giladi, J Healy, G Myers, C Hart, P Kapranov, D Lipson, S Roels, E Thayer, S Letovsky. 2010. Error tolerant indexing and alignment of short reads with covering template families. J Comput Biol 17: 1279–1293.
- ↵A Goren, F Ozsolak, N Shoresh, M Ku, M Adli, C Hart, M Gymrek, O Zuk, A Regev, PM Milos, . 2010. Chromatin profiling by directly sequencing small quantities of immunoprecipitated DNA. Nat Methods 7: 47–49.
- ↵PB Hackett, DA Largaespada, LJ Cooper. 2010. A transposon and transposase system for human application. Mol Ther 18: 674–683.
- ↵TD Harris, PR Buzby, H Babcock, E Beer, J Bowers, I Braslavsky, M Causey, J Colonell, J Dimeo, JW Efcavitch, . 2008. Single-molecule DNA sequencing of a viral genome. Science 320: 106–109.
- ↵DJ Hedges, T Guettouche, S Yang, G Bademci, A Diaz, A Andersen, WF Hulme, S Linker, A Mehta, YJ Edwards, . 2011. Comparison of three targeted enrichment strategies on the SOLiD sequencing platform. PLoS ONE 6: e18595. doi: 10.1371/journal.pone.0018595.
- ↵JR Hutton. 1977. Renaturation kinetics and thermal stability of DNA in aqueous solutions of formamide and urea. Nucleic Acids Res 4: 3537–3555.
- ↵A Kahvejian, J Quackenbush, JF Thompson. 2008. What would you do if you could sequence everything? Nat Biotechnol 26: 1125–1133.
- ↵MC King, JH Marks, JB Mandell. 2003. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 302: 643–646.
- ↵C Lerman, J Seay, A Balshem, J Audrain. 1995. Interest in genetic testing among first-degree relatives of breast cancer patients. Am J Med Genet 57: 385–392.
- ↵D Lipson, T Raz, A Kieu, DR Jones, E Giladi, E Thayer, JF Thompson, S Letovsky, P Milos, M Causey. 2009. Quantification of the yeast transcriptome by single-molecule sequencing. Nat Biotechnol 27: 652–658.
- ↵Y Miki, J Swensen, D Shattuck-Eidens, PA Futreal, K Harshman, S Tavtigian, Q Liu, C Cochran, LM Bennett, W Ding, . 1994. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266: 66–71.
- ↵PM Milos. 2009. Emergence of single-molecule sequencing and potential for molecular diagnostic applications. Expert Rev Mol Diagn 9: 659–666.
- ↵JE Morgan, IM Carr, E Sheridan, CE Chu, B Hayward, N Camm, HA Lindsay, CJ Mattocks, AF Markham, DT Bonthron, . 2010. Genetic diagnosis of familial breast cancer using clonal sequencing. Hum Mutat 31: 484–491.
- ↵D Pushkarev, NF Neff, SR Quake. 2009. Single-molecule sequencing of an individual human genome. Nat Biotechnol 27: 847–852.
- ↵C Schroeder, F Stutzmann, BH Weber, O Riess, M Bonin. 2010. High-throughput resequencing in the diagnosis of BRCA1/2 mutations using oligonucleotide resequencing microarrays. Breast Cancer Res Treat 122: 287–297.
- ↵JR ten Bosch, WW Grody. 2008. Keeping up with the next generation: Massively parallel sequencing in clinical diagnostics. J Mol Diagn 10: 484–492.
- ↵MH Tsai, H Yan, X Chen, GV Chandramouli, S Zhao, D Coffin, CN Coleman, JB Mitchell, EY Chuang. 2005. Evaluation of hybridization conditions for spotted oligonucleotide-based DNA microarrays. Mol Biotechnol 29: 221–224.
- ↵T Tucker, M Marra, JM Friedman. 2009. Massively parallel sequencing: The next big thing in genetic medicine. Am J Hum Genet 85: 142–154.
- ↵T Walsh, S Casadei, KH Coats, E Swisher, SM Stray, J Higgins, KC Roach, J Mandell, MK Lee, S Ciernikova, . 2006. Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA 295: 1379–1388.
- ↵T Walsh, MK Lee, S Casadei, AM Thornton, SM Stray, C Pennil, AS Nord, JB Mandell, EM Swisher, MC King. 2010. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc Natl Acad Sci 107: 12629–12633.
Notes
[1] [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.122192.111.