
Novel roles for KLF1 in erythropoiesis revealed by mRNA-seq
- Michael R. Tallack1,
- Graham W. Magor1,
- Benjamin Dartigues2,3,
- Lei Sun2,4,
- Stephen Huang1,5,
- Jessica M. Fittock1,5,
- Sally V. Fry1,
- Evgeny A. Glazov6,
- Timothy L. Bailey2 and
- Andrew C. Perkins1,5,7,8
- 1Mater Medical Research Institute, Mater Hospital, Brisbane, Queensland 4101, Australia;
- 2Institute for Molecular Bioscience, The University of Queensland, Brisbane, Queensland 4072, Australia;
- 3University of Bordeaux, 33400 Talence, France;
- 4School of Information and Electrical Engineering, China University of Mining and Technology, Xuzhou, Jiangsu 221008, China;
- 5School of Biomedical Sciences, The University of Queensland, Brisbane, Queensland 4072, Australia;
- 6The University of Queensland Diamantina Institute, The University of Queensland, Brisbane, Queensland 4102, Australia;
- 7The Princess Alexandra Hospital, Brisbane, Queensland 4102, Australia
Abstract
KLF1 (formerly known as EKLF) regulates the development of erythroid cells from bi-potent progenitor cells via the transcriptional activation of a diverse set of genes. Mice lacking Klf1 die in utero prior to E15 from severe anemia due to the inadequate expression of genes controlling hemoglobin production, cell membrane and cytoskeletal integrity, and the cell cycle. We have recently described the full repertoire of KLF1 binding sites in vivo by performing KLF1 ChIP-seq in primary erythroid tissue (E14.5 fetal liver). Here we describe the KLF1-dependent erythroid transcriptome by comparing mRNA-seq from Klf1+/+ and Klf1−/− erythroid tissue. This has revealed novel target genes not previously obtainable by traditional microarray technology, and provided novel insights into the function of KLF1 as a transcriptional activator. We define a cis-regulatory module bound by KLF1, GATA1, TAL1, and EP300 that coordinates a core set of erythroid genes. We also describe a novel set of erythroid-specific promoters that drive high-level expression of otherwise ubiquitously expressed genes in erythroid cells. Our study has identified two novel lncRNAs that are dynamically expressed during erythroid differentiation, and discovered a role for KLF1 in directing apoptotic gene expression to drive the terminal stages of erythroid maturation.
The erythroid transcription factor KLF1 (formerly known as EKLF) is essential for erythropoiesis in mouse and contains pathogenic mutations in some human cases of congenital dyserythropoietic anemia (CDA; OMIM 613673) (Nuez et al. 1995; Perkins et al. 1995; Arnaud et al. 2010; Singleton et al. 2011). Loss-of-function mutations in human KLF1 also underlie the InLu blood group phenotype, result in elevated levels of fetal hemoglobin, and lead to increased levels of zinc protoporphyrin (Singleton et al. 2008; Borg et al. 2010; Satta et al. 2011). Studies of global gene regulation by KLF1 using traditional oligonucleotide microarray technology and, more recently, ChIP-seq technology have provided valuable insights into the KLF1-dependent erythroid transcriptome (Drissen et al. 2005; Hodge et al. 2006; Tallack et al. 2010; Pilon et al. 2011). These studies have begun to reveal the extensive repertoire of KLF1 target genes that includes not only genes responsible for the production of hemoglobin but also regulators of the cell cycle, membrane and cytoskeletal components, and regulators of apoptosis (Tallack and Perkins 2010; Siatecka and Bieker 2011).
The expression of Klf1 is restricted to erythroid cells and their precursor, the megakaryocyte-erythrocyte progenitor (MEP). Klf1−/− mice succumb to anemia and die by E15, which can be partly explained by its roles in the activation of gene expression for Hbb-b1 (also known as β-globin), the cell cycle regulators E2f2 and E2f4, and components of the erythroid cytoskeleton such as Epb4.9 (also known as dematin) (Miller and Bieker 1993; Nuez et al. 1995; Perkins et al. 1995; Drissen et al. 2005; Hodge et al. 2006; Frontelo et al. 2007; Pilon et al. 2008; Tallack et al. 2009). Loss of these together results in reduced hemoglobin production, impaired proliferation, and erythrocyte fragility, respectively. However, the few direct KLF1 target genes that have been described are not sufficient to explain all of the roles that KLF1 plays in erythroid differentiation and the pathology of human CDA resulting from KLF1 mutations.
The control of erythroid differentiation has been extensively studied, particularly with regard to the roles of extracellular cytokine signaling and transcription factor proteins (Hattangadi et al. 2011). However, it is only recently that we have begun to understand some of the processes that are unique to erythroid development such as the process of enucleation (Ji et al. 2011). Recent studies have described that enucleation is controlled by genes that regulate histone deacetylation to condense chromatin such as Hdac2, genes that underpin vesicle trafficking to create a cleft between the nucleus and cytoplasm, and genes that govern the formation of a contractile actin ring to “pinch off” the nucleus (Ji et al. 2008, 2010; Keerthivasan et al. 2010). Interestingly, some of these processes are not unique to enucleation but are also important steps in the process of apoptosis (Testa 2004; Hattangadi et al. 2011). Additionally, the loss of components of the apoptotic machinery such as Bcl2l1 (also known as BclX) leads to a failure in terminal erythroid differentiation (Motoyama et al. 1995; Wagner et al. 2000).
The aim of this study was to build upon our previous ChIP-seq study describing the in vivo occupancy of KLF1 in erythroid cells (Tallack et al. 2010). We have used next-generation DNA sequencing to describe a more comprehensive list of likely KLF1 target genes than was previously possible using oligonucleotide microarrays. By combining these data sets, we have gained deeper insights into how KLF1 works as a transcription factor in vivo, and have described for the first time a set of novel erythroid-specific promoters that are KLF1-dependent. Our data have also revealed two novel long noncoding RNAs (lncRNAs). We suggest that these, along with regulation of the apoptotic machinery, might be critical KLF1-regulated processes driving terminal erythroid differentiation.
Results
KLF1 activates an extensive erythroid gene expression program
In order to comprehensively define the set of direct KLF1 target genes during erythroid development, we previously performed KLF1 ChIP-seq on E14.5 primary fetal liver (Tallack et al. 2010). However, a limitation of this study was that at the time of publication, the set of differentially regulated KLF1 genes for comparison had been determined using oligonucleotide microarray technology and contained only the “lowest hanging fruit” (Drissen et al. 2005; Hodge et al. 2006; Pilon et al. 2008; Tallack et al. 2010). Hence we have now used next-generation DNA sequencing, a highly sensitive and unbiased approach, to define for the first time the comprehensive KLF1-dependent erythroid transcriptome.
We performed mRNA-seq using the Illumina GAII sequencing platform for three independent biological samples of Klf1+/+ and Klf1−/− fetal liver. We collected greater than 20 million sequence reads for each sample and mapped these to the mouse genome (mm9). To determine a highly sensitive set of KLF1-dependent genes, we performed a differential gene expression analysis using Cuffdiff software (Trapnell et al. 2010) and RefSeq gene annotations (Pruitt et al. 2007) that allowed us to describe 690 KLF1 “activated” genes (significantly greater expression in Klf1+/+, Benjamini-Hochberg–corrected P < 0.05) and 118 KLF1 “repressed” genes (significantly lower expression in Klf1+/+; Benjamini-Hochberg–corrected P < 0.05) (Fig. 1A; Supplemental Table S1). The use of next-generation DNA sequencing has allowed us to identify likely KLF1 target genes across five orders of magnitude of gene expression as judged by the FPKM (fragments per kilobase of exon per million fragments mapped) metric (Fig. 1A). This analysis identified previously known KLF1 target genes such as Epb4.9, Klf3 (previously known as Bklf), and E2f2 as differentially expressed; however, the power of this approach is likely to be in the identification of genes fitting into the LOW expression category (mean FPKM < 10 for Klf1+/+) as KLF1-dependent (Fig. 1A; Drissen et al. 2005; Hodge et al. 2006; Funnell et al. 2007; Pilon et al. 2008; Tallack et al. 2009).
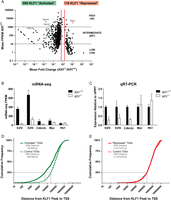
mRNA-seq reveals a comprehensive set of KLF1 activated genes. (A) A scatter plot representation of mRNA-seq expression data. Each data point represents a single RefSeq gene plotted according to the mean fold change between Klf1 genotypes (x-axis) and the mean Klf1+/+ expression (y-axis). The identity of several data points is indicated for reference. Vertical red lines indicate the 1.5-fold differential expression cutoff to define the KLF1 “activated” (decreased expression in Klf1−/− samples) and KLF1 “repressed” (increased expression in Klf1−/− samples). Genes were further sorted according to their mean Klf1+/+ expression into high, intermediate, and low expression categories. This data set is available as Supplemental Table S1. (B) mRNA-seq quantification by FPKM values. Each bar represents the mean FPKM + 95% confidence interval for three independent biological replicates. (C) qRT-PCR validation of mRNA-seq data. Each bar represents the mean + SEM for at least three independent biological replicates. Gene expression was normalized to the housekeeping gene HPRT and wild-type expression level. Klf1+/? is used to indicate that both Klf1+/+ and Klf1+/− samples were considered together. (D) Cumulative frequency distribution for the distances between KLF1 “activated” TSSs and KLF1 ChIP-seq peak locations (solid green line). The average distribution for 10 randomly simulated controls is also shown (dashed green line). (E) Cumulative frequency distribution for the distances between KLF1 “repressed” TSSs and KLF1 ChIP-seq peak locations (solid red line). The average distribution for 10 randomly simulated controls is also shown (dashed red line).
Our identification of differentially expressed genes by mRNA-seq is consistent with our previous oligonucleotide microarray study (Supplemental Fig. S1); however, it is worth noting that of the 808 genes identified by this approach (690 KLF1 “activated” and 118 KLF1 “repressed”), only 122 were also identified as differentially expressed by Hodge et al. (2006; Supplemental Table S2). We conclude that this is likely to be due to the increased sensitivity of mRNA-seq as well as the biases in gene representation and probe sensitivity that are inherent for oligonucleotide microarrays. We further validated our mRNA-seq data by comparing it to previously performed qRT-PCR for the direct KLF1 target genes E2f2, E2f4, and Cdkn2c (also known as p18INK4c) (Fig. 1B,C; Tallack et al. 2007, 2009). We also performed additional qRT-PCR for genes not found to be significantly differentially expressed, Myc and Rb1, and gained a similar result (Fig. 1B,C).
KLF1 has been suggested to operate as both a transcriptional activator and a transcriptional repressor depending on both the cellular context and target gene (Feng et al. 1994; Chen and Bieker 2001, 2004; Drissen et al. 2005; Hodge et al. 2006; Siatecka et al. 2007). Interestingly, we found overrepresentation of the CACC motif (specifically the JASPAR KLF4 motif) at both KLF1 “activated” and KLF1 “repressed” gene promoters (within 500 bp) (Supplemental Fig. S2). We additionally found an overrepresentation of an NFYA motif, resembling a CCAAT box, in both sets of gene promoters (Supplemental Fig. S2). However, ChIP-seq for KLF1 shows occupancy within the promoter (1000 bp) of a single KLF1 “repressed” gene (out of 118). Thus, we suggest CACC motifs in these promoters are likely to bind repressor KLFs such as KLF3, KLF8, and KLF12. Indeed, Klf3 and Klf8 are known direct targets of KLF1 (Funnell et al. 2007; Eaton et al. 2008). We have not been able to successfully ChIP these repressor KLFs due to lack of antibodies.
We sought to gain additional insight into how KLF1 might operate as an activator or a repressor by pairing each of the 945 high-confidence in vivo KLF1 binding sites (peaks) we previously identified with the transcription start sites (TSSs) of the closest “activated” or “repressed” gene (Supplemental Table S3). Each of these KLF1 peak–TSS pairs represents a possible trans interaction regulating the erythroid transcriptome. We calculated and plotted the cumulative frequency distributions of the genomic distances for each of these interactions compared with the distributions for 10 randomly selected sets of TSSs of the same size as the controls (Fig. 1D,E). We noted that the distribution for KLF1 peak–“activated” TSS pairs was significantly different to the controls; however, the KLF1 peak–“repressed” TSS pairs distribution was identical to the control simulated distributions. This suggests that the distance between the KLF1 peaks and genes that show a reduced expression in Klf1−/− erythroid tissue is on average much closer than would be expected by chance, consistent with the hypothesis that KLF1 acts as an activator (Fig. 1D). The fact that the same distribution calculated between KLF1 peaks and “repressed” genes is identical to the control does suggest KLF1 acts as a repressor very infrequently in erythroid cells. It does not exclude occasional repression of specific transcripts or repression of megakaryocyte genes such as Fli1 in MEPs that represent just 6% of E14.5 fetal liver cells (Fig. 1E; Starck et al. 2003; Papathanasiou et al. 2010).
Insights into KLF1 cooperation in vivo
We previously described a significant overlap in in vivo binding between KLF1 and another critical erythroid transcription factor, GATA1 (Tallack et al. 2010). At the time of this publication, the same analysis was also performed for an additional erythroid transcription factor, TAL1 (formerly known as SCL) using the best available ChIP-seq data set (Wilson et al. 2009). A more suitable ChIP-seq data set for erythroid TAL1 binding has since been published, and we assessed the overlap between this data set and the previously used data sets for KLF1 and GATA1 (Cheng et al. 2009; Kassouf et al. 2010; Tallack et al. 2010). We found a set of 119 regions in the mouse genome (mm9) that are occupied by these three factors (Fig. 2A; Supplemental Table S4). We suggest that these regions are likely to be promoters and/or enhancers that drive specific and potent erythroid gene expression.
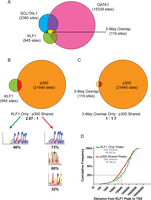
KLF1 works co-operatively with GATA1, TAL1, and EP300 to activate genes. (A) Venn diagram showing the overlap between in vivo binding of KLF1 (Tallack et al. 2010), GATA1 (Cheng et al. 2009), and TAL1 (Kassouf et al. 2010) from erythroid tissue ChIP-seq. (B) Venn diagram showing the overlap between in vivo binding of KLF1 (Tallack et al. 2010) and EP300 (Birney et al. 2007) from erythroid tissue ChIP-seq. KLF1 peaks were separated into two categories, KLF1 only and EP300 shared, as indicated, and subjected to motif enrichment analysis using MEME-ChIP. Significantly enriched motifs for each set are shown together with the percentage of peaks in that category containing the motif. (C) Venn diagram showing the overlap between three-way overlap peaks (KLF1-GATA1-TAL1, shown in A) and EP300 from erythroid tissue ChIP-seq. (D) Cumulative frequency distribution for the distances between KLF1 “activated” TSSs and either KLF1 only peaks (green line) or EP300 shared peaks (red line) as defined in B.
The activation of target genes by KLF1 is likely to be largely dependent on its direct recruitment of transcriptional coactivators such as EP300/CREBBP (Zhang et al. 2001). We took advantage of a recent study describing EP300 coactivator occupancy in murine erythroleukemia (MEL) cells to determine sites of in vivo KLF1 binding that are shared by EP300 (Fig. 2B; Birney et al. 2007). Interestingly, of the 945 KLF1 binding sites, only around a one-third (305 of 945) are also bound by EP300 (Fig. 2B). This was irrespective of whether the KLF1 binding site was at a “classical promoter” location (≤1000 bp upstream of a TSS, eight of 23 sites shared with EP300) or at a nonpromoter regulatory element (299 of 915 sites shared with EP300).
This EP300 ChIP-seq data also provided some useful insight into the function of particular KLF1 bound regions. For example, we have previously described intragenic regions of the E2f2 and E2f4 loci that act as erythroid enhancers (Tallack et al. 2009). The EP300 ChIP-seq data of Birney et al. (2007) shows that this is likely to be in part via recruitment of EP300 since EP300 binding is strong at these regions (data not shown). Hence we decided to test a KLF1-bound region that was not found to bind EP300 by ChIP-seq. We cloned and tested an intergenic region 40 kb downstream from the Klf3 locus for erythroid enhancer activity (Supplemental Fig. S3). Although this region binds KLF1 very strongly (peak height, 30), it fails to act as an enhancer in MEL cells (unlike the enhancers of E2f2 and E2f4). It will be interesting to determine the function of KLF1 at this region (Klf3 +40e) (Supplemental Fig. S3) and similar regions that do not cobind EP300, although this is not trivial.
We also examined the representation of specific transcription factor DNA motifs in the two sets of peaks (KLF1 Only and EP300 Shared) using MEME-ChIP (Fig. 2B; Machanick and Bailey 2011). Not surprisingly, we found the presence of the CACC motif (5′-CCMCRCCCN-3′) in both sets of peaks, suggesting that KLF1 truly binds both sets. The EP300 Shared set of peaks also contained a significant overrepresentation of the GATA1 (5′-WGATAR-3′) binding motif and a motif resembling an E-Box, presumably capable of binding TAL1 (Fig. 2B). Central motif enrichment analysis (CMEA) using CentriMo also showed that GATA1 and TAL1 binding motifs are more highly enriched in the EP300 Shared set of KLF1 binding sites compared with the KLF1 Only sites (Supplemental Fig. S4; Bailey and Machanick 2012). These results suggest that the cooperative binding of KLF1 together with GATA1 and/or TAL1 serves to more strongly recruit EP300 to the DNA as might be expected. We further tested this hypothesis by looking at the overlap between the KLF1-GATA1-TAL1 three-way sites (three-way overlap, 119 sites) with EP300 (Fig. 2C). The majority of the three-way overlap sites are also bound by EP300 (75 of 119), consistent with the hypothesis that regions containing more than one of the factors can more effectively recruit EP300. These regions are likely to represent a core cis-regulatory module driving erythroid-specific gene expression via the KLF1-GATA1-TAL1 transcription factor network and associated factors. This DNA-bound complex is likely to be a giant complex of proteins that includes LMO2, LDB1, EP300, and probably many more associated proteins (Soler et al. 2010). We found no overrepresentation of other cis-motifs, so other DNA binding transcription factors are not likely to participate in the function of this core erythroid enhancer complex.
Since KLF1 peaks are on average closer to “activated” TSSs than expected (Fig. 1D), we sought to determine if the presence of EP300 affected this distribution, or in other words, “are KLF1–EP300 Shared sites closer to TSSs than KLF1 Only sites?”. We measured the distance from each peak to the closest “activated” TSS for the KLF1 Only (Fig. 2D, green line) and KLF1–EP300 Shared (Fig. 2D, red line) sets of peaks and plotted the distributions. This data indicates that on average KLF1–EP300 Shared peaks are closer to “activated” genes than are KLF1 Only peaks (Fig. 2D). Since KLF1 binding sites that do not recruit EP300 tend to be further from TSSs, this suggests at least a subset of these sites might not be involved in transcriptional activation at all. They are unlikely to be artifacts as they frequently contain an extended CACC motif at a high frequency (46%) (Fig. 2B).
KLF1 mRNA-seq reveals a set of novel erythroid gene promoters
We and others have previously identified that KLF1, GATA1, and TAL1 frequently bind target genes at sites other than the promoter, particularly within introns (Cheng et al. 2009; Kassouf et al. 2010; Tallack et al. 2010). We used our list of 119 three-way overlap sites as a starting point to look for novel erythroid promoters that might serve to drive high levels of erythroid-specific expression (Supplemental Table S4). We identified novel promoters for seven genes using our mRNA-seq data, particularly making use of splice junction reads (reads that are split when mapped to the genome), some of which are further supported by mouse ESTs or CAGE tag clusters (Table 1; Supplemental Fig. S5). This list includes the gene encoding the first two enzymes of the heme synthesis pathway, Alas2 and Alad (Table 1). The start of translation for these two genes begins in exon 2 of the RefSeq transcripts, allowing for the use of novel erythroid-specific promoters that will produce the same protein (Fig. 3A,B). Indeed, ChIP-seq data for KLF1, GATA1, and TAL1 show binding of all three factors within the first intron for both Alas2 and Alad (Fig. 3A,B, vertical green stripes).
Alternative erythroid TSSs uncovered by Klf1 mRNA-seq
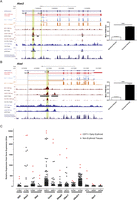
Identification of a novel set of KLF1-GATA1-TAL1–dependent erythroid promoters. (A) An image of murine Alas2 from the UCSC Genome Browser with tracks depicting a novel mechanism of erythroid gene regulation whereby transcription begins at RefSeq exon 2 (Ery TSS). The RefSeq gene annotations (dark blue) are shown together with mRNA-seq transcripts defined using Cuffdiff (red). Representative tracks for a single Klf1−/− and Klf1+/+ mRNA-seq sample are shown in light blue and orange, respectively, with both mapped read density (wiggle) and splice junction information (all tracks are shown in Supplemental Fig. S5). ChIP-seq tracks for KLF1, GATA1, and TAL1 are shown in maroon with peak calls underneath. CTCF and EP300 ChIP-seq from the ENCODE consortium are shown in blue as well as a vertebrate conservation track (phyloP). Gray and green vertical lines are included to point out RefSeq TSSs (Ref) and novel erythroid TSSs (Ery), respectively. Regions of multitranscription factor occupancy are also highlighted by a vertical green line. (Bar chart) Quantification of relative promoter usage describing the ratio of RefSeq exon 1 counts or erythroid exon 1 counts relative to exon 2 for six biological replicates. Data shown are the mean + SEM. (***) P < 0.001 by Student's t-test. (B) An image of murine Alad from the UCSC Genome Browser depicting a novel mechanism of erythroid gene regulation whereby transcription begins at a novel first exon residing within RefSeq intron 1 (Ery TSS). All tracks are shown as above in A. The bar chart also shows quantification of relative promoter usage for either RefSeq exon 1 or the novel erythroid exon 1 compared to exon 2 for six biological replicates. Data shown are the mean ± SEM. (***) P < 0.001 by Student's t-test. (C) Relative gene expression obtained from Gene Expression Atlas for each of the genes described to use novel erythroid promoters (Table 1). Red dots are used to show the expression in the two CD71+ early erythroid samples, while black dots show the expression in all other tissues of the atlas. The HG-U133A probe numbers are shown for each gene since some genes are represented by multiple probes. Klf3 serves as a positive control since it has previously been shown to be ubiquitously expressed with greatest expression in erythroid cells (Funnell et al. 2007).
Our mRNA-seq data for Alas2 indicate that the majority of erythroid transcription begins at exon 2 and that in this instance the transcription factor binding within the first intron resembles canonical promoter binding for these transcription factors (Fig. 3A, Klf1+/+ in orange, Klf1−/− in light blue, declared transcripts in red). Very few reads from our mRNA-seq map to RefSeq exon 1 of Alas2 compared with the large number mapping to exon 2 (Fig. 3A, exons 1 and 2 highlighted by vertical stripes). To support this observation, we determined the ratio of reads mapping to either the RefSeq exon 1 (Fig. 3A, labeled Ref) or the novel erythroid exon 1 (Fig. 3A, labeled Ery, RefSeq exon 2) compared with exon 2 of each transcript, and found these ratios to be significantly different (Fig. 3A, bar chart). Our mRNA-seq data also confirm the KLF1 dependance of Alas2 gene expression, which is roughly threefold lower in Klf1−/− fetal liver samples (Fig. 3A, cf. light blue track and orange track; Table 1). We also noted that the coactivator EP300 is recruited to this novel promoter, presumably by the binding of KLF1, GATA1, and TAL1 (Fig. 3A, dark blue track).
Similarly, erythroid expression of Alad does not begin at exon 1 of the RefSeq transcript (Fig. 3B). However, in the case of Alad, we have uncovered a completely novel first exon beginning immediately upstream of KLF1-GATA1-TAL1 binding (Fig. 3B, vertical green stripe, declared transcripts in red). Likewise, expression of Alad is highly dependent of KLF1, being fivefold lower in the Klf1−/− samples (Fig. 3B, cf. light blue track and orange track; Table 1). We also found the exon 1:exon 2 ratios for the RefSeq exon 1 and novel erythroid exon 1 to be significantly different, suggesting that the novel erythroid exon 1 was likely to be dominant in erythropoiesis (Fig. 3B, bar chart).
In the cases of Alas2 and Alad, these genes are broadly expressed since heme is a cofactor for a large number of hemoproteins, such as myoglobin and the cytochromes (Reedy et al. 2008). However, their demand in erythroid tissues is far greater since hemoglobin is so abundant. Hence it seems reasonable that these genes have evolved an alternative “switch” to control their erythroid expression. This has also been shown for the transcriptional repressor Klf3 (Funnell et al. 2007). To test this hypothesis in human tissue, we analyzed the expression of each of the genes we identified as having novel erythroid promoters using the human gene atlas gene expression set containing 79 different tissues, one of which was “CD71 positive early erythroid” (Su et al. 2004; Vaquerizas et al. 2009). The gene atlas data support our hypothesis since each of the genes shows a fairly broad range of expression, with the two erythroid samples clearly expressed most highly and often for multiple probes on the microarray (Fig. 3C, erythroid as red dots, all other tissues black dots). We anticipated genes such as Alas2, Alad, Urod, and Steap3 would show this pattern of expression due to their crucial roles in heme synthesis and iron procurement by erythroid cells. However, we have also uncovered three additional genes, Ubac1, Ube2v1, and Itsn1 that are highly expressed in erythroid tissue, dependent on KLF1, GATA1, and TAL1 for their expression and have a relatively unknown function (Fig. 3C; Table 1). It will be of great interest to determine the roles that these genes play in erythropoiesis.
Erythroid lncRNAs controlled by KLF1
While a great deal is known of the contribution of microRNAs (miRs) to erythropoiesis, very little is known of the contribution of other species of nonprotein coding RNAs, particularly those classified as lncRNAs (Rasmussen et al. 2010; Hattangadi et al. 2011; Zhang et al. 2011). We have used our mRNA-seq data to identify two novel lncRNAs that are previously uncharacterized, KLF1-dependent, and expressed at the terminal stages of erythroid differentiation. We have designated these lncRNAs Lincred1 and Lincred2 for long intergenic noncoding RNA of erythroid differentiation (Fig. 4A,B; Supplemental Fig. S6). The presence of ChIP-seq peaks for KLF1, GATA1, and TAL1 at or near the promoters of Lincred1 and Lincred2 suggests they are likely to be functional during erythroid differentiation (Fig. 4A,B). The transcripts we have declared to be Lincred1 and Lincred2 were compiled using Cufflinks software using splice junction tags to define exons (Fig. 4A,B, red tracks). Lincred1 has two alternative transcripts we designated Lincred1-giant and Lincred1-dwarf to reflect their relative lengths.
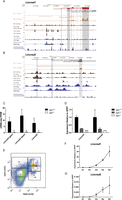
KLF1 regulates two novel erythroid-specific long noncoding RNAs (lncRNAs). (A) An image from the UCSC Genome Browser with tracks depicting a novel long intergenic noncoding RNA of erythroid differentiation defined as Lincred1. (Red) Erythroid RNAs present in this region as defined by Cufflinks. There appears to be two separate isoforms of the one gene; hence, they have been named “giant” and “dwarf” for the long and short isoforms, respectively. (Black) Mouse mRNAs and ESTs deposited into GenBank for this same region. Representative mRNA-seq tracks for a single Klf1−/− and Klf1+/+ are shown in light blue and orange, respectively (all mRNA-seq data are presented in Supplemental Fig. S6). ChIP-seq data for KLF1, GATA1, and TAL1 (maroon tracks), as well as CTCF and EP300 (dark blue tracks), from erythroid tissues are shown together with peak calls. (Bottom) A vertebrate conservation track (phyloP). Exons of the novel lncRNAs are highlighted by vertical gray bars. (B) An image from the UCSC Genome Browser for the novel erythroid lncRNA Lincred2. Tracks in the browser image are as described above for A. (C) Quantification of mRNA-seq data for Lincred1-giant, Lincred1-dwarf, and Lincred2 by the FPKM method. Each bar represents the mean FPKM for three independent biological replicates + 95% confidence interval. (*) Benjamini-Hochberg–corrected P < 0.05. (D) Expression validation by qRT-PCR using specific primers for Lincred1 and Lincred2. Each bar represents the mean + SEM for at least three independent biological replicates normalized to the housekeeping gene HPRT and to the Klf1+/+ expression level. (***) P < 0.001 by Student's t-test. Note that primers for Lincred1 amplify both the giant and dwarf isoforms. (E) Representative FACS plot for sorting E13.5 fetal liver cells into R1–R5 populations using the cell surface markers CD71 and TER119. Gates are shown together with the proportion of cells in each population. (F) Expression of Lincred1 throughout erythroid differentiation (R1–R5) as determined by qRT-PCR. Each point shown is the mean ± SEM for three independent biological replicates normalized to expression of the housekeeping gene HPRT. (G) Expression of Lincred2 throughout erythroid differentiation shown as above in F.
The quantification of mRNA-seq data indicates that Lincred1 (giant and dwarf) and Lincred2 are both KLF1-dependent transcripts (Fig. 4C). We tested the Lincred transcripts for possible open reading frames (ORFs) to determine if they truly were likely to be nonprotein coding. Neither Lincred1 nor Lincred2 contains any significant ORFs that would encode a protein of greater than 100 amino acids, providing further evidence that they are lncRNAs (data not shown). We confirmed the KLF1 dependence of Lincred1 and Lincred2 expression by qRT-PCR using specific primers (Lincred1 primers amplify both isoforms) and observed a Klf1 gene dosage effect on their expression (Fig. 4D).
In order to infer possible functions for Lincred1 and Lincred2 in erythroid differentiation, we sorted E13.5 fetal liver cells into five populations (R1–R5) using the cell surface markers CD71 and TER119, as performed previously (Fig. 4E; Zhang et al. 2003). Both Lincred1 and Lincred2 have almost an undetectable expression in erythroid progenitors (R1 and R2) and a pattern of increasing expression in the terminal stages of erythroid differentiation (highest in R5) (Fig. 4F,G). This expression pattern mirrors the expression pattern of Klf1 itself and provides some evidence that Lincred1 and Lincred2 might be involved in regulating processes at play during terminal erythroid differentiation, although loss- and gain-of-function studies are required to confirm this (Supplemental Fig. S7).
KLF1 as a regulator of the apoptotic program
mRNA-seq has provided many additional KLF1-dependent genes compared to previous oligonucleotide microarray studies, so we sought to look broadly for unappreciated functions for KLF1 in erythropoiesis. We performed Gene Ontology (GO) analysis using the online tool DAVID to look for enriched terms in the 690 KLF1 “activated” and 118 KLF1 “repressed” genes (Supplemental Table S5). In addition to terms we expected to find such as erythropoiesis and heme biosynthesis, we found terms relating to apoptosis were enriched in our KLF1 “activated” gene set (Supplemental Table S5).
This result prompted us to look closer at specific apoptotic genes that might be direct KLF1 targets. We previously reported that Bcl2l1 was likely to be a direct KLF1 target gene; hence, we sought to confirm this by qRT-PCR (Tallack et al. 2010). Additionally, previous studies of apoptotic genes and erythropoiesis had suggested that Bcl2l11 (formerly Bim) expression levels, in this instance regulated by Zbtb7a (also known as Lrf and Pokemon), were critical determinants of erythroid cell survival (Maeda et al. 2009). We found by qRT-PCR that expression of the anti-apoptotic gene Bcl2l1 was significantly reduced in Klf1−/− erythroid cells (approximately twofold) (Fig. 5A) and that expression of the pro-apoptotic gene Bcl2l11 was elevated (approximately 2.5-fold) (Fig. 5A). Both of these genes have been previously shown to play a role in erythropoiesis (Wagner et al. 2000; Maeda et al. 2009). This suggested that the apoptotic gene expression program might be perturbed in the absence of Klf1.
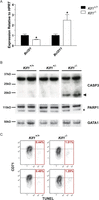
Loss of Klf1 disrupts the normal apoptotic program. (A) Expression of Bcl2l1 and Bcl2l11 by qRT-PCR in E14.5 fetal liver normalized to the housekeeping gene HPRT. Each bar shown is the mean + SEM for at least three independent biological replicates. (*) P < 0.05 by Student's t-test. Klf1+/? is used to indicate that both Klf1+/+ and Klf1+/− samples were considered together. (B) Western blot analysis of whole-cell protein extracts for different Klf1 genotypes as indicated. Protein levels of caspase 3 (CASP3) and PARP1 were investigated using specific antibodies with GATA1 as a loading control. Expected cleavage products of caspase 3 and PARP1 are shown by a left-facing arrow and asterisk, respectively. (C) Representative TUNEL assay FACS plots for Klf1+/+ and Klf1−/− genotypes as indicated. Cells were co-stained for TUNEL and the erythroid marker CD71. The percentages of cells staining TUNEL positive are indicated.
We sought to determine if aberrant induction of apoptosis was also a contributor to the severe anemia in Klf1−/− mice. We found evidence of the cleaved active 17-kD form of caspase 3 in Klf1−/− erythroid cells that was not present in either Klf1+/+ or Klf1+/− cells (Fig. 5B). Traditionally, active caspase 3 drives apoptosis, partly via cleavage of poly (ADP-ribose) polymerase family, member 1 (PARP1). However, we did not find any evidence of increased cleavage of PARP1 in Klf1−/− cells (Fig. 5B). We additionally looked for cleavage of GATA1 and TAL1 since these have been reported to be targets of activated caspases in erythroid cells, but we found no evidence that this was occurring (Supplemental Fig. S8; De Maria et al. 1999; Zeuner et al. 2003). This might be due to protection of GATA1 and TAL1 by the heat shock proteins HSPA1A and HSPA1B, which have increased expression in Klf1−/− cells (Supplemental Fig. S8; Ribeil et al. 2007). Neither is a direct KLF1 target gene so up-regulation is indirect (data not shown). The latter observation strongly suggests Klf1−/− cells “sense” activated caspases and up-regulate genes indirectly (such as Hspa1a and Hspa1b) to provide protection against cell death.
Since we had observed Klf1−/− cells to have a distorted apoptotic genetic program, we looked for further signs that apoptosis might be occurring. We found no significant difference in Annexin V staining at the cell surface between Klf1−/− and Klf1+/+ cells (data not shown). We also used the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay combined with an erythroid cell surface marker (CD71) to determine if apoptotic cell numbers were increased in Klf1−/− tissue (Fig. 5C). We found a slight increase in TUNEL-positive erythroid cells in Klf1−/− embryos (∼0.5% Klf1+/+ vs. ∼1.1% Klf1−/−) (Fig. 5C). Despite an absence of significant apoptosis, we believe caspase 3 cleavage might derail normal erythroid differentiation since these activated proteases could potentially digest many other protein targets.
Discussion
In this study we have described the use of next-generation DNA sequencing to gain a comprehensive description of KLF1-dependent erythroid genes (Fig. 1A; Supplemental Table S1). The sensitivity of mRNA-seq has allowed us to identify differentially expressed genes between the Klf1−/− and Klf1+/+ genotypes that were previously missed by oligonucleotide microarray, particularly those that we have assigned to the “low” expression category (Fig. 1A; Supplemental Table S1). We have compared these data to our previously published work describing the in vivo chromatin occupancy of KLF1 to begin to define KLF1 peak-TSS pairs, many of which are likely to reflect in vivo KLF1 gene regulatory activities (Supplemental Table S3; Tallack et al. 2010). However, the assignment of these KLF1 peak–TSS pairs is based upon a linear model of the genome and does not account for three-dimensional DNA looping or the presence of nuclear transcription factories seeded by KLF1 and RNA polymerase II (Drissen et al. 2004; Schoenfelder et al. 2010; Eskiw and Fraser 2011). Nevertheless, this method has revealed that KLF1 is likely to predominantly function as a transcriptional activator in vivo and that in most cases the closest gene to an occupied chromatin segment is unlikely to be the KLF1 target gene, at least in terms of a linear genome (Fig. 1D,E). We did not compare our mRNA-seq data to the recently published KLF1 ChIP-seq data of Pilon and colleagues (2011) since our bioinformatic analysis of these data showed no evidence of direct, sequence-specific binding by KLF1 in the ChIP-seq regions they reported, (Supplemental Figs. S9, S10). In contrast, the same analysis on our own previously published ChIP-seq study reveals an enrichment of the KLF1 binding motif in regions declared as peaks (Supplemental Fig. S10; Tallack et al. 2010). These analyses are discussed in greater detail in the Supplemental Material accompanying this article.
We have taken advantage of other erythroid ChIP-seq studies to define a core set of cis-regulatory modules, regions with KLF1, GATA1 and TAL1 occupancy in close proximity (Fig. 2A; Supplemental Table S4). The majority of these regions are also occupied by the transcriptional coactivator EP300 as might be expected (Fig. 2C). However, we were surprised to find that many genomic sites bound by KLF1 are not co-occupied by EP300 (Fig. 2B). These sites still contain a significant overrepresentation of the KLF1 binding motif (46%), suggesting that they are real and that KLF1 binding is specific (Fig. 2B). We suggest KLF1 functions at these sites in a EP300-independent manner. This is consistent with the recent study of Mas et al. (2011) that describes the N-terminal transactivation domain of KLF1 can bind to additional coactivator proteins besides EP300.
We also undertook this study in part to address some of the prevailing questions that have arisen from recent studies describing KLF1 in vivo chromatin occupancy on a global scale by ChIP-seq (Tallack et al. 2010; Pilon et al. 2011). In particular, our previous study as well as similar studies of GATA1, TAL1, and associated proteins has shown that many sites of chromatin occupancy by erythroid transcription factors do not occur in proximal promoter regions (Cheng et al. 2009; Kassouf et al. 2010; Soler et al. 2010; Tallack et al. 2010). In particular, these studies have identified a significant portion of the occupied chromatin regions are actually the first introns of target genes. We hypothesized that these regions might act as either novel erythroid-specific gene promoters, or that these regions might act as enhancers that cooperate with the gene promoter by DNA looping, as has been suggested elsewhere (Magklara and Smith 2009; Alotaibi et al. 2010). Our mRNA-seq data have suggested that the former hypothesis is true for many instances of intron 1 chromatin occupancy (Table 1; Fig. 3). In the cases of Alas2, Alad, and Steap3, this allows for a unique erythroid-specific transcriptional regulatory input to drive high-level erythroid expression while still producing an identical protein output since the start of translation falls downstream (Table 1; Fig. 3; Supplemental Fig. S5). The latter hypothesis is also likely to be true since in the case of the cell cycle regulator E2f2, a well-known KLF1 target gene, chromatin occupancy occurs within intron 1, yet we cannot find any evidence that this leads to transcription from a novel TSS by mRNA-seq (Tallack et al. 2009).
ChIP-seq studies for KLF1, GATA1, and TAL1 have also described a number of regions of chromatin occupancy for which a target gene cannot be easily assigned due to their location in regions devoid of likely target genes. In many cases, these regions of chromatin occupancy are likely to be involved in long-range chromatin looping, such as is well understood for the β-globin LCR, or they might be essential to seed the formation of transcription factories within the nucleus (Drissen et al. 2004; Schoenfelder et al. 2010). A comprehensive description of the three-dimensional structure of the erythroid progenitor nucleus by chromosome conformation capture–based technologies will be necessary to gain global insights into these phenomena (Lieberman-Aiden et al. 2009). Nevertheless, we hypothesize that some of these sites of chromatin occupancy where the target gene was ambiguous might represent the promoters of previously uncharacterized erythroid transcripts. Surprisingly, recent efforts to survey transcription across different tissues, such as the Illumina Human Body Map version 2, have not included erythroid progenitor cells in the tissue sets interrogated (Cabili et al. 2011). We identified from our mRNA-seq data two previously undescribed genes we have designated Lincred1 and Lincred2 (Fig. 4). We hypothesize that these genes are likely to produce noncoding RNAs based on the absence of substantial ORFs in their sequences. While we are yet to describe their precise function or mechanism of action, the up-regulation of Lincred1 and Lincred2 during the terminal stages of erythropoiesis suggests function. The fact that these noncoding genes are also regulated by KLF1, GATA1, and TAL1 indicates that they are likely to play a significant role in erythropoiesis. Not unexpectedly another class of noncoding RNAs, the miRs, have recently been shown to perform critical roles in terminal erythroid differentiation (Rasmussen et al. 2010; Zhang et al. 2011). It is likely that KLF1 also plays a role in regulating mammalian miRs since the zebrafish Klf, Klfd, has been shown to regulate expression of zebrafish mir144; however, small molecule RNA-seq would be required to properly address this question (Fu et al. 2009).
Klf1−/− embryos have previously been shown to have defects in β-like globin gene expression, formation of an appropriate cell membrane and cytoskeletal structure, and a distorted cell cycle program (Nuez et al. 1995; Perkins et al. 1995; Drissen et al. 2005; Hodge et al. 2006; Tallack et al. 2007, 2009; Pilon et al. 2008). Each of these phenotypes contributes to the severe anemia of Klf1−/− embryos; however, the perturbations to gene expression are more dramatic than these pathways alone (Supplemental Table S5). In particular, we noticed that the loss of Klf1 was associated with the decreased expression of apoptotic genes (Supplemental Table S5). Since we observed a decrease in expression of Bcl2l1 and an increase in Bcl2l11, we expected to find increased apoptosis in the fetal livers of Klf1−/− embryos (Fig. 5A,C). This was not found to be the case and seems at odds with the increased levels of the 17-kD form of caspase 3 that we observed in Klf1−/− fetal livers (Fig. 5B). Similar perturbations to the apoptotic pathway are found in Gata1−/− and Zbtb7a−/− mice; however, these embryos do have increased numbers of apoptotic cells (Maeda et al. 2009). It is particularly interesting that in addition to regulating apoptosis, caspase 3, BCL2L1, and BCL2L11 all appear to play critical roles in erythropoiesis (Wagner et al. 2000; Zermati et al. 2001; Maeda et al. 2009). We suggest that in the absence of Klf1, genes of the apoptotic program have been disrupted sufficiently to prevent terminal erythroid differentiation, but perhaps not to a level sufficient to complete the final apoptosis pathway. This phenotype of failed terminal erythroid differentiation in Klf1−/− mice is reminiscent of the pathology of CDA in human patients resulting from KLF1 mutations whereby immature nucleated erythroid cells are released into the blood stream (Arnaud et al. 2010).
In summary, we have presented here a comprehensive set of KLF1-dependent erythroid genes in addition to novel insights into KLF1's mechanism of action as a transcriptional activator. We suggest that KLF1 cooperates with the transcription factors GATA1, TAL1 and the coactivator EP300 at 75 sites in the erythroid genome to regulate erythroid-specific levels of gene expression, in some cases via previously uncharacterized erythroid promoters. We also report here the discovery of two novel lncRNAs with erythroid-specific expression that are KLF1-dependent and describe that perturbation to the apoptotic gene expression program in Klf1−/− mice does not result in apoptosis but is likely to contribute to a failure of terminal erythroid differentiation.
Methods
mRNA-seq library preparation and sequencing
Fetal liver tissue from E14.5 Klf1+/− × Klf1+/− timed matings was collected, homogenized, and preserved in TRIzol reagent (Invitrogen). Genotyping was performed by PCR according to the method previously described (Hodge et al. 2006). Total RNA was collected as per the manufacturer's recommendations and depleted of alpha-globin and beta-globin mRNA using the Mouse/Rat GLOBINclear Kit (Ambion) starting with 10 μg DNase I (Invitrogen) treated RNA. Three Klf1+/+ and three Klf1−/− samples from two independent litters were selected and used to prepare mRNA-seq libraries using the mRNA-Seq Sample Prep Kit (Illumina). Each library was sequenced in a separate lane using the GAII sequencing platform (Illumina) to obtain >20 × 106 single-end 76-bp reads. Raw reads have been deposited in the Gene Expression Omnibus (GEO) under accession no. GSE33979.
mRNA-seq mapping, expression, and transcript construction
mRNA-seq reads were mapped to the mm9 genome of UCSC (ftp://ftp.cbcb.umd.edu/pub/data/bowtie_indexes/mm9.ebwt.zip) to generate six BAM files individually using Tophat-1.3.1 (http://tophat.cbcb.umd.edu/downloads/) (Trapnell et al. 2009). Subsequently, the six BAM files were imported to Cuffdiff of Cufflinks-1.0.3 (http://cufflinks.cbcb.umd.edu/downloads/) with a set of RefSeq annotated transcripts in GTF format (ftp://igenome:G3nom3s4u@ftp.illumina.com/Mus_musculus/UCSC/mm9/Mus_musculus_UCSC_mm9.tar.gz) to conduct differential gene expression tests (Trapnell et al. 2010; Roberts et al. 2011a,b).
We defined differentially expressed genes as either KLF1 “activated” or KLF1 “repressed” for genes with significantly greater expression in the Klf1+/+ or Klf1−/− samples, respectively. We used a mean Klf1+/+ FPKM cutoff of 0.5 to ensure genes in our lists were sufficiently expressed in the E14.5 fetal liver, and only reported genes with a greater than 1.5-fold difference in mean FPKM between the Klf1+/+ and Klf1−/− genotypes. All of the genes reported in our gene lists were found to be significantly differentially expressed between the genotypes by Cuffdiff after a Benjamini-Hochberg correction for multiple-testing errors (P < 0.05).
The lncRNAs Lincred1-giant, Lincred1-dwarf, and Lincred2 were assembled using the de novo transcript assembly tool Cufflinks-1.0.3. Differential gene expression data for these transcripts was also obtained using the Cuffdiff component of the Cufflinks suite.
Quantitative real-time RT-PCR
Fetal liver total RNA was collected using TRIzol reagent (Invitrogen) and reverse transcribed to cDNA using Superscript III reverse transcriptase (Invitrogen). qRT-PCR was performed using the 7900HT Fast Real-Time PCR System (Applied Biosystems) using SYBR Green chemistry (Applied Biosystems) with transcript-specific primers described in Supplemental Table S6.
Western blotting
Fetal liver whole-cell protein extracts were prepared using RIPA buffer and resolved on 4%–12% Bis-Tris precast gels (Invitrogen). Blotting was performed by transferring proteins to PVDF membrane (GE Healthcare), blocking, and incubating with the appropriate primary and ECL-conjugated secondary antibodies. The primary antibodies used were as follows: anti-caspase 3, 9661 Cell Signaling Technology; anti-GATA1, sc-265 Santa Cruz Biotechnology; and anti-PARP, MCA1522G AbD Serotec.
TUNEL assay
Homogenized E14.5 fetal livers were assayed for apoptosis using the TUNEL method. Cells were prepared according to the manufacturer's recommendations using the In Situ Cell Death Detection Kit, Fluorescein (Roche); co-stained with CD71-PE (BD Pharmingen); and analyzed using a FACSCanto II flow cytometer (BD) and FlowJo software (Treestar).
Fetal liver cell sorting (R1–R5)
Homogenized E13.5 fetal livers from wild-type BALB/c timed matings were dual stained with CD71-FITC (BD Pharmingen) and TER119-PE (BD Pharmingen) and sorted into R1–R5 populations according to the method described by Zhang et al. (2003) using an Influx Cell Sorter (BD Cytopeia). Greater than 500,000 cells were collected for each population on three separate occasions and used to generate cDNA for qRT-PCR as described above.
ChIP-seq peak overlap analysis
Analysis of ChIP-seq peak overlap for KLF1, GATA1, TAL1, and EP300 was performed according to the method previously described (Tallack et al. 2010) using the data sets of Tallack et al. (2010), Cheng et al. (2009), Kassouf et al. (2010), and ENCODE/Stanford/Yale, respectively. Essentially we considered peaks to “overlap” where the ends of the features were within 500 bp of each other.
ChIP-seq peak to TSS lists and distributions
In order to match-up a KLF1 binding event (peak) with a corresponding function, we paired KLF1 peaks (all peaks or sets of peaks selected based on EP300 co-occupancy) to the closest TSS for differentially expressed genes identified by mRNA-seq. We performed these analyses for the KLF1 “activated” (690 genes) and KLF1 “repressed” (118 genes) sets of genes considering all RefSeq annotated TSSs for these genes. By use of the online Galaxy Genomics Server (http://main.g2.bx.psu.edu/) (Giardine et al. 2005; Blankenberg et al. 2010; Goecks et al. 2010), we started with each of the KLF1 peak centers and searched for the closest TSS in either direction from the appropriate gene list (either “activated” or “repressed”). This allowed us to generate a list of putative KLF1 activation and KLF1 repression genomic interactions including the specific distance of such interaction. By use of these lists, we generated cumulative frequency distributions to gain global insights into KLF1 function. In creating KLF1 activation and KLF1 repression lists and distributions, we performed the same analysis 10 times on randomly selected lists of 690 or 118 (for activation and repression, respectively) genes and generated the mean distribution of these simulated analyses to serve as a control. This analysis allowed us to ascertain whether a proximity relationship between KLF1 binding (ChIP-seq peaks) and gene activation or repression existed or whether the distribution of distances was random. For the EP300 analysis, the KLF1 peaks were separated into two sets, KLF1 only and EP300 shared, and each of these sets was used to determine the closest “activated” TSS and plot the distributions.
Gene Expression Atlas analysis
We used the ArrayExpress Archive (http://www.ebi.ac.uk/arrayexpress/) to download the human gene expression atlas representing 79 different cell and tissue types each in duplicate (E-TABM-145) (Su et al. 2004; Vaquerizas et al. 2009). This study was performed using the HG-U133A GeneChip human genome array (Affymetrix) to survey for tissue specific gene expression. The sample designated “CD71+ early erythroid” was the best indicator of erythroid-specific gene expression. We used these data to look for genes that were broadly expressed at a background level with higher levels of expression in the duplicate “CD71 early erythroid” samples. Expression of the transcription factor Klf3, which is known to follow this pattern served as a good example (Funnell et al. 2007).
GO enrichment analysis
The online GO tool DAVID (http://david.abcc.ncifcrf.gov/) was used to determine enriched GO terms in the KLF1 “activated” and KLF1 “repressed” gene sets (Huang da et al. 2009a,b). We used the Gene Functional Classification tool from the DAVID suite to cluster enriched GO terms (biological process, molecular function, and cellular compartment) with the classification stringency set to medium and all other options default.
Motif enrichment analysis using MEME-ChIP
We performed a motif enrichment analysis using the de novo motif identification tool MEME-ChIP (http://meme.sdsc.edu/) for the KLF1 only and EP300 shared sets of peaks (Machanick and Bailey 2011). Our extended peak regions (described above in “ChIP-seq Peak Overlap Analysis”) served as the input for MEME-ChIP and were “trimmed” to obtain the central 100 bp of sequence for each peak. We used the following parameters: distribution of motif occurrences, zero or one per sequence; number of different motifs, 5; minimum motif width, 4; and maximum motif width, 30.
Data access
mRNA-seq data reported in this study have been deposited into the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE33979. The previously published ChIP-seq data for KLF1 are also available in the same database under accession number GSE20478 (Tallack et al. 2010). The sequences of the novel lncRNAs described here have been deposited into GenBank (http://www.ncbi.nlm.nih.gov/genbank) with the following accession numbers: Lincred1-giant, JQ173108; Lincred1-dwarf, JQ173109; Lincred2, JQ173110.
Acknowledgments
This work was supported by an Australian Research Council Discovery Grant (DP0770471/ACP), an Australian National Health and Medical Research Council Project Grant (APP1030143), and a grant from the Cancer Council Queensland (519718/ACP). We thank Geoff Osborne and Virginia Nink from the Queensland Brain Institute Flow Cytometry Facility for assistance with cell sorting. We thank Kate Stacey, Jasmyn Cridland, and Marcel Dinger for experimental advice. We acknowledge the use of mouse ENCODE project data (EP300 and CTCF ChIP-seq) provided by the laboratories of Michael Snyder (Stanford University) and Sherman Weissman (Yale University) and accessed through the UCSC Data Co-ordination Center.
Author contributions: M.R.T., L.S., G.W.M., S.H., S.V.F., and E.A.G. performed the experiments described in the study. M.R.T. prepared the figures and tables. M.R.T., T.L.B., and A.C.P. designed the study. M.R.T. and A.C.P. wrote the manuscript. All authors contributed to discussion and approved the final manuscript.
Footnotes
-
↵8 Corresponding author
E-mail aperkins{at}mmri.mater.org.au
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.135707.111.
- Received December 1, 2011.
- Accepted July 23, 2012.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported License), as described at http://creativecommons.org/licenses/by-nc/3.0/.








