
Dynamics of enhancer chromatin signatures mark the transition from pluripotency to cell specification during embryogenesis
- Ozren Bogdanović1,
- Ana Fernandez-Miñán1,3,
- Juan J. Tena1,3,
- Elisa de la Calle-Mustienes1,
- Carmen Hidalgo1,
- Ila van Kruysbergen1,
- Simon J. van Heeringen2,
- Gert Jan C. Veenstra2 and
- José Luis Gómez-Skarmeta1,4
- 1Centro Andaluz de Biología del Desarrollo (CABD), CSIC-UPO-JA, 41013 Sevilla, Spain;
- 2Radboud University Nijmegen, Department of Molecular Biology, Faculty of Science, Nijmegen Centre for Molecular Life Sciences, Nijmegen 6525 GA, The Netherlands
-
↵3 These authors contributed equally to this work.
Abstract
The generation of distinctive cell types that form different tissues and organs requires precise, temporal and spatial control of gene expression. This depends on specific cis-regulatory elements distributed in the noncoding DNA surrounding their target genes. Studies performed on mammalian embryonic stem cells and Drosophila embryos suggest that active enhancers form part of a defined chromatin landscape marked by histone H3 lysine 4 mono-methylation (H3K4me1) and histone H3 lysine 27 acetylation (H3K27ac). Nevertheless, little is known about the dynamics and the potential roles of these marks during vertebrate embryogenesis. Here, we provide genomic maps of H3K4me1/me3 and H3K27ac at four developmental time-points of zebrafish embryogenesis and analyze embryonic enhancer activity. We find that (1) changes in H3K27ac enrichment at enhancers accompany the shift from pluripotency to tissue-specific gene expression, (2) in early embryos, the peaks of H3K27ac enrichment are bound by pluripotent factors such as Nanog, and (3) the degree of evolutionary conservation is higher for enhancers that become marked by H3K27ac at the end of gastrulation, suggesting their implication in the establishment of the most conserved (phylotypic) transcriptome that is known to occur later at the pharyngula stage.
During the course of embryonic development, spatio-temporal specificity in gene expression is achieved through the action of cis-acting regulatory DNA. By far, the most widely studied population of these regulatory sequences is transcriptional enhancers. They contain multiple binding sites for a plenitude of transcription factors that can interact with promoters in cis, independently of their genomic location (Tjian and Maniatis 1994; Ong and Corces 2011). Genomic studies performed in various model systems helped in identifying the chromatin environment these elements reside in. Enhancers correlate with the enrichment of histone H3 lysine 4 monomethylation (H3K4me1), are located within DNase hypersensitive sites, and are able to recruit transcriptional activators such as EP300 (also known as p300) and CREBBP (also known as CBP) (Heintzman et al. 2007, 2009; Visel et al. 2009; Kim et al. 2010). Moreover, it has been proposed that enhancers can be divided into distinct functional classes based on their epigenetic makeup. So far, the best predictor of active enhancers is the coenrichment of H3K4me1 and H3K27ac (Creyghton et al. 2010; Hawkins et al. 2011; Rada-Iglesias et al. 2011; Bonn et al. 2012), even though chromatin marks such as H3K4me3 and H3K36me3 have also proven useful in active enhancer annotation (Pekowska et al. 2011; Zentner et al. 2011). Next-generation sequencing (coupled to ChIP or FAIRE) has been described as the method of choice for the identification of gene regulatory elements during embryogenesis (Visel et al. 2009; Blow et al. 2010; Aday et al. 2011) or within the context of human diseases (Gaulton et al. 2010; Sakabe and Nobrega 2010). Indeed, the zebrafish (Danio rerio) model system was recently employed in an H3K4me1/me3-based genome-wide screen to identify putative cis-regulatory features in a vertebrate embryo (Aday et al. 2011). Similarly, a recent study performed on Drosophila embryos provided substantial insights into how chromatin modifications relate to spatio-temporal enhancer activity (Bonn et al. 2012). Notwithstanding these considerable efforts to deduce the genomic cis-regulatory logic within a developmental context, not much is known on how many of these sequences are actually active during different stages of vertebrate embryogenesis and what their functions are in vivo. Using the ChIP-seq approach, we have generated genomic tracks of H3K4me3, H3K4me1, and H3K27ac histone modifications at four developmental time-points of zebrafish embryogenesis. Based on these genomic signatures, we have identified stage-specific collections of putative distal regulatory elements (PDREs). Stable transgenesis experiments demonstrate that the majority of PDREs act as transcriptional enhancers in vivo. Within the identified PDREs, we find sites displaying significant differences in H3K27ac enrichment between subsequent developmental stages. These differentially acetylated regions associate with genes involved in stage-specific developmental processes and provide binding sites for pluripotency factors and tissue-specific transcriptional regulators during early and late embryogenesis, respectively. Indeed, we show that the pluripotent factor Nanog binds to sites of H3K27ac enrichment in the early embryo and that stage-specific H3K27 acetylation correlates with temporal expression intensities of nearby genes. In total, we have examined the embryonic epigenomes associated with early zebrafish development and generated a collection of stage-specific enhancer elements that modulate the embryonic transcriptome. In addition to expanding on the prior knowledge derived from cell culture research, this study will contribute to the identification of gene regulatory networks associated with early developmental processes.
Results
Identification of putative distal regulatory elements in the zebrafish genome
To interrogate the genomic landscape of putative regulatory elements in zebrafish embryos, embryonic chromatin originating from dome, 80% epiboly, 24 h post-fertilization (hpf) and 48 hpf embryos (Kimmel et al. 1995) was immunoprecipitated with H3K4me1, H3K4me3, and H3K27ac antibodies (Fig. 1A). The reads obtained from sequencing of immunoprecipitated DNA were then aligned to the D. rerio genome (Ensembl version Zv8) (Supplemental Table S1). H3K4me3, as expected (Santos-Rosa et al. 2002), marked promoters, whereas H3K4me1 and H3K27ac, besides marking promoters, displayed broader profiles in line with their role of open chromatin and enhancer marks (Fig. 1B; Heintzman et al. 2007; Creyghton et al. 2010; Hawkins et al. 2011; Rada-Iglesias et al. 2011). Both H3K4me1 and H3K27ac correlate well with temporal expression patterns and the identified enhancers of genes such as neurog1 (Fig. 1B,C; Blader et al. 2004). Actually, the enhancer mark deposition in some cases precedes transcriptional activation, determined by H3K4me3 enrichment (Fig. 1B). Genomic peaks of H3K4me1/me3 and H3K27ac were identified by the MACS algorithm (Zhang et al. 2008) and validated by ChIP-qPCR (Supplemental Fig. S1). In addition, the comparison of our data with previously published data corresponding to a single-stage zebrafish ChIP (Aday et al. 2011), revealed almost identical genomic patterns for H3K4me1 and H3K4me3 between the two studies (Supplemental Fig. S2). Using a similar approach as described previously (Creyghton et al. 2010), a stage-specific PDRE collection was generated based on the presence of the H3K4me1 signature (Fig. 2A,B; Supplemental Table S2). In agreement with the genomic intersections (Supplemental Fig. S3), we observe a genomic colocalization of H3K4me1 and H3K27ac marks (Fig. 2A). Keeping in mind that H3K4me1 is a constitutive enhancer mark whereas H3K27ac is associated with tissue-specific enhancer activation (Bonn et al. 2012), we do not exclude the possibility that these two marks might mark different embryonic cell populations. When clustered together over stage-specific PDRE collections, H3K4me1 and H3K27ac display similar but not identical dynamics (Supplemental Fig. S4). The number of PDREs identified at the dome (blastula) stage is considerably (∼10-fold) smaller than the number of PDREs identified at other stages, possibly because of reduced tissue complexity at blastula stages (Fig. 2B). Upon closer inspection of the PDREs, a substantial shift in PDRE size was discovered; dome-specific PDREs are more than twofold wider than PDREs identified at the 80% epiboly stage, followed by further decrease in size at 24 hpf and 48 hpf (Fig. 2C). These temporal changes in PDRE size suggest a relatively open and permissive regulatory landscape in early embryos. A significant proportion of PDREs is conserved among other teleost species such as stickleback, whereas this conservation is reduced, but still present, in humans (Fig. 2D). To establish a link between identified PDREs and developmental processes, a merged collection of PDREs from all four examined stages was mapped to their closest genes, and the distribution of PDREs around genes with gene ontology related to development was compared to randomly selected genes (Fig. 2E). The developmental genes were significantly (Kolmogorov-Smirnov test, P-value < 0.00001) enriched for PDREs when compared to their random controls, supporting a role for PDREs in developmental processes.
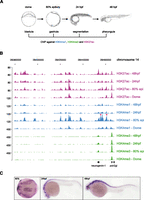
Genomic locations of H3K27ac, H3K4me1, and H3K4me3. (A) A schematic representation of zebrafish early development. H3K4me1/me3 and H3K27ac were immunoprecipitated at the described stages. (B) UCSC Genome Browser view of H3K27ac, H3K4me1, and H3K4me3 tracks obtained for four zebrafish developmental stages: dome, 80% epiboly, 24 hpf, 48 hpf. (Red asterisks) Previously described neurog1 enhancers. (C) Endogenous expression pattern of neurog1 at 80% epiboly, 24-hpf, and 48-hpf zebrafish embryos driven by H3K4me1/H3K27ac-marked transcriptional enhancers. The transcript is localized along the central nervous system (forebrain, midbrain, hindbrain, and spinal cord).
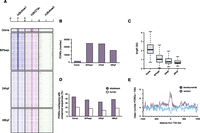
Characteristics of identified putative distal regulatory elements. (A) Heat maps showing the distribution of H3K4me1/me3 and H3K27ac tags −5kb/+5kb relative to the PDRE center at four developmental stages. (B) The number of PDREs identified at dome (blastula) stage is considerably lower than the number of PDREs identified at 80% epiboly or 24/48 hpf, in line with reduced tissue complexity of blastula embryos. (C) Box plots showing the distribution of PDRE length at four examined developmental stages. The PDREs identified at dome stage are considerably larger than the ones identified at subsequent stages, possibly due to a more relaxed chromatin conformation present during early embryogenesis. (D) Conservation of identified PDREs as judged by genomic overlaps with regions conserved to teleosts (sticklebacks) and humans. Approximately 40% and 20% of PDREs overlap with regions conserved in sticklebacks and humans, respectively. (E) The identified PDREs are significantly enriched (Kolmogorov-Smirnov test, P-value < 0.00001) for developmental genes, indicative of a role these sequences might play in early development.
Differentially H3K27-acetylated regions provide binding sites for stage-specific transcription factors
Following the observation that deposition of H3K27ac is more dynamic than that of H3K4me1 (Creyghton et al. 2010; Rada-Iglesias et al. 2011; Bonn et al. 2012), we decided to identify regions displaying significant changes in H3K27ac enrichment between subsequent developmental stages. These differentially acetylated regions (DARs) were identified using Fisher's exact test (for details, see Methods) and visualized by k-means clustering (k = 3) (Supplemental Fig. S5A–D; Supplemental Tables S3–S5). The first two clusters (Supplemental Tables S6–S8) correspond to regions either gaining or losing H3K27ac between two subsequent developmental stages (Fig. 3A,B; Supplemental Figs. S5, S6). The dome−80% epiboly DARs (Fig. 3A) form two clusters; the first cluster (DAR1) is highly enriched for H3K27ac at the dome stage followed by a loss of this mark at 80% epiboly. H3K4me1 also follows this pattern with, however, a noticeable delay. Cluster 2 corresponds to DARs that gain both H3K27ac and H3K4me1 at 80% epiboly and remain enriched for both marks during early development (DAR2). The 80% epiboly−24 hpf DARs (DAR3 and DAR4) (Fig. 3A) are marked in a similar fashion. In both clusters, the H3K4me1 mark follows the dynamics of H3K27ac and even precedes H3K27ac at 80% epiboly (Supplemental Fig. S6D). The differential enrichment of H3K27ac has been confirmed in a series of independent ChIP experiments and validated by qPCR (Supplemental Fig. S7). In addition, only a small number of DARs was identified for 24-hpf and 48-hpf stages (Supplemental Fig. S5D; Supplemental Table S8). This observation is not surprising since in 24- and 48-hpf embryos, most tissues are already formed and enhancer activity is likely localized to reduced areas of gene expression, making changes in enhancer makeup more difficult to detect in whole embryos. To identify genes potentially regulated by DARs, genomic locations of each cluster were mapped to their nearest genes, and a gene ontology (GO) analysis was performed (Supplemental Fig. S8; Supplemental Tables S9–S12). DARs enriched for H3K27ac at dome but not later were not associated with any specific GO category (Supplemental Table S9). However, DARs that become marked by H3K27ac at 80% epiboly as well as DARs that lose or gain H3K27ac at 24 hpf were highly enriched for developmental processes (Supplemental Fig. S8). Interestingly, DARs that lose H3K27ac at 24 hpf represent fairly specific GO categories associated with developmental processes operating mainly during epiboly stages.
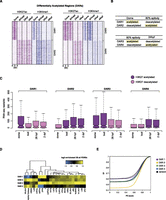
H3K27 differentially acetylated regions. (A) K-means clustering (k = 2) of regions differentially acetylated between subsequent developmental stages. The H3K4me1 mark follows the dynamics of H3K27ac; however, it disappears after H3K27ac has already disappeared (DAR1) (left panel). Similarly to regions differentially acetylated between dome and 80% epiboly, in later DARs, the H3K4me1 mark follows H3K27ac and even precedes H3K27ac on certain genomic locations (DAR4). (B) A schematic representation of different DARs; terms “acetylated” and “deacetylated” correspond to genomic locations identified by Fisher's exact test as differentially enriched for H3K27ac between two subsequent stages. (C) Distribution of gene expression intensities (RNA-seq reads normalized for gene length) displays an overall positive correlation with H3K27ac deposition on enhancer elements at all stages. The H3K27 acetylated DARs are found in the vicinity of highly expressed genes. (D) Hierarchical clustering of motif occurrence in differentially acetylated regions. The frequency of all JASPAR vertebrate motifs in the four DAR clusters was compared to the frequency in all PDREs. All motifs significantly overrepresented (P < 0.05, hypergeometric test in combination with Benjamini-Hochberg correction) in at least one DAR cluster were combined and clustered. (Yellow) Overrepresentation compared to all PDREs; (blue) underrepresentation. (E) Cumulative frequency (Cf) of DARs was plotted against average phastCons (PC) conservation scores that were calculated for each DAR. Each point on the graph represents the frequency of DARs with that of a lower PC score. The frequency of dome (−) 80% epi (+) DARs with a PC score 0 is 0.43, whereas that number is considerably higher (0.59) for dome (+) 80% epi (−) DARs, making them, therefore, less conserved. The black dotted lines correspond to the range of conservation of random DNA.
To test how differential H3K27ac acetylation of putative enhancers influences the expression status of neighboring genes, our H3K27ac data was integrated with previously published expression (RNA-seq) profiles of similar zebrafish developmental stages (Fig. 3C; Pauli et al. 2012). The number of RNA-seq reads was determined for all genes (Ensembl models) that reside in the vicinity of H3K27ac DARs. Indeed, the temporal profiles of H3K27ac display a noticeable positive correlation with gene expression data throughout the early development; the regions marked with H3K27ac at the dome stage (DAR1) associate with genes that are more highly expressed at that stage than other stages where the putative elements are deacetylated (Fig. 3C). A similar positive correlation was observed for other DARs. We next wanted to determine whether the identified DARs differ in the composition of transcription factor binding sites. Sequences corresponding to each cluster were analyzed for the presence of known vertebrate transcription factor signatures (Sandelin et al. 2004). Strikingly, DARs acetylated at dome and 80% epiboly stages but not later (DARs 1 and 3) were found significantly enriched for pluripotency factor motifs such as Pou5f1 and Sox2 as opposed to regions that acquire H3K27ac at 80% epiboly or 24 hpf that were enriched for tissue-specific transcription factor motifs (Fig. 3D; Supplemental Tables S13–S16). Furthermore, we wanted to establish whether DARs display differences in sequence conservation. To that end, we have calculated average conservation scores (phastCons) (Siepel et al. 2005) for each DAR present in each cluster and plotted their cumulative frequencies (Fig. 3E). Whereas the phastCons scores of earliest DARs acetylated in dome fall within the range of values observed for randomly chosen genomic regions, DARs that become acetylated at 80% epiboly and stay marked through the early development, are by far the most conserved ones. The other two groups marked at 80% epiboly and 24 hpf display conservation scores similar to the ones calculated for random genomic DNA. It has been recently described that the most conserved (oldest) zebrafish transcriptomes are the ones expressed during segmentation/pharyngula stages (Domazet-Loso and Tautz 2010; Kalinka et al. 2010). Our results indicate that the cis-regulatory state needed for the expression of such conserved transcriptomes is possibly established at the epiboly stage, prior to segmentation. Altogether, these findings suggest that H3K27ac marks the transition from pluripotency to cell specification and that regions differentially marked by H3K27ac can be divided into distinct classes based on their evolutionary conservation scores and in line with their developmental function.
Nanog binds to putative early enhancers that are deacetylated during gastrulation
As discussed above, DARs acetylated at dome and 80% epiboly (DARs 1 and 3), but not later, were found significantly enriched for pluripotency factor Pou5f1 and Sox2 binding sites. A recent ChIP-seq study of a Nanog-like homolog in zebrafish (Xu et al. 2012) enabled us to further explore this finding and provide more substantial proof that early DARs might be involved in the regulation of pluripotency. In mammals, the Nanog gene codes for a homeobox transcription factor implicated in self-renewal of embryonic stem cells (Chambers et al. 2007). As predicted, we indeed observe an enrichment of the Nanog-like protein in DARs that are acetylated early on (DAR1 and DAR3) and potentially associated with other pluripotent factors (Supplemental Fig. S9A). Furthermore, we observe a direct link between the Nanog-like protein and early H3K27-acetylated enhancers found in the vicinity of pluripotent genes such as pou5f1 (Supplemental Fig. S9B). To reconcile our observations and provide a better insight into the dynamics of enhancer makeup during early development, we have clustered the genomic profiles of Nanog-like protein, H3K27ac and H3K27me3 (Pauli et al. 2012), another mark proposed to label poised and inactive enhancers (Fig. 4A; Supplemental Fig. S10; Rada-Iglesias et al. 2011; Bonn et al. 2012). The clustering was performed over PDREs enriched for H3K4me1 and depleted of H3K27ac at 80% epiboly, as these regions would enable us to detect the proportion of active (H3K27-acetylated) and inactive (H3K27-deacetylated) enhancers before and after this stage. In total, four clusters can be identified. The first cluster (1) corresponds to PDREs marked with H3K27me3 at the shield stage (between dome and 80% epiboly). These PDREs are deacetylated and show no particular Nanog-like enrichment. However, these regions display an increase in H3K27ac at the 24-hpf and 48-hpf stages. The second cluster (2) is potential poised enhancers, marked only by H3K4me1. The third cluster (3) corresponds to PDREs, H3K27-acetylated at the dome stage but not later. These regions are highly enriched in the Nanog-like factor at the dome stage as predicted by the genomic intersections (Supplemental Fig. S9A). Cluster four (4) consists of regions that become acetylated after gastrulation, displaying the highest H3K27ac enrichment at 48 hpf. We next wanted to explore whether the nearby genes of the described PDREs are enriched in certain GO categories (Fig. 2B). Indeed, we find PDREs from the first cluster linked to genes enriched in neural development. The genes belonging to the second cluster display enrichment for processes related to brain development, known to occur in later stage embryos. The third and the fourth cluster are enriched in processes corresponding to early embryos (developmental process, multicellular organismal process) or late embryos (regulation of cell differentiation), respectively. To demonstrate a link between differential PDRE makeup and gene expression, we have obtained expression intensities (Pauli et al. 2012) of nearby genes at corresponding developmental stages (Fig. 4C). We again observe a positive correlation of H3K27ac and gene expression. Accordingly, the genes associated with PDREs marked only by H3K4me1 (cluster 2), displayed lower expression intensities than those marked by both H3K4me1 and H3K27ac. Altogether, we find that only a small proportion of PDREs are marked by H3K27me3, in line with previous observations in human ESCs (Hawkins et al. 2011; Rada-Iglesias et al. 2011). These regions appear to gain H3K27ac during later stages; probably the PDREs marked by H3K27me3 remain marked in that fashion whereas the ones that gain H3K27ac belong to different nucleosomal populations. Finally, we observe a strong coenrichment of H3K27ac and the Nanog-like protein on early developmental enhancers, thereby supporting the notion of early DARs playing a role in pluripotency networks.
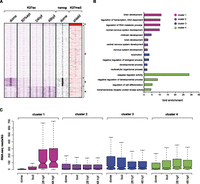
Clustering of H3K27ac, H3K27me3, and Nanog over poised (only H3K4me1-marked) enhancers at 80% epiboly. (A) K-means clustering (k = 4) identifies four groups of regulatory elements. The first group is enhancers marked by H3K27me3 that gain H3K27ac as the development proceeds. The second group corresponds to elements that remain poised during early development. The third and the fourth group correspond to elements that gain or lose H3K27ac, respectively. Zebrafish Nanog-like factor displays strong enrichment over early active (H3K27ac) enhancers. (B) GO analysis of nearest neighbor genes associated to each cluster. Only the four entries displaying highest enrichments are shown (P-value cut-off = 0.01). (C) Distribution of gene expression intensities (RNA-seq reads normalized for gene length) associated with each cluster. As previously shown, an overall positive correlation exists between expression intensity and enhancer H3K27ac levels.
Most PDREs function as enhancers in stable transgenic assays
We next aimed to validate the enhancer activity of identified PDREs. We therefore examined the genomic region encompassing zic3 and fgf13a genes. This region was chosen for the following reasons: (1) the zic3 and fgf13a genes are evolutionarily linked in all vertebrates (Keller and Chitnis 2007); (2) this genomic region contains a large number of PDREs with different degrees of evolutionary conservation that could control the expression of zic3, fgf13a or both genes; and (3) both zic3 and fgf13a play essential roles during development. The zinc finger transcription factor encoded by the zic3 gene is associated with pluripotency, heterotaxy, and cardiac and neural tube defects in humans (Grinberg et al. 2004; Ware et al. 2004, 2006; Lim et al. 2007), whereas fgf13 that encodes an intracellular FGF molecule is required for processes such as neuronal differentiation and cardiac conductivity (Nishimoto and Nishida 2007; Wang et al. 2011). We have selected 18 PDREs distributed along 600 kb harboring the zic3 and fgf13a genes (Fig. 5A; Supplemental Table S17) and tested them using the zebrafish enhancer detection (ZED) vector (Bessa et al. 2009) for enhancer activity in stable transgenic assays (Fig. 5B–M). This vector has been specifically designed for enhancer assays and, as such, contains a positive control for transgenesis efficiency (Bessa et al. 2009). Whereas most of these PDREs (12 of 18, 67%) displayed enhancer activity, 33% of them were negative in our enhancer assay (Supplemental Table S16), indicating that either not all H3K4me1 and H3K27ac-marked regions behave as functional enhancers or that the tested enhancers act in concert with other sequences not included in these assays. Most of these PDREs reside within the 300 kb surrounding the zic3 locus, suggesting that they are likely regulating this gene. Accordingly, in stable transgenic embryos, these PDREs activated reporter gene expression in tissues that are more reminiscent of the domains expressing zic3 and not fgf13a (Fig. 5N,O; Sprague et al. 2006). Enhancer activity was independent of the degree of sequence conservation of these PDREs (Fig. 5A). These data support previous observations that H3K4me1/H3K27ac-marked regions contain active enhancers and that such activity can be reproduced in vivo. In addition, we demonstrate that enhancers associated with a certain genomic locus can, by and large, reproduce the expression pattern of the nearby gene.
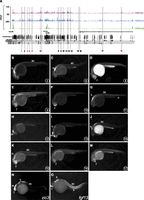
Enhancer activity of PDREs analyzed in stable (F1) zebrafish transgenic lines. (A) Distribution of H3K4me3, H3K4me1, and H3K27ac tracks along 500 kb spanning the zic3 locus at 24 hpf. Shaded in gray are 18 PDREs assayed for enhancer activity in zebrafish transgenic embryos. These PDREs show different degrees of evolutionary conservation as indicated by the conservation tracks below. Out of 18 tested regions, six regions did not exhibit enhancer activity (red numbers). (B–O) Lateral views of 24-hpf zebrafish embryos. (B–M) GFP expression driven by the PDREs indicated in the lower right corner of each panel. (N,O) Expression patterns of zic3 and fgf13a genes at the same stage. Forebrain (f); midbrain (m); hindbrain (h); otic vesicle (ov); spinal cord (sc), notochord (n) and somites (s).
Discussion
In the present study, we set out to investigate the regulatory landscapes associated with early vertebrate development. Recent reports have addressed the dynamics of chromatin marks associated with enhancer elements during stem cell differentiation (Creyghton et al. 2010; Hawkins et al. 2011; Rada-Iglesias et al. 2011). However, it is still unclear how the identification of stem cell enhancers can be extrapolated to mammalian/vertebrate development. Even though such studies provide valuable insights into the mechanisms of cis regulation, their in vivo significance remains to be determined. For example, ES cells are derived from the inner cell mass (ICM), which is one of the two products of the first lineage differentiation event in mammals. This event is accompanied by the asymmetric deposition of the H3K27me3 mark (Dahl et al. 2010). In addition, it has been demonstrated that the very process of ESC derivation causes robust changes in both H3K4me3 and H3K27me3 methylation (Dahl et al. 2010). Similarly, differences in transcriptional programs between ES cells and ICM outgrowths have also been detected by RNA-seq profiling (Guo et al. 2010; Tang et al. 2010). Taken together, these findings suggest that ES cells and the embryonic tissues they are derived from differ in their modes of transcriptional regulation as well as in their intrinsic pluripotency. Studies performed on early vertebrate embryos such as those of zebrafish (Danio rerio), might therefore provide less biased insights into the mechanisms of transcriptional regulation associated with early development.
Our genome-wide analysis of H3K4me1 and H3K27ac at four different stages allowed us to identify ∼50,000 potential cis-regulatory elements operating during the first 48 h of zebrafish development. This number is similar to the total number of enhancers found in nine different cell types in humans (Ernst et al. 2011). In contrast to the relatively high number of potential cis-regulatory elements found in gastrula (24,600), 24-hpf (24,000), and 48-hpf (15,000) embryos, we identify only 2000 regions at the blastula stage. This demonstrates that the high complexity of gene expression associated with the transition from blastula to gastrula, necessary to generate multiple cellular types, is driven by a robust increase in cis-regulatory activity. Interestingly, we also find that the most conserved cis-regulatory elements are those that are turned on during the transition from blastula to gastrula. Our results correlate well with the degree of trans-vertebrate transcriptome conservation during development (Domazet-Loso and Tautz 2010; Irie and Kuratani 2011); transcriptome conservation is higher during vertebrate segmentation and minimal at the blastula stages, supporting the “hourglass” model of developmental evolution, which states that maximum morphological similarity between vertebrates occurs at the phylotypic stage. Our results suggest that the regulatory state necessary to achieve such a conserved transcriptome at the phylotypic stage can be detected somewhat earlier, during late gastrulation, by means of the most evolutionary conserved sets of potential cis-regulatory elements.
We hereby provide a powerful resource that will allow for the dissection of gene regulatory networks, in which genes that operate during different embryonic stages are immersed. Our stable zebrafish transgenics demonstrate that most of the regions displaying H3K4me1 and H3K27ac histone marks are associated with enhancers. Nevertheless, one third of the H3K4me1/H3K27ac-marked regions assayed exhibited no signs of enhancer activity. These sequences could be promoter-specific enhancers or enhancers that act in concert with other sequences not tested in our transgenic assays. Alternatively, H3K4me1/H3K27ac may also be associated with other types of cis-regulatory regions. Furthermore, we observe that H3K4me1 and H3K27ac largely overlap but that their dynamics of deposition and removal are somewhat different. Thus, as previously reported in mammalian stem cells and Drosophila embryos (Creyghton et al. 2010; Hawkins et al. 2011; Rada-Iglesias et al. 2011; Bonn et al. 2012), H3K4me1 deposition precedes that of H3K27ac at many sites. These regions likely correspond to poised enhancers, as proposed for mammalian stem cells (Creyghton et al. 2010; Hawkins et al. 2011; Rada-Iglesias et al. 2011). However, these studies have shown contradictory results on whether H3K27me3 is a mark of poised or inactive enhancers (Hawkins et al. 2011; Rada-Iglesias et al. 2011; Bonn et al. 2012). Here, we demonstrate that H3K27me3 can only be found in a small fraction of H3K4me1 positive/H3K27ac negative regions. These regions seem to acquire H3K27ac along the developmental period examined, in line with their proposed role for poised enhancers. However, it is not clear whether H3K27ac deposition occurs in the same cells where these regions were previously marked by H3K27me3. It is therefore still plausible that H3K27me3-marked regions may correspond to inactive enhancers, as previously reported in Drosophila (Bonn et al. 2012). Nevertheless, we were not able to detect the H3K27me3 mark on early active enhancers (H3K27ac/Nanog) during later stages, even though these regions lost H3K27ac. One explanation could be that the H3K27me3 deposition requires more time. If so, the H3K27me3-marked subgroup of enhancers could represent very early enhancers that are already silenced at the gastrula stage. Our results also indicate that H3K4me1 precedes H3K27ac and that the removal of H3K4me1 is somewhat delayed when compared to the removal of H3K27ac. These data could be compatible with a temporal scenario in which poised enhancers marked by H3K4me1 acquire H3K27ac when activated and lose this mark when inactivated. Some of these enhancers could be further inactivated in a more permanent way by the Polycomb complex causing the deposition of H3K27me3. However, the confirmation of this scenario will require further studies.
The unbiased analysis of our data precisely identifies cis-regulatory elements, and their potential target genes, associated with the transition from a pluripotent state, at the blastula stage, to more committed states during gastrulation and organogenesis. Enhancers that lose H3K27ac at blastula and gastrula stages are enriched for pluripotent factor binding sites, and the majority of them actually overlap with genomic peaks of the Nanog-like factor (Xu et al. 2012) at blastula stages. This strongly suggests that Nanog and other pluripotent factors regulate these regions during pluripotency stages and that these enhancers probably need to be inactivated in order to facilitate developmental progression. Indeed, these early enhancers flank pluripotent genes and genes involved in early developmental processes such as germ layer formation, in which some degree of pluripotency is still maintained. On the contrary, the enhancers that incorporate H3K27ac at gastrula stages or later are associated with genes that control pattern formation. Moreover, these enhancers display a significant enrichment in tissue-specific transcription factor signatures and are likely participating in the acquisition of more differentiated states. We therefore expect that the coordination between processes that lead to the deposition of H3K27ac at some enhancers and the removal of that mark in other enhancers constitute an essential mechanism for the transition of pluripotency to cell differentiation.
Methods
ChIP-seq
Chromatin immunoprecipitation (ChIP) was performed following the protocol described in Wardle et al. (2006) with minor modifications. For dome, 80% epiboly, and 24-hpf stages, we used 5000, 3000, and 1000 embryos, respectively. The samples were sonicated using the Diagenode Bioruptor device with the following cycling conditions: 15 min high–30 sec on, 30 sec off; 15 min on ice; 15 min high–30 sec on, 30 sec off. The size of sonicated DNA was in the range of 100–500 bp. The anti-H3K4me1 (CS-037-100) and anti-H3K4me3 (pAB-033-050) antibodies were obtained from Diagenode. The anti-H3K27ac (ab4729) antibody was purchased from Abcam. Immunoprecipitated DNA was purified with QIAquick columns (Qiagen). The DNA ends were repaired, and the adaptors ligated. The size-selected (300 bp) library was then amplified in a PCR reaction and sequenced using the Genome Analyzer (Illumina). The sequenced reads were mapped to the reference zebrafish genome (Ensembl version Zv8 or Zv9) with the ELAND (v.2) software.
Peak analysis
Highly enriched regions (peaks) of histone methylation and acetylation were obtained by the MACS (v.1.3.3) algorithm (Zhang et al. 2008) using standard settings with one modification (mfold = 20). The genomic intersections of H3K4me1/me3 and H3K27ac peaks were performed using the Galaxy platform with minimum (1-bp) overlap (Blankenberg et al. 2007; Taylor et al. 2007). Randomly selected peaks were verified by Q-PCR and compared to their random controls. The PCRs were performed on 1:50 dilutions of the ChIP samples using the C1000 Thermal Cycler (BioRad). The genomic distribution of H3K4me1/me3 and H3K27ac peaks was calculated using the PinkThing tool (http://pinkthing.cmbi.ru.nl).
Analysis of putative distal regulatory elements
The PDREs were identified using an approach similar to the one described previously (Creyghton et al. 2010). Essentially, H3K4me1 peaks were filtered for H3K4me3-enriched regions and TSS sites (−1kb/+1kb) belonging to RefSeq and Ensembl models. The heat maps were generated by the seqMINER program (Ye et al. 2011) using standard settings. The sequenced reads were mapped to a region spanning −5kb/+5kb from the PDRE center. Box plots were generated in R using default settings. To generate average profiles of PDREs over developmental and random genes, our merged collection of PDREs was mapped to −1Mb/+1Mb surrounding the transcription start site. The bin size was 10 kb. The developmental genes were selected based on the GOA description entry “embryo development” (http://www.biomart.org). To compare distributions of PDREs over developmental and random genes, the Kolmogorov-Smirnov test was used. This test is nonparametric, and it makes no assumption on the distribution of the data analyzed. The results were highly significant (D = 0.5400, P-value < 0.00001). The PDREs corresponding to the Zv9 genome assembly (Supplemental Tables S19–S22) have been generated by converting Zv8 PDRE locations with the UCSC “liftOver” tool (http://genome.ucsc.edu/cgi-bin/hgLiftOver).
Analysis of H3K27 differentially acetylated regions
The differentially acetylated regions were identified using Fisher's exact test (Q-value cut-off = 0.05). For each PDRE, a 2 × 2 contingency table was constructed. The number of reads mapped to a PDRE and the number of total sequenced reads were compared between two subsequent developmental stages. These DARs were then filtered for regions with greater than fourfold differences in read counts. K-means clustering (k = 3) was performed by the seqMINER program (Ye et al. 2011) using standard settings. The sequenced reads were mapped to a region spanning −5kb/+5kb from the DAR center. Entries belonging to clusters containing DARs with low read counts (cluster 3) were eliminated from further analysis, whereas the clusters with both significant and abundant changes (clusters 1 and 2) were analyzed further. The selected DARs were then mapped to their nearest genes using the PinkThing tool (http://pinkthing.cmbi.ru.nl). Gene ontology analysis was performed using the Ontologizer program (Bauer et al. 2008). For the GO analysis, the Parent-Child union method was used in combination with the Benjamini-Hochberg multiple testing correction and a corrected P-value threshold was set at 0.01. The motifs were obtained from the JASPAR vertebrate motif database (Sandelin et al. 2004). For the motif analysis, motifs enriched in every cluster were compared to the entire collection of PDREs by a hypergeometric test, followed by a Benjamini-Hochberg multiple testing correction (P-value cut-off = 0.05). Hierarchical clustering of motifs significantly enriched in at least one cluster was performed in R. For the evolutionary analysis of DARs, the vertebrate phastCons track was obtained from the UCSC repository. The values ranging from 0 = nonconserved to 1 = highly conserved, were assigned to each nucleotide in each DAR, and the mean phastCons value was calculated. Different DARs were validated by qPCR. The PCRs were performed on 1:10 dilutions of ChIP samples using the C1000 Thermal Cycler (BioRad). For primers, see Supplemental Table S18. The DARs corresponding to the Zv9 genome assembly (Supplemental Tables S23–S25) have been generated by converting Zv8 DAR locations with the UCSC “liftOver” tool (http://genome.ucsc.edu/cgi-bin/hgLiftOver).
Integration of ChIP-seq and RNA-seq data
To integrate H3K27ac and H3K4me1 ChIP-seq data with previously published RNA-seq profiles (Pauli et al. 2012), genomic locations corresponding to clusters of differentially acetylated regions or putative distal regulatory elements were converted from the zebrafish Zv8 to Zv9 assembly using the UCSC “liftOver” tool (http://genome.ucsc.edu/cgi-bin/hgLiftOver). The RNA-seq reads were then mapped to the nearest gene corresponding to each genomic location. The y-axis represents the number of mapped RNA-seq reads divided by gene length (kb).
Zebrafish transgenesis
All PDREs were amplified by PCR from the zebrafish genome (Supplemental Table S17). The PCR fragments were subcloned in the PCR8/GW/TOPO vector and, using Gateway technology, transferred to the corresponding ZED vector (Bessa et al. 2009). Zebrafish transgenic embryos were generated using the Tol2 transposon/transposase method (Kawakami 2004) with minor modifications. One-cell embryos were injected with: 2 nl of 25 ng/μl of transposase mRNA, 20 ng/μl of phenol/chloroform-purified ZED constructs, and 0.05% phenol red solution. Three or more independent stable transgenic lines were generated for each construct. All animal experiments were conducted following the guidelines established and approved by the local government and the Institutional Animal Care and Use Committee, always in accordance with best practices outlined by the European Union.
Zebrafish in situ hybridization
Antisense RNA probes were prepared from cDNA using digoxigenin or fluorescein (Boehringer Mannheim) as labels. Zebrafish specimens were prepared, hybridized, and stained as described before (Jowett and Lettice 1994).
Data access
The ChIP-seq data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession no. GSE32483.
Acknowledgments
We thank F. Casares, Miguel Manzanares, and Jaime Carvajal for helpful discussions and comments on the manuscript. We thank the Spanish and Andalusian Governments for grants (BFU2010-14839, CSD2007-00008, and Proyecto de Excelencia CVI-3488) for funding this study.
Footnotes
-
↵4 Corresponding author
E-mail jlgomska{at}upo.es
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.134833.111.
- Received November 14, 2011.
- Accepted May 7, 2012.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported License), as described at http://creativecommons.org/licenses/by-nc/3.0/.








