
Genome-wide detection of novel regulatory RNAs in E. coli
- 1Department of Ecology and Evolutionary Biology, Yale University, New Haven, Connecticut 06516, USA;
- 2Microbial Diversity Institute, Yale University, New Haven, Connecticut 06516, USA;
- 3Howard Hughes Medical Institute, Section of Microbial Pathogenesis, Yale School of Medicine, New Haven, Connecticut 06516, USA
Abstract
The intergenic regions in bacterial genomes can contain regulatory leader sequences and small RNAs (sRNAs), which both serve to modulate gene expression. Computational analyses have predicted the presence of hundreds of these noncoding regulatory RNAs in Escherichia coli; however, only about 80 have been experimentally validated. By applying a deep-sequencing approach, we detected and quantified the vast majority of the previously validated regulatory elements and identified 10 new sRNAs and nine new regulatory leader sequences in the intergenic regions of E. coli. Half of the newly discovered sRNAs displayed enhanced stability in the presence of the RNA-binding protein Hfq, which is vital to the function of many of the known E. coli sRNAs. Whereas previous methods have often relied on phylogenetic conservation to identify regulatory leader sequences, only five of the newly discovered E. coli leader sequences were present in the genomes of other enteric species. For those newly identified regulatory elements having orthologs in Salmonella, evolutionary analyses showed that these regions encoded new noncoding elements rather than small, unannotated protein-coding transcripts. In addition to discovering new noncoding regulatory elements, we validated 53 sRNAs that were previously predicted but never detected and showed that the presence, within intergenic regions, of σ70 promoters and sequences with compensatory mutations that maintain stable RNA secondary structures across related species is a good predictor of novel sRNAs.
Small regulatory RNAs function in the transcriptional and post-transcriptional control of gene expression in organisms from all domains of life. Unlike protein-coding regions, which are specified by a genetic code, regulatory RNAs, as a group, have no clear-cut signatures that denote their boundaries or even their occurrence in a genome. In enteric bacteria, which includes species for which the most comprehensive information is available, these regulatory elements are typically on the order of 50 to 200 nt in length, can act in cis or trans, and have been shown to control a variety of processes, including stress responses, metabolic reactions, and pathogenesis (Romby et al. 2006; Lee and Groisman 2010; Mandin and Gottesman 2010; Park et al. 2010). Moreover, regulatory RNAs have been detected in a wide variety of non-enteric species, including Pseudomonas aeruginosa (Livny et al. 2006), Helicobacter pylori (Sharma et al. 2010), and Vibrio cholerae (Liu et al. 2009).
Since their initial characterization (Hindley 1967), the repertoire of bacterial small RNAs (sRNAs) has been expanding (Wassarman et al. 1999). About 80 sRNA transcripts have been experimentally verified in Escherichia coli; however, computational methods suggest the presence of hundreds of other sRNAs within its genome. These computational predictions have been based largely on (1) formation of stable RNA secondary structures, (2) proximity to σ70 promoters and Rho-independent terminators, and (3) conservation across species (Vogel and Sharma 2005). Each of these methods has defined somewhat different sets of putative sRNAs. However, because there is no consistent model of sequence evolution for these elements, it is difficult to assess the accuracy of these predictions without experimental validation. Thus, many of the computationally predicted noncoding elements may not be authentic.
In addition to sRNAs, bacteria contain regulatory elements within the 5′-leader regions of several mRNAs (Tucker and Breaker 2005; Smith et al. 2010). These regulatory elements (such as riboswitches) control transcription elongation, mRNA stability, and initiation of translation in response to specific stimuli (Coppins et al. 2007). In E. coli, eight experimentally validated riboswitches and three putative riboswitch-like elements are known (Griffith-Jones et al. 2003). But, similar to sRNAs, regulatory leaders are difficult to identify based on primary sequence conservation alone. Covariance models that detect RNA secondary-structure conservation can circumvent this constraint, but they generally detect only those elements conserved in a large number of bacterial species (Yao et al. 2007).
In this study, we interrogate the transcriptome of E. coli by applying a deep sequencing technology that overcomes many of the technical limitations of previous experimental approaches (i.e., low expression levels and the small size and/or complex secondary structures of sRNAs that make them poor substrates for microarrays, Northern blots, copurification, and cloning techniques). This method requires no prior knowledge of sequence or structural conservation and offers a powerful means to identify novel regulatory RNAs in bacteria (Sittka et al. 2008). Moreover, it allowed us to detect and quantify transcripts of all known E. coli sRNAs, detect riboswitch-mediated transcription termination, and identify several new sRNAs and 5′ leaders that were not previously recognized by any experimental or computational approach. In addition, we experimentally validate numerous computationally predicted sRNAs, thereby vastly increasing the number of known regulatory RNAs in E. coli.
Results
Interrogation of intergenic transcripts at great depths
To characterize the transcriptome of wild-type E. coli, we obtained a total of 62.4 million 36-nt reads from cultures harvested during exponential phase in N-minimal media supplemented with 10 mM or 10 μM MgCl2, which are repressing and inducing conditions, respectively, for the PhoQ/PhoP two-component system (Groisman 2001). Of these reads, a total of 61.7 million were of sufficient quality to be mapped onto the E. coli K-12 MG1655 genome. Structural RNAs typically account for the vast majority of RNAs in a cell, but on account of the mechanical removal of 16S and 23S rRNAs, these sequences represented <3% of the total reads in our sample. In contrast, 5S rRNAs and tRNAs, which were not excluded because they overlap in length with known sRNAs, constituted 68% and 6% of the reads, respectively. Importantly, ∼4.3 million reads mapped back to intergenic sequences, indicating that the approach could identify even low-abundant transcripts from these regions.
The remaining 10.3 million reads corresponded to transcripts originating from annotated open reading frames (ORFs) and known noncoding sRNAs. The size-selected libraries were dominated by transcripts shorter than 330 nt in length—there was 70× higher coverage for ORFs in this size class—but even with this enrichment, we detected transcripts from ∼97% of annotated ORFs regardless of their lengths, similar to that shown previously by Selinger et al. (2000).
Validation of transcripts mapped to intergenic regions and identification of Pho- and Mg2+-dependent sRNAs
To test the accuracy and specificity of our assays, we analyzed (1) expression of the sRNA mgrR and (2) transcription elongation controlled by the leader region of the magnesium transporter mgtA gene in wild-type and phoP-deleted strains of E. coli grown both at high and low Mg2+ concentrations (for complete read stats, see Supplemental Table S1). As predicted by the PhoP dependence of these transcripts, and in agreement with previous reports (Cromie et al. 2006; Moon and Gottesman 2009), we observed high expression of mgrR at low Mg2+ concentrations in a PhoP-dependent manner and increased transcription of the mgtA coding sequence in a PhoP- and Mg2+-concentration-dependent manner (Supplemental Table S2).
In addition to mgrR (Moon and Gottesman 2009), we identified four additional sRNAs that appear to be under the control of the PhoP/PhoQ two-component system (Supplemental Table S3). Two of these, isrC and sokX, are repressed in the presence of PhoP, whereas the other two, glmY and gcvB, are expressed at higher levels in the wild-type strain than in the phoP mutant. Interestingly, the expression patterns of isrC, sokX, and gcvB were independent of Mg2+ levels, suggesting that the PhoP-mediated regulation of these sRNAs responds to an additional cue, as shown earlier for acid-resistance genes (Zwir et al. 2005). Transcript levels of three sRNAs (sraA, eyeA, and ryjB) increased at higher Mg2+ concentrations, but the opposite pattern was observed for rprA. These results show that this approach will effectively detect and quantify sRNAs from E. coli grown in different conditions; however, for the rest of this article, we present data only from the wild-type E. coli MG1655.
Detection of transcription termination by known riboswitches
Riboswitches present in the 5′ ends of some mRNAs are known to regulate the expression of the downstream gene by transcriptional and/or translational control (Barrick and Breaker 2007). We examined the mRNA levels between the leader and the corresponding coding region of eight known riboswitches in E. coli to try to detect transcription elongation control. We expected to detect an accumulation of prematurely terminated transcripts in riboswitches that exert transcriptional control and not in riboswitches that act exclusively by regulating translation. The level of transcription at each riboswitch and its 3′ ORF was quantified by determining the number of reads overlapping the region followed by normalization to both the total number of reads in each library and the length of the region. The average of the values calculated from the two conditions (10 mM or 10 μM MgCl2) is denoted as the Mean Expression Value (MEV). Except for the riboswitch upstream of the lysC gene, all riboswitches displayed substantially higher levels of transcripts in the leader region when compared to the downstream coding region (Fig. 1). We determined that in E. coli, all three TPP riboswitches control transcription elongation with the riboswitch 5′ of thiC mRNA producing more prematurely terminated transcripts than the TPP riboswitches upstream of thiM and thiB (Fig. 1), when grown in defined media. This observation is in agreement with earlier studies that showed that the thiC riboswitch is a much stronger attenuator of transcription and translation than the thiM riboswitch (Winkler et al. 2002a). Similar to the transcription termination observed in this study, the Mg2+-sensing riboswitch has been shown to terminate transcription of mgtA in Salmonella at high Mg2+ concentrations (Supplemental Table S2; Cromie et al. 2006).

Transcription termination at known riboswitches. (A) Ratios of mean expression values (MEV) of upstream leader regions to those of their corresponding ORFs. Positive values show that the MEVs of riboswitches are markedly higher than the MEVs of their downstream coding regions. (B) Expression profiles of leader sequences and downstream coding regions. The y-axes denote coverage at each nucleotide position, limited to a maximum coverage of 1000 (dashed lines). Numerical positions of transcription start sites (black arrows) follow the coordinates for the E. coli K12 genome (NC_000913.2). Wide gray arrows on x-axes, depicting ORFs, are not drawn to scale.
The FMN riboswitch is thought to function as a transcriptional attenuator in Bacillus subtilis (Winkler et al. 2002b), and a short transcript (Vogel et al. 2003), presumably ascribed to Rho-dependent transcriptional termination within the 5′-leader sequence of ribB (Peters et al. 2009) has been observed previously in E. coli. The lysine riboswitch (5′ of lysC) is known to be a translational attenuator in Gram-negative bacteria (Serganov and Patel 2009) as opposed to functioning as a transcriptional attenuator in Gram-positive bacteria, which might account for the lack of transcription termination control in E. coli. The riboswitches upstream of btuB and moaA genes regulate translation by blocking ribosome-binding sites (Nahvi et al. 2002; Regulski et al. 2008). However, similar to our results, transcription termination within riboswitches has been observed even in the absence of sequences resembling classical intrinsic transcriptional terminators (Irnov et al. 2010; Livny and Waldor 2010). This suggests the presence of noncanonical intrinsic terminators or the involvement of the transcription factor Rho in the control of these riboswitches (Peters et al. 2009). From our results, it is clear that RNA-seq is a useful and sensitive method to identify leader sequences that regulate transcription elongation.
Identification of novel regulatory leader regions
We selected 28 transcripts that appeared to originate at least 100 nt upstream of the 3′ ORF in order to identify new cis-acting regulatory sequences that affect mRNA levels. By examining the MEVs of the 5′-leader regions and the 3′ ORFs, we found nine leader regions that have MEVs at least seven times greater than the MEVs for their 3′ ORFs (Fig. 2; Table 1), a pattern similar to that of known riboswitches. We next used RNAz (Gruber et al. 2007), which identifies RNA structures based on both structural conservation and thermodynamic stability, to detect potential regulatory elements within these nine transcripts. RNAz predicted the presence of functional RNAs in five of the transcripts (Table 1), whereas there was no evidence of conserved regulatory elements in the leader sequences of yahM, ybjM, ynaE, and ydfK. Each of these leader sequences displayed a dramatic reduction in the number of reads mapped to its 3′ ORF when compared to its 5′ leader region (Fig. 2), suggesting that the expression of their corresponding coding regions is regulated at the level of transcription elongation/termination.
New regulatory leader sequences in E. coli identified by RNA-seq
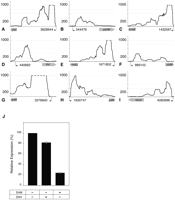
Transcription termination at putative regulatory leader sequences. (A–I) Expression profiles of leader sequences and downstream coding regions. The y-axes denote coverage at each nucleotide position, limited to a maximum coverage of 1000 (dashed lines). Putative transcription start sites (black arrows) and their locations in the E. coli genome (NC_000913.2) are shown on x-axes. Lengths of the corresponding ORFs (wide gray arrows) are not drawn to scale. (J) In vitro transcription of thiI coding region by T7 RNA polymerase in the presence of 100 μM S-adenosylmethionine (SAM) or S-adenosylhomocysteine (SAH) relative to its expression in the absence of SAM and SAH. Data represent means of three experiments ± standard deviations.
Many genes involved in sulfur metabolism are known to carry S-adenosylmethionine (SAM)–responding riboswitches (Weinberg et al. 2008). Because thiI is a sulfur transferase and its leader sequence regulates gene expression in Vibrio cholerae (Livny and Waldor 2010), we tested the ability of the thiI leader sequence to control transcription elongation in an in vitro system. As shown for other SAM-responding riboswitches (Winkler et al. 2003), we found that the presence of SAM retards transcript elongation into the coding region of thiI by 76% (Fig. 2J). S-Adenosylhomocysteine (SAH), an analog of SAM, reduced transcription only by 18%, demonstrating that the putative riboswitch can discriminate between the two ligands. In contrast, the leader sequence of mdtJ, which transports spermidine, a polyamine synthesized from SAM, did not show transcription control in response to either SAM or SAH.
Because regulatory elements often function only in specific environments, it is possible that the lack of evidence for transcriptional elongation control reflects the particular growth condition and not the absence of a regulatory element. Therefore, we analyzed all remaining intergenic transcripts using RibEx (Abreu-Goodger and Merino 2005), which identifies conserved sequences upstream of orthologous genes in multiple genera. Based on this analysis, we detected riboswitch-like elements upstream of 10 additional genes. We further analyzed these candidate elements using RNAz to determine whether they have the potential to fold into secondary structures that are conserved across genera. We detected conserved structural RNAs upstream of three of the genes (Supplemental Table S4). These 10 elements, together with the nine others recognized by the RNA-seq analysis, yields a total of 19 new leader sequences with potential regulatory functions in the E. coli genome.
Expression levels of known sRNAs
A total of about 3 million reads from wild-type E. coli mapped to the 80 experimentally validated sRNAs (Supplemental Table S5). The level of transcription of each sRNA was quantified by determining its MEV, as described above. We also determined the percentage of nucleotides within each gene containing at least one mapped sequencing-read (PRM, percentage of region mapped). Applying a cutoff of MEV ≥ 1 and PRM ≥ 50% for considering a gene as transcriptionally active, we detected substantial transcript production (MEV ≥ 1 and PRM ≥ 50%) from 78 of the known sRNAs, with 68 of them having an MEV ≥ 2 times higher than either of its 50 flanking nucleotides. The two experimentally validated sRNAs for which transcripts were not detected were (1) sroC, an sRNA element in the gltIJKL operon that was deleted from our laboratory strain during serial culture, and (2) rseX, which reduces levels of the outer membrane proteins OmpA and OmpC when overexpressed from a plasmid; however, it is noteworthy that transcripts from this genomic location have never been detected previously (Chen et al. 2002; Douchin et al. 2006).
Most sRNAs (59/80) have MEVs between 1 and 10,000, with a mean of 821. Ten sRNAs, including the housekeeping 6S RNA and tmRNA, were expressed at MEVs >10,000, whereas 11 sRNAs had MEVs <10. However, similar to what is observed for protein-coding regions, these known sRNAs are expressed at very different levels (Supplemental Fig. S1; Supplemental Table S5).
Detection of novel sRNAs
To uncover novel regulatory elements operating at the RNA level, we analyzed transcripts from every intergenic region (IGR) of E. coli. We determined that the vast majority (3087/3683; 86%) of them are transcriptionally active (at a cutoff of MEV ≥ 1 and PRM ≥ 50%) at the tested growth conditions. Of those, we focused our attention on the 1145 IGRs that were longer than 150 nt, the typical lengths of known sRNAs and riboswitches. We examined the levels of transcription from these IGRs and found 171 to contain substantial transcripts compared to their flanking sequences. After removing IGRs that contained repeat palindromic elements, including RIPs/REPs and ERICs (Wilson and Sharp 2006), 119 IGRs with potential regulatory elements remained (Supplemental Table S6). We discovered that 51 of these had transcripts that originated from sites that are different from the transcription start sites (TSSs) described for the flanking genes in EcoCyc (Keseler et al. 2009).
To identify new sRNAs, we selected 17 transcripts that (1) had good read coverage, (2) originated from previously undefined TSSs, and (3) appeared to terminate within the same IGR when examined on Artemis (Rutherford et al. 2000). The coding strand and boundaries of each of these transcripts were determined by a modified 3′-RACE procedure and by visualization of the transcripts on Artemis. The 3′ ends of seven transcripts extended into the downstream ORF, and hence were considered to be leader regions, whereas the other 10 transcripts, ranging in size from 72 to 255 nt, appear to be bona fide sRNAs (Fig. 3; Table 2). Some of the sRNAs produced multiple bands in the 3′-RACE analysis (Supplemental Fig. S3), presumably due to processing of the transcript, as observed for other sRNAs (Argaman et al. 2001; Vogel et al. 2003). A majority of sRNAs in E. coli are known to require Hfq, an RNA-binding protein, for optimal regulation of target-gene expression (Gottesman 2004). To test whether Hfq stabilizes the putative sRNAs identified in this study, we measured their abundance during exponential growth in both wild-type and hfq-deleted strains of E. coli. As shown in Figure 4, five of these sRNAs were significantly more abundant in wild-type E. coli than in the mutant strain that lacked Hfq. In sum, we were able to identify 10 novel intergenic sRNAs, five of which exhibited higher stability in the presence of Hfq, denoting similarity in their regulation to many of the previously characterized sRNAs in E. coli.
New sRNAs in E. coli identified by RNA-seq
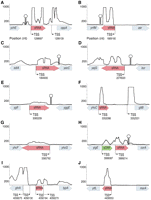
Detection of novel sRNAs in intergenic regions. (A–J) Expression profiles of intergenic regions. The y-axes denote coverage at each nucleotide, limited to a maximum of 1000 (dashed lines). Positions of transcription start sites (TSS) and terminators (stem–loop structures) found within each intergenic region are depicted. Nucleotide positions follow the numbering of the E. coli genome (NC_000913.2). Wide arrows on x-axes contain named ORFs (gray), sRNAs (pink), and an unnamed small ORF (green). Lengths of flanking genes are not drawn to scale.
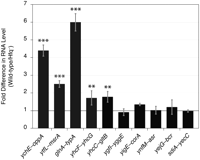
Hfq stabilization of sRNAs. Abundances of sRNAs in wild-type E. coli relative to the amounts in an isogenic hfq-deleted strain (normalized to 1, black line). The intergenic location of each sRNA is indicated on the x-axis. For each sRNA tested, data represent the means of three experiments ± standard deviations. Statistically significant differences from expression in hfq-deleted strain are indicated by (***) p ≤ 0.001 and (**) p ≤ 0.01 (unpaired t-test).
Validating previously predicted sRNAs
Computational methods that predict the presence of sRNAs within bacterial genomes are usually based on sequence conservation, structural homology, and/or the presence of common features such as promoters and terminators. We compared the intergenic transcripts detected by RNA-seq to those predicted computationally in five genome-wide studies (Table 3) and detected substantial transcripts (≥1 MEV, ≥50% PRM, and ≥2× MEV than either of its 50 flanking nucleotides) from 133 candidate sRNAs, of which 58 have not been experimentally validated before (Supplemental Table S7). Thirty-eight of the newly validated sRNAs were recognized in two studies that used locations with σ70 promoter and Rho-independent terminator combinations in IGRs (Chen et al. 2002; Yachie et al. 2006), whereas 19 sRNAs were predicted by identifying IGRs that contained RNAs that maintained their secondary structures across related bacteria by compensatory mutations (Rivas et al. 2001). From these results, we conclude that while the presence of promoter–terminator combinations in intergenic regions is the most reliable predictor of sRNAs, a comparative approach that detects structure-preserving compensatory mutations is also a useful strategy.
RNA-seq-based detection of previously predicted sRNAs
Distinguishing sRNAs from unrecognized ORFs
Because nucleotide substitutions in protein-coding regions that change the amino acid (nonsynonymous) are usually more detrimental than those that do not (synonymous or silent substitutions), the number of synonymous substitutions per synonymous sites (Ks) far exceeds the number of nonsynonymous substitutions at nonsynonymous sites (Ka) in regions that encode functional proteins. Hence, for most proteins, the Ka/Ks ratios are significantly <1, whereas for non-coding regions, this ratio approaches 1 (Ochman 2002). When comparing orthologous sequences from E. coli and Salmonella enterica serovar Typhimurium, the Ka/Ks ratio for mgrB, a small protein-coding gene, is 0.07 in the reading frame, but the ratio is close to 1 in all other frames. Similarly the Ka/Ks ratio for arcZ, a known sRNA that does not encode a protein, is ∼1 in all six reading frames. The Ka/Ks ratios for sgrS and sibA, two sRNAs that encode small proteins, were 0.3 and 0.6, respectively, in the ORFs and approached or exceeded 1 in all the other frames.
To test the possibility that some of newly identified regulatory elements might actually correspond to protein-coding regions, we calculated Ka/Ks ratios for five sRNAs and seven regulatory leaders newly identified in this study, and for 10 previously predicted sRNAs that are present in both E. coli and Typhimurium. For all of the regulatory leader regions and previously predicted sRNAs, Ka/Ks ratios were not significantly different from one in any of the six possible reading frames. In addition, for two of the sRNAs (those present in the yejG-bcr and yhcC-gltB IGRs), Ka/Ks ∼1, indicating that these regions are unlikely to encode proteins. The sRNAs identified in the yigE-corA, glnA-typA, and ytfL-msrA IGRs have Ka/Ks values between 0.1 and 0.2 in one of the potential reading frames, suggesting the presence of small ORFs; but on closer examination, there was no clear presence of start and stop codons, or the putative proteins were interrupted by stop codons in one or the other species. For example, a 15-amino-acid ORF in the yigE-corA IGR of E. coli was disrupted in Typhimurium due to a 2-nt indel, and an 18-amino-acid, serine-rich ORF in Typhimurium in this same IGR was shortened to 10 amino acids in E. coli due to a 1-nt deletion. Similarly, a 43-amino-acid ORF in the glnA-typA IGR of Typhimurium was reduced to a 21-amino-acid ORF in E. coli due to a stop mutation, and a 31-amino-acid putative ORF in the ytfL-msrA IGR in E. coli was shortened to only eight amino acids in Typhimurium due to the deletion of 1 nt. Thus, these IGRs most probably contain new noncoding regulatory elements rather than unannotated ORFs.
Discussion
We identified new regulatory RNAs (10 sRNAs and 19 regulatory leader sequences) and experimentally validated 53 previously predicted sRNAs by RNA-seq. Five of these novel sRNAs were destabilized in cells lacking the RNA-binding protein Hfq, as expected of regulatory sRNAs (Moon and Gottesman 2009), and we further validated one of the newly detected regulatory leader sequences by an in vitro transcription assay. Ka/Ks ratios for those newly identified elements present in both E. coli and Typhimurium indicated that these elements do not encode small protein-coding regions; and by quantifying all previously known sRNAs, we demonstrated that the regulation of many are PhoP- or Mg2+-dependent. These results validate the use of deep sequencing to uncover new regulatory RNAs, especially those that are either not conserved among related bacteria or are expressed at very low levels.
In E. coli, detection of new sRNAs has progressed in concert with experimental techniques that allow the detection of sRNAs expressed at successively lower levels (Table 4). The first sRNAs (6S, 4.5S, spf, ssrA, and rnpB) were discovered after the development of polyacrylamide gel electrophoresis (PAGE) (Ikemura and Dahlberg 1973), and the introduction of Northern blots (Alwine et al. 1977) and, more recently, microarrays (Schena et al. 1995), has increased dramatically the number of known sRNAs to about 80 (Hershberg et al. 2003). Concurrent with these methods, comparative genomics and computational approaches have fostered the identification of large numbers of putative sRNAs (Argaman et al. 2001; Rivas et al. 2001; Chen et al. 2002; Yachie et al. 2006; Tran et al. 2009). However, only a small fraction of the sRNAs that were recognized computationally has been experimentally validated, a disparity usually attributed to the limiting resolution of Northern blots, which had previously been the traditional method for detecting bacterial transcripts. Our application of a high-throughput RNA-seq approach has expanded the known repertoire of sRNAs in E. coli. In addition to detecting transcripts from all but one of the previously known sRNAs, we confirmed the presence of 10 novel sRNAs and 53 sRNAs that were predicted but never experimentally validated.
Detection of low abundance sRNAs by RNA-seq
RNA-seq allows the simultaneous quantification of all known sRNAs in E. coli. A large majority of the sRNA pool (84%) consisted of transcripts corresponding to just 10 sRNAs (Supplemental Fig. S1), with RyhB, a regulator of genes involved in iron metabolism, being the most highly expressed. This sRNA is transcribed in low-iron conditions and promotes the production of siderophores (Massé and Gottesman 2002; Salvail et al. 2010). RyhB has been detected in abundant quantities in minimal media with glucose as the carbon source (Argaman et al. 2001; Wassarman et al. 2001), but not when grown in rich media, which parallels what we observed (Supplemental Table S5). ssrS (6S RNA), the other sRNA expressed at a high level, binds to the RNA-polymerase holoenzyme, down-regulating σ70-dependent transcriptions during stationary phase (Wassarman 2007), and recent studies suggest additional possible regulatory roles during exponential phase (Neusser et al. 2010).
We were able to detect transcripts from nearly all of the 80 known sRNAs; however, 10 of the sRNAs did not reach our cutoff of having MEVs that were appreciably higher than their flanking sequences. This observation is in agreement with previous studies showing that at least seven of these sRNAs are not expressed by E. coli during exponential growth in glucose-supplemented minimal media. These seven sRNAs are the CRP-dependent cyaR, which is repressed during growth in glucose (Johansen et al. 2008; De Lay and Gottesman 2009); rybB and ohsC, which are induced in stationary phase (Vogel et al. 2003; Kawano et al. 2005); istR-2 which is produced during the SOS response (Vogel et al. 2004); rydC, which is minimally expressed during log-phase growth (Antal et al. 2005); DicF, which is not expressed during exponential growth (Bouché and Bouché 1989); and sokA, an antisense sRNA (Fozo et al. 2008), which is a partially deleted pseudogene in E. coli MG1655. By examining wild-type and phoP-deleted strains grown at high and low Mg2+ concentrations, we identified sRNAs whose expression patterns were associated with the presence of PhoP and/or with the amount of Mg2+ in the growth medium, and experiments are under way to understand the physiological significance of these findings.
Five of the newly identified sRNAs are present in significantly higher amounts in wild-type E. coli than in an isogenic strain that lacks hfq (Fig. 4). This conservation suggests that, like other sRNAs (Sledjeski et al. 2001; Massé et al. 2003), these five sRNAs also require Hfq for stability and for optimal function. The sRNA detected in the ychE-oppA IGR overlaps a transcript previously reported to be part of the 5′-leader sequence of the oppA gene (Kawano et al. 2005), but its dependence on Hfq for stability suggests that it acts as a trans-acting sRNA. Of the 10 novel sRNAs, three—the sRNAs detected between ynfM and asr genes, the sRNA detected between the sdiA and yecC coding regions, and the sRNA between yigE and corA—overlap three putative sRNAs predicted computationally based on the occurrence of a σ70 promoter and a Rho-independent terminator (Chen et al. 2002). Additionally, an intrinsic terminator is present at the 3′ end of the sRNA identified between the ygfI and yggE genes (Kingsford et al. 2007). Because Rho-independent terminators were not detected at the 3′ end of the six other novel sRNAs, termination of transcription of these elements may rely on noncanonical intrinsic terminators, be associated with paused RNA polymerase, or use Rho-dependent terminators. Unlike intrinsic terminators, the locations where the Rho protein act cannot be readily identified by sequence analysis, suggesting that experimental approaches (Peters et al. 2009) will be required to reveal the mechanisms by which transcription termination is controlled at these sites.
Putative σ70 promoters are present upstream of all 10 newly recognized sRNAs, and the TSSs determined for eight of the sRNAs match TSSs identified in a recent study that mapped genome-wide RNA polymerase binding sites (Cho et al. 2009). The TSSs we identified for the sRNAs between sdiA and yecC genes, and the sRNA between yejG and bcr genes, were not detected in that study probably because of the differences in growth conditions between the two studies. The sRNA found between glnA and typA genes validates the prediction by Rivas et al. (2001) that this IGR contains an sRNA. In addition, our analyses confirmed 52 other sRNAs that were previously predicted by various computational approaches, thereby vastly increasing the number of experimentally verified sRNAs in E. coli. A majority of these sRNAs (Tables 2; Supplemental Table S7) have associated σ70 promoters, indicating that the presence of a σ70 promoter that does not correspond to a known ORF is the best indicator of novel sRNAs. However, computational methods tend to produce numerous false positives due to low signal strength of bacterial promoters (Mendoza-Vargas et al. 2009). Our analyses also showed that the identification of regions that have undergone compensatory mutations to preserve RNA secondary structures in related organisms is also a good strategy to predict the presence of new sRNAs (Table 3; Supplemental Table S7; Rivas et al. 2001), but this strategy also produces numerous false positives due to the presence of cis-regulatory elements in the untranslated regions of mRNAs (Supplemental Table S8). Hence, by using a high-throughput analysis in conjunction with computational predictions, sRNAs can be identified rapidly at a genome-wide scale.
As with the computational detection of sRNAs, many of the current methods used to identify regulatory elements in the 5′-leader regions of genes also rely on nucleotide sequence or secondary structure conservation (Weinberg et al. 2010). This approach, while effective, will only detect those elements that are maintained across a spectrum of bacterial species. In contrast, by directly detecting transcription termination on a genome-wide scale, we have identified regulatory elements in the leader sequences of numerous genes without the need to consider sequence or secondary structure. This allows the identification of regulatory leader RNAs that control species-specific genes. In addition, this experimental approach has the advantage of both detecting and experimentally validating regulatory leader sequences in a single step.
Most of the new regulatory leader regions detected in this study are restricted to members of the family Enterobacteriaceae. In one case, a suspected regulatory element was reported to occur in the 5′-leader region of the rplM gene in a Gram-positive Firmicute (Yao et al. 2007), and the element we discovered at the 5′ end of the E. coli rplM gene has a very different sequence but similar secondary structure to the one in Firmicutes. Although computational tools did not detect conserved structural RNAs in the 5′-leader regions of the yahM, ybjM, ynaE, and ydfK genes, we observed strong control of transcriptional elongation at each of these sites (Fig. 2). Interestingly, prematurely terminated transcripts were detected from the 5′-leader sequences of the ybjM, ynaE, ydfK, mdtJ, typA, yhiI, and dinQ genes, similar to earlier studies that detected small transcripts from within riboswitches (Vogel et al. 2003; Kawano et al. 2005; Mendoza-Vargas et al. 2009). We were able to identify a putative SAM-responding regulator in the 5′-leader sequence of the thiI gene. Homologs of this thiamine biosynthesis and tRNA modification gene in several Gram-negative bacteria have been reported to contain a conserved motif in their 5′-leader sequences (Livny and Waldor 2010), suggesting that a similar regulatory mechanism might be present in related bacteria as observed in the E. coli thiI leader region. Additionally, we observed an anti-sense RNA, which has been previously reported (Dornenburg et al. 2010) at this genomic region. The other regulatory RNAs identified in this study require further experiments to better understand their modes of action, and newer studies using alternative approaches will be needed to identify those regulatory RNAs that escaped detection due to the size selection (30–330 bp) we performed to generate the sequencing libraries. By quantifying the transcripts from exponentially growing E. coli on a genome-wide scale, we were able to detect several new sRNAs and riboswitch-like regulators yielding new insights into the highly dynamic regulatory RNA pool of E. coli.
Methods
RNA preparation, library construction, and sequencing
E. coli K-12 MG1655 (NC_000913.2) and phoP-deleted strain EG12976 were grown in N-minimal media (pH 7.7) supplemented with 10 mM or 10 μM MgCl2, 0.1% casamino acids, and 0.4% glucose to mid-log phase (OD600 = 0.4), as previously described (Zwir et al. 2005). Cells were chilled on ice and harvested by centrifugation at 4°C. Total RNA was isolated by a modified phenol/guanidine thiocyanate extraction procedure using TRI reagent (ABI) but without filtration steps in order to retain all small RNAs. Samples were rid of genomic DNA by treatment with DNase, and 16S and 23S rRNAs were removed with the MICROBExpress kit (ABI). Sequencing libraries were prepared using the Illumina mRNA-seq sample preparation kit as per the supplier's instructions except that (1) total RNA was not fragmented and (2) double-stranded cDNA was size-selected (100 to 400 bp) to maximize the recovery of small RNAs. After accounting for the added adapters (∼70 bp), this size-selected library is enriched in reads originating from transcripts that are <330 nt in length. Libraries were amplified by 10 cycles of PCR with Phusion polymerase (Finnzymes) and assayed for quality on a Bioanalyser (Agilent Technologies). Each library was diluted to 8 pM and loaded onto a single lane of an Illumina GA flow-cell. Sequencing was performed at the Center for Genome Sciences (Washington University, St. Louis, MO) and the Keck Center (Yale University, New Haven, CT) with a cycle number (read length) of 36. The raw reads from all four sequencing runs have been deposited in the NCBI Sequence Read Archive (accession number SRP006793), and the read stats are available in Supplemental Table S1.
Measuring Hfq dependence
An hfq-deleted strain of E. coli K-12 (JW4130-1) and its isogenic parent strain (BW25113) (Baba et al. 2006) were grown to mid-log phase (OD600 = 0.4) in N-minimal medium, as described above. Total RNA was DNase-treated, and 1 μg was used as a template for preparing cDNA. Quantitative PCR was performed, and the abundances of each sRNA in the wild-type and the hfq-deleted strains were calculated from Ct (threshold cycle) values.
Mapping and visualization of sequence reads
Sequencing reads were plotted onto the E. coli genome using MAQ (Li et al. 2008) allowing up to two mismatches between a 36-nt read and the published E. coli K-12 MG1655 sequence. The number of piled sequences at each base was determined from the out.mapview file using an in-house Perl script that outputs a file with a column showing the coverage at each genomic base. This file was supplied to Artemis (Rutherford et al. 2000) as a graph in order to visualize the sequence coverage at each genomic position.
Transcript quantification
Coordinates of all intergenic regions (IGRs) and protein-coding genes in E. coli MG1655 were downloaded from EcoGene (Rudd 2000). The level of transcription of each region reported in Supplemental Tables S2 and S3 was quantified by determining the number of reads overlapping the region followed by normalization to both the total number of reads in each library and the length of the region. For the rest of the manuscript, expression values determined for the wild-type E. coli from both high and low magnesium conditions were averaged and are denoted as the Mean Expression Value (MEV). MEV cutoffs for designating a region as being expressed above background levels were based on sets of reference genes whose expression levels are known to be (1) absent, (2) very low, or (3) induced when E. coli is propagated in the presence of glucose. For example, the MEVs of both the mhpABCDFE and paaABCDEFGHIJK operons (which enable E. coli to use aromatic acids as carbon and energy sources in the absence of glucose) (Ferrández et al. 2000; Torres et al. 2003) were 0.80. The MEV of the lacAYZ operon, which is suppressed in the presence of glucose (but known to be leaky), was 1.7, whereas that of the lacI inhibitor, which is expressed in the presence of glucose, was 9.9. Furthermore, only 32% of nucleotides within the mhp and paa operons had at least one sequencing-read mapped (PRM, percentage of region mapped) onto it, as opposed to 62% of bases within the lacAYZ operon and 86% within the lacI gene. Based on these results, we applied a cutoff of MEV ≥ 1 and PRM ≥ 50% for considering a region as transcriptionally active.
Discovery of intergenic regions with potential regulatory RNAs
To identify novel intergenic sRNAs and regulatory leader regions, we inspected all IGRs >150 nt in length in the E. coli MG1655 genome (n = 1145). To eliminate reads that overlap the junction between the IGR and a flanking gene, MEVs and PRMs were calculated after excluding the first and last 50 nt of each IGR.
Due to the possibility of read-through from upstream genes, the MEV of each IGR was evaluated in the context of its flanking genes. If flanking genes are divergent (i.e., transcribed in opposite directions away from the IGR), we required the MEV of the IGR to be at least two times higher than the MEV of either of the flanking genes to consider the IGR as being more highly expressed. For IGRs whose flanking genes are transcribed in the same direction, we required the MEV to be at least two times that of each flanking gene to consider the IGR as being more highly expressed. For IGRs whose flanking genes are convergent (transcribed toward one other), we require its MEV to be at least two times higher than the sum of the MEVs of the flanking genes to be considered as being more highly expressed. In each of these cases, the MEV of the IGR is compared to the 50 flanking nucleotides from each neighboring gene.
Detection and quantification of sRNAs
The locations of experimentally verified sRNA genes and other regulatory RNA elements in the E. coli genome were obtained from Rfam and EcoCyc (Keseler et al. 2009). The coordinates for candidate sRNAs that were predicted but not experimentally confirmed in earlier studies were assembled from the original sources (Argaman et al. 2001; Rivas et al. 2001; Chen et al. 2002; Yachie et al. 2006; Tran et al. 2009) and from selab.janelia.org. For predictions that were made using the NC_000913.1 assembly, the coordinates of the candidate sRNAs were mapped to the NC_000913.2 version of the genome prior to RNA-seq analyses. Expression levels of these sRNA genes were determined by calculating the MEVs and PRMs for each predicted sRNA. sRNAs with an MEV that is two or more times greater than that of its 50 flanking nucleotides were considered to be more highly expressed.
Confirmation of coding strand and 3′ ends of candidate sRNAs
We applied a modified RACE procedure (Kawano et al. 2005) to determine the 3′ ends of candidate sRNAs. In brief, total RNA, depleted of 16S and 23S rRNA, was dephosphorylated with alkanine phosphatase (New England Biolabs), and a short oligonucleotide adapter was ligated to 3′ ends using T4 RNA ligase (NEB). The 3′-adapter-ligated RNA was reverse-transcribed using a primer complementary to the adapter, and the resulting cDNA was used as the template in PCR reactions using primers specific to a candidate-sRNA along with an adapter-complementary primer. Amplicons were resolved on 3% low-range ultra agarose (Bio-Rad) gels to determine their lengths. To confirm that candidate sRNAs are not part of transcripts from flanking coding sequences, PCR with primers that anneal to the sRNAs in combination with primers that anneal to flanking ORFs was performed.
Secondary structure conservation analysis
Candidate regulatory RNAs were searched against the Rfam database to confirm that they were not homologs of previously identified elements. Nucleotide sequence alignments were built for RNAs having homologs that were identified by BLAST (E-value ≤1 and ≥50% identity). Sequences were screened with the RNAz program (Gruber et al. 2007) to detect conserved secondary structures.
Prediction of terminators and promoters
Rho-independent terminators were predicted using RNAMotif (Macke et al. 2001) and TransTermHP (Kingsford et al. 2007). PPP (http://bioinformatics.biol.rug.nl/websoftware) and BDGP (Reese 2001) were used to predict promoters.
In vitro transcription assay
The leader and coding regions of the thiI and mdtJ genes were PCR-amplified using primers that introduced a T7 promoter sequence 5′ of the leader sequences. The PCR products were purified using MinElute columns (QIAGEN), and 100 ng was used as template for in vitro transcription using a MEGAscript kit (ABI). In vitro transcription was performed for 30 min in the presence or absence of 100 μM SAM or SAH as indicated in Figure 2J and then treated with Turbo DNase (ABI) for 15 min to remove the DNA templates. cDNA was synthesized from in vitro transcripts and used as templates for qPCR using primers that map to the coding regions of thiI and mdtJ.
Ka/Ks ratios
Homologs in Typhimurium for regulatory RNAs in E. coli were identified by BLAST (E-value ≤1 and ≥50% identity), confirmed by synteny, and aligned by ClustalW. Ka/Ks ratios were obtained with the program KaKs_Calculator (Zhang et al. 2006), which applies a codon-based, maximum-likelihood method (Goldman and Yang 1994). A Fisher's exact test was used to assess the statistical significance of Ka and Ks values.
Data access
The raw reads from all four sequencing runs have been deposited in the NCBI Sequence Read Archive (accession number SRP006793).
Acknowledgments
We thank John McCutcheon, Chih-Horng Kuo, and Patrick Degnan for helpful discussions. We are grateful to the Coli Genetic Stock Center at Yale University for providing the E. coli strains, Kim Hammond and Michelle Dawn for assistance with figures, and the reviewers for helpful comments. This research was supported in part by NIH grants GM74738 and GM56120 to H.O. and by AI49561 to E.A.G., who is an investigator at the Howard Hughes Medical Institute.
Footnotes
-
↵4 Corresponding author.
E-mail howard.ochman{at}yale.edu.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.119370.110.
- Received December 17, 2010.
- Accepted June 8, 2011.
- Copyright © 2011 by Cold Spring Harbor Laboratory Press








