
Unamplified cap analysis of gene expression on a single-molecule sequencer
- Mutsumi Kanamori-Katayama1,
- Masayoshi Itoh1,
- Hideya Kawaji1,
- Timo Lassmann,
- Shintaro Katayama,
- Miki Kojima,
- Nicolas Bertin,
- Ai Kaiho,
- Noriko Ninomiya,
- Carsten O. Daub,
- Piero Carninci,
- Alistair R.R. Forrest2 and
- Yoshihide Hayashizaki2
-
↵1 These authors contributed equally to this work.
Abstract
We report the development of a simplified cap analysis of gene expression (CAGE) protocol adapted for single-molecule sequencers that avoids second strand synthesis, ligation, digestion, and PCR. HeliScopeCAGE directly sequences the 3′ end of cap trapped first-strand cDNAs. As with previous versions of CAGE, we better define transcription start sites (TSS) than known models, identify novel regions of transcription and alternative promoters, and find two major classes of TSS signal, sharp peaks and broad regions. However, using this protocol, we observe reproducible evidence of regulation at the much finer level of individual TSS positions. The libraries are quantitative over 5 orders of magnitude and highly reproducible (Pearson's correlation coefficient of 0.987). We have also scaled down the sample requirement to 5 μg of total RNA for a standard HeliScopeCAGE library and 100 ng for a low-quantity version. When the same RNA was run as 5-μg and 100-ng versions, the 100 ng was still able to detect expression for ∼60% of the 13,468 loci detected by a 5-μg library using the same threshold, allowing comparative analysis of even rare cell populations. Testing the protocol for differential gene expression measurements on triplicate HeLa and THP-1 samples, we find that the log fold change compared to Illumina microarray measurements is highly correlated (0.871). In addition, HeliScopeCAGE finds differential expression for thousands more loci including those with probes on the array. Finally, although the majority of tags are 5′ associated, we also observe a low level of signal on exons that is useful for defining gene structures.
The advent of high-throughput next-generation sequencing has given rise to a plethora of short read methods to profile the transcriptional output of the genome. RNA-seq (shotgun transcriptome sequencing), digital gene expression (DGE), short RNA libraries, and cap analysis of gene expression (CAGE) have all been adapted to the current high-throughput, short read, second-generation sequencing platforms (454 Genome Sequencer FLX System [Roche], Applied Biosystems [Life Technologies] SOLiD, and Illumina) RNA-seq (Cloonan et al. 2008; Marioni et al. 2008; Mortazavi et al. 2008; Nagalakshmi et al. 2008; Sultan et al. 2008; Wilhelm et al. 2008), DGE ('t Hoen et al. 2008; Matsumura et al. 2010), and CAGE (Suzuki et al. 2009; Hestand et al. 2010). These have proven to both outperform microarrays in terms of sensitivity and specificity (Marioni et al. 2008) and especially in the case of RNA-seq and CAGE are finding new transcripts and additional transcriptional complexity (Cloonan et al. 2008).
Despite these advances, second-generation sequencers use PCR amplification to generate clonal populations of molecules restricted to a bead, cluster, or nanowell to provide sufficient template to allow fluorescent-based imaging. The majority of transcriptome protocols also use pre-amplification of the sample at a library stage to generate enough material for easy visualization and loading. These two amplification steps can generate potential biases in the library population and the population of molecules that are actually sequenced. Here we describe HeliScopeCAGE, the adaptation of cap analysis of gene expression to a third-generation sequencer (the first true single-molecule sequencer to market [the HeliScope Genetic Analysis System; Helicos Biosciences]) that completely avoids amplification.
The HeliScope system images the growth of individual DNA molecules using a DNA polymerase- and template-dependent extension from an oligo(dT) primer on the HeliScope flow cell surface (Harris et al. 2008). High-resolution optics means that the system can monitor strand extension on a single molecule without the need of clonal amplification. To date, the HeliScope platform has been used for RNA-seq and DGE (Lipson et al. 2009; Ozsolak et al. 2009, 2010a,b) applications, and most recently used in a survey of small RNAs to identify a putative novel class of 3′ UTR (untranslated region)–associated antisense poly(U) short RNAs (Kapranov et al. 2010) that are thought to be products of an RNA-dependent RNA polymerase activity. They independently confirm their existence using strand-specific Northern blots and RNase protection assays against the antisense RNAs.
Cap-trapping was developed to overcome the problem of partial cDNA sequences and enrich full-length cDNAs (Kawai et al. 2001; Okazaki et al. 2002; Carninci et al. 2005). In the protocol the cap found on 5′ end–complete mRNA molecules is chemically biotinylated to allow streptavidin capture of full-length capped RNAs. cDNA sequences that have completely copied the 5′ end of the starting mRNA are selected for during an RNase treatment step, whereas incomplete cDNAs are removed by cleavage of the single-stranded RNA section tethering the cDNA. This approach was then used to develop a 5′ tag technology, cap analysis of gene expression (CAGE), as a method to globally map transcription start site usage within a biological sample (Shiraki et al. 2003; Kodzius et al. 2004). CAGE was used extensively in FANTOM3 to globally map transcription start sites in mouse tissues (Carninci et al. 2006). With the advent of second-generation sequencers, CAGE was adapted to the Genome Sequencer FLX System (454) and used to study TSS dynamics in a human myeloid leukemia cell line (THP-1) undergoing PMA-induced monocytic differentiation. The location of the signal, proximal predicted transcription factor binding sites, and the level of expression at each TSS were then used to build a transcriptional regulatory network model of the key factors involved in the differentiation (Suzuki et al. 2009).
The 454 version of the protocol, however, requires second-strand synthesis, digestion, linker ligation, concatenation, and multiple rounds of PCR, which significantly adds to the handling of the library and potential for artifacts, both from PCR and from ligation and digestion. The simplified HeliScopeCAGE protocol presented here is the first CAGE protocol to sequence first-strand cDNA, thereby avoiding second-strand synthesis, amplification, ligation, and digestion. It uses random primed cDNA synthesis, cap-trapping of 5′ end complete cDNAs, poly(A) tailing, and direct sequencing of the tailed first-strand cDNA. We demonstrate that HeliScopeCAGE outperforms Illumina microarrays as a robust expression profiling platform and also avoids PCR biases inherent in previous protocols. The protocol also reduces sample requirements from the original 50-μg version to <5 μg (with a low-quantity version demonstrated to work from 100 ng of total RNA). In principle, this protocol is amenable to any new third-generation, single-molecule sequencer that appears on the market in the coming years.
Results
A simplified CAGE protocol for the HeliScope single-molecule sequencer
The original CAGE library protocol involved cDNA synthesis, cap-trapping of 5′ complete cDNA/capped RNA hybrids, second-strand synthesis, linker ligation, full-length cDNA cloning in bacteria, digestion of 5′ tags, and concatenation and subcloning of concatemers prior to capillary sequencing (Shiraki et al. 2003; Kodzius et al. 2004). For the FANTOM4 project, the protocol was adapted to the Genome Sequencer FLX System (454) second-generation sequencer (Suzuki et al. 2009). The adaptation enabled us to sequence more deeply and avoided the bacterial cloning steps. However, the protocol still required linker ligation, digestion, and concatenation, as well as several rounds of PCR amplification at multiple stages (tag amplification, concatemer amplification, 454 clonal amplification).
Here we have developed a new, much simpler CAGE protocol for single-molecule sequencing (Fig. 1). First-strand cDNA is generated from 5 μg of total RNA using an excess of random primer. 5′ end complete first-strand cDNAs are captured for capped RNAs. First-strand cDNA is poly(A)-tailed and blocked and then loaded directly onto the HeliScope flow cell for sequencing. Five micrograms of total RNA typically yields 10–15 ng of HeliScopeCAGE library, of which 20% is typically loaded on the flow cell to generate on average 15 million reads (Supplemental Table 1).
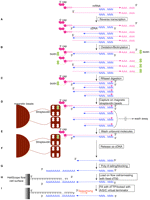
HeliScope CAGE protocol workflow. (A) Reverse transcription. cDNA is synthesized using SuperScript III and random N15 primer. (B) Oxidation/biotinylation. The cap structure is oxidized with sodium peroxide and biotinylated with biotin (long arm) hydrazide. (C) RNase I digestion. Single-strand RNA is digested with RNase I. (D) Capture on magnetic streptavidin beads. Biotinylated RNA/cDNA hybrid molecules are captured using magnetic streptavidin beads. (E) Wash unbound molecules. Unbound RNA/DNA hybrid molecules are washed away. (F) Release ss-cDNA. Captured RNA/DNA hybrid molecules are treated with RNase H and RNase I, then heat-treated. (G) Poly(A) tailing/blocking. Released cDNA is poly(A)-tailed using terminal deoxynucleotidyl transferase and dATP, then blocked with biotin-ddATP. (H) Load on flow cell. Blocked poly(A)-tailed cDNA is loaded on the HeliScope flow cell channel and anneals with the dT 50 surface. (I) Fill with dTTP/locked with A/G/C virtual terminator. After annealing of cDNA, the single-strand poly(A) tail part is filled with DNA polymerase, dTTP, and an A/G/C virtual terminator that is used in HeliScope sequencing to lock the poly(T) termini. The library is then ready for sequencing.
HeliScopeCAGE is a reproducible expression profiling technology linear over 5 orders of magnitude
To assess the reproducibility of HeliScopeCAGE, we generated technical triplicate libraries using 5 μg of total RNA from the human myeloid leukemia cell line THP-1 (Tsuchiya et al. 1980). Each library was run on a single channel of the HeliScope sequencer, and we obtained 12–18 million mappable reads to the reference sequence of the human genome. We confirmed their enrichment to the 5′ end of mRNAs based on the genomic coordinates of the reference full-length transcripts of RefSeq (Maglott et al. 2000). Typically for these and other libraries, we observe ∼50% of the signal is within 500 bp of the 5′ end of a reference transcript in the sense orientation (Fig. 2A). Technical replicates were highly reproducible between libraries (Fig. 2B; Supplemental Fig. 1), where the Pearson's correlation coefficient of gene expression (log2 of tpm, tags per million) between any two libraries is above 0.98. The scatterplot indicates that the dynamic range of this protocol is over 5 orders, from 0.1 to 10,000 tpm with the current depth of sequencing.

HeliScopeCAGE is a highly quantitative reproducible technology. (A) Average distribution of HeliScopeCAGE tags on annotated regions of the genome for the THP-1 and HeLa libraries. (B) Scatterplot of gene expressions between two technical replicates of HeliScopeCAGE on THP-1 RNA (5 μg of total RNA as starting material). The CAGE tag counts mapped within ±500 bp from the RefSeq transcription starting site are normalized as TPM (tags per million) with the library sizes. (C) (i) Gene expressions between different starting materials, 5 μg and 100 ng of total RNA of THP-1. Scatterplots of the two profiles with read counts. (ii) The number of detected genes with each profile. A gene is considered detected when five or more reads are obtained. Note: Given that mRNA is present at ∼1% of total RNA, A indicates a 300–500-fold enrichment of signal at promoters compared to rRNAs.
To facilitate profiling of rare cell populations, we also developed a low-quantity version and tested this on 1 μg, 500 ng, 200 ng, and 100 ng of THP-1 total RNA. A comparison between the genes detected by the 5-μg and 100-ng versions is shown in Figure 2C. Considering only those genes with at least five tags, 8214 genes are detected in common by both the 5-μg and 100-ng versions. This equates to ∼60% of the loci measured at this threshold in the 5-μg version. Extended comparisons between the 5-μg and low-quantity versions are shown in Supplemental Figure 2. The libraries are highly correlated, with a minimum correlation of 0.92 between any two. For the 5-μg and 1-μg versions, we found little difference in the total number of genes detected at 1 tpm or greater (11%—1324 out of 11,635) (Supplemental Fig. 2); however, as we reduced the amount of starting material, weakly expressed genes were lost from the bottom of the profile, reducing the dynamic range from 5 orders for the 5-μg version down to 3.5 orders for the 100-ng version. This means that for the top 8–10,000 mid to highly expressed genes, we can still reliably measure expression and map promoters in samples containing as little as 100 ng without PCR amplification.
Gene expression analysis using HeliScopeCAGE
To demonstrate the utility of HeliScopeCAGE and determine its limitations in gene expression analysis, we also generated a set of triplicate libraries for the HeLa cell line (Scherer et al. 1953) and compared them to the THP-1 profiles. The same RNAs were also applied to Illumina Sentrix 6 version 3 microarrays. The performance of HeliScopeCAGE for detecting differential expression was then assessed by only considering genes that were on the array. Gene expression tables for the THP-1 and HeLa libraries were generated by taking any mapped reads within 500 bases of a RefSeq 5′ end. For comparisons, we took the further more stringent requirement that the gene had to be detected in all three replicates for both THP-1 and HeLa. Using this stringent requirement, 6506 genes were detected by both platforms in all six samples. Plotting fold-change measurements between HeLa and THP-1 assessed by HeliScopeCAGE and microarray showed that they were highly correlated (Pearson's correlation of 0.871) (Fig. 3A). Running differential gene expression analysis with edgeR (Robinson et al. 2009) and limma (Smyth et al. 2005) R packages on the HeliScopeCAGE and the microarray data, respectively, identified sets of genes differentially expressed between THP-1 and HeLa. Using equivalently strict thresholds for significance for both packages (FDR [false discovery rate] < 0.001 [0.1%] for edgeR and a Bstatistic >0 for the microarrays), we identified an overlapping set of 2022 genes detected as differentially expressed by both platforms. A further 323 and 2280 genes were detected as differentially expressed in only the microarray or HeliScopeCAGE protocols, respectively (Fig. 3A; Supplemental Table 2).
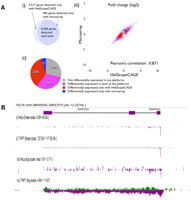
Differential expression using HeliScopeCAGE. (A) Comparison between HeliScopeCAGE and microarray. (i) the number of genes detected in both of THP-1 and HeLa RNA with each platform (detected all of the three technical replicates). (ii) The number of genes detected as differentially expressed. False discovery rate (FDR) <0.001 for HeliScopeCAGE and Bstatistics >0 are used as criteria for the differential expression. (B) Genomic view of a novel Human Endogenous retrovirus (HERV) related transcript highly expressed in THP-1 but not detected in HeLa. (i) and (ii) On linear scale; (iii) and (iv) log scale for HeLa and THP-1, respectively. (Green) Plus strand; (purple) minus strand relative to genome assembly.
Of the genes not detected by both platforms, 3517 were only detected by HeliScopeCAGE. A fraction of these can be explained by the poly(A) status of the transcripts. As the microarrays use an oligo(dT) primer non-poly(A) transcripts are missed. This is most obvious from the 37 members of the histone gene family that are detected at high counts with HeliScopeCAGE but missed by the arrays (Supplemental Table 2). The majority of the differences, however, appear to be due to the detection limits of the microarrays. As CAGE is a digital expression measurement, we can rank the order of expression for each gene and ask whether they are detected by the microarrays or not. Considering the THP-1 replicates, we find that for loci with more than 100 tags per million, 86% are called as detected by all three microarray replicates (among the 14% that are missed are the majority of histone genes, as mentioned above). However, if we consider those loci with 100 or less tpm, the number detected by the arrays drops to 31%, and if we consider the lowest count range of 5 to 10 tpm, only 14% of the loci are called as detected in all three replicates of the microarray, indicating a clear relationship between expression level and the ability of the microarrays to detect expression.
Conversely, 465 genes were called as detected on the microarrays that were not detected by HeliScopeCAGE within 500 bases of a known RefSeq 5′ end. Manual inspection of all 465 loci confirmed that 236 of these are actually expressed in either THP-1 or HeLa but use a previously recorded alternative promoter to that of the RefSeq transcript (Supplemental Table 3), indicating deficiencies in RefSeq. Interestingly, we may also be able to explain signal for a further 18 of these loci. For four loci (KRT10, CNFN, CD163L1, and DLL3), we observe a strong peak of signal within a known internal exon, suggesting promoter activity in the adjacent intron (Supplemental Fig. 3a); this architecture has previously been observed for loci such as NTRK1 (Forrest et al. 2006). For five loci, we observe a strong peak upstream of the annotated 5′ end with no EST support (PLXNB2, PLXNA1, ADCY3, CHD1, and MORC2) (Supplemental Fig. 3b). Finally, for nine loci, we find no evidence of signal originating from the annotated RefSeq 5′ end; however, for all of them, intergenic fusion transcripts have been recorded, and we find strong expression of the upstream partner. (These include C10ORF32-AS3MT, BLOC1S1-RDH5, ABCB8-ACCN3, PMF1-BGLAP, FGFR1OP-CCR6, APITD1-CORT, SFT2D2-TBX19, VAMP8-VAMP5, and MYO18A-TIAF1 [Supplemental Fig. 3c].)
One possible explanation for the remaining 229 loci detected by microarray but missed by HeliScopeCAGE is that the 5′ end of these transcripts is generated from duplicated regions of the genome. Using the mappability track available in the UCSC Genome Browser, we extracted the average mappability within 1 kb of the annotated 5′ end of RefSeq transcripts for these loci. Only 81 of these had average mappability scores of <90%. (A mappability of 50% would indicate two mapping locations.) The remaining 148 loci had 90% or better mappability, but no recorded transcript could be found to link a promoter signal to the microarray signal, suggesting possible false positives on the array by cross-hybridization.
Differential promoter usage in THP-1 and HeLa and novel transcripts
The above analysis focuses only on the set of genes for which there was a microarray probe; however, a distinct advantage of CAGE data over microarrays is that we can measure expression naive of gene models. Assignment to known genes, alternative promoters, and novel transcripts can then be carried out after genome mapping. To address this, we extensively analyzed the 5-μg libraries of THP-1 and HeLa. We aggregated neighboring CAGE tags on the genome into tag clusters (TC) and picked up the union of the top 50,000 TCs with highest maximum expression in any of the six libraries (89,976 highly expressed TCs in total). Of these, 26,918 (29%) fell outside of the Entrez gene boundaries (farther than 1 kb upstream or downstream of the 5′ and 3′ ends), and of those, 8381 had significant differential expression with FDR <0.1% (Supplemental Table 4). One example of differentially expressed regions is shown in Figure 3B. This copy of a human endogenous retrovirus (HERV) is highly active in THP-1 cells (Fig. 3B), which is consistent with a previous publication demonstrating the selective activity of HERV in leukemia lines (Patzke et al. 2002). More examples of differentially expressed novel loci are shown in Supplemental Figure 4. This demonstrates that the HeliScopeCAGE identified a significant number of novel loci differentially regulated between the two cell types.
The above analysis also identified 7108 genes with expression coming from more than one TC, and for 4905 of these, at least one of the promoters was differentially expressed between HeLa and THP-1. Three hundred eight genes showed alternative TCs with discordant regulation between the two biological states, while 4597 were concordant, suggesting that the vast majority of alternative TCs are co-regulated between these two cellular states (see Supplemental Fig. 5 for examples). To further focus on the subject of alternative promoters, we plotted the log fold change for HeLa versus THP-1 for the top two most highly expressed promoter-associated TCs from the same gene. The majority of alternative TCs associated to a single gene are co-regulated (Pearson correlation of 0.558, compared to 0.041 for randomized pairs of promoters) (Supplemental Fig. 6). If we consider the distance between these clusters, we find that 48% of the pairs are separated by <100 bases, suggesting that they are likely to be regulated by the same (or at least overlapping subsets) transcription factor binding sites (TFBS) in the core promoter; the correlation of fold change for such closely separated pairs increases to 0.627, while TCs at greater than 500 bases separation (600 pairs) are less correlated (0.373), suggesting divergent regulation, and for pairs >2 kb apart, the correlation dropped further to 0.219 (Supplemental Table 5).
Brief comment on previous CAGE protocols
We have previously published THP-1 CAGE libraries that were sequenced on the 454 Life Sciences (Roche) Genome Sequencer FLX System. The 454 libraries required 50 μg of total RNA, took multiple weeks to prepare, multiple rounds of PCR amplification and concatenation, yielded on average 1 million useable tags per sample, were 21 bases long, and had correlations for technical replicates of 0.903. The HeliScopeCAGE libraries, on the other hand, yielded ∼15 million mapped tags from 5 μg of total RNA, required no amplification, take 1–2 d per sample to prepare, and have a correlation of 0.987. In addition, the mapped HeliScope reads have a median length of 33 bases, with many >40 bases long (Supplemental Fig. 7). Given that tags shorter than 20 bases are much less unique in the genome than tags above 20, the longer reads from HeliScopeCAGE greatly improve single mapping compared to the 454 version and largely remove the need for multimap correction (see comments in Faulkner et al. 2008 on length and multimap rescue). These differences have significantly improved the reproducibility of the CAGE protocol (see Supplemental Fig. 8a, which shows overlaid scatterplots of 454 and HeliScopeCAGE technical replicates). In addition, comparison of CAGE signal on HeliScopeCAGE, 454CAGE, and Illumina microarray signals for THP-1 to qRT-PCR (quantitative reverse-transcriptase polymerase chain reaction) measurements for a set of approximately 2000 transcription factors confirm that HeliScopeCAGE outperforms both Illumina microarrays and 454CAGE in correlation of absolute counts (Supplemental Fig. 8b). Finally, our laboratory has also recently published on nanoCAGE, a PCR-based CAGE protocol that uses template switching and semisuppressive PCR to generate 5′ enriched signal from small amounts of RNA (Plessy et al. 2010). The nanoCAGE protocol, which does not use cap-trapping, was demonstrated to generate TSS enriched signal for as little as 10 ng of total RNA; however, this required high numbers of PCR cycles, meaning that fewer unique promoter regions are detected, and, in addition, more replicates are needed to reliably call differential expression between samples. We still recommend use of nanoCAGE for very small starting amounts of RNA; however, for samples with 100 ng or more RNA, HeliScopeCAGE is currently the method of choice.
Shape analysis of transcription start site signal in unamplified CAGE libraries
In FANTOM3 we examined the distribution of individual transcription start site frequencies within transcription initiation regions and observed two major classes of signal shape—broad CpG-associated promoters and sharp TATA-box-associated promoters (Carninci et al. 2006). We revisited this analysis using the HeliScopeCAGE deep sequencing data sets. We find that for the top 5000 most highly expressed clusters in each of the THP-1 and HeLa replicates, respectively, 14%–15% or 16%–18% of the tag clusters fall into the sharp peak category (Fig. 4A; Supplemental Fig. 9; Supplemental Table 6). Consistently, the TATA-box motif was only associated with the sharp peak category; however, at least within these two states (HeLa and THP-1), we find that CpG is strongly associated with both broad and sharp peak categories. In FANTOM3 we also observed two additional classes that appeared to be superimposed combinations of broad with a sharp peak or multiple closely associated sharp peaks; with the depth of the HeliScopeCAGE data, we can easily identify regional peaks and troughs within broader transcription initiation regions, including peaks that are specific to either the HeLa or THP-1 states (Fig. 4B; Supplemental Figs. 10, 11).
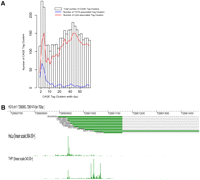
Distribution of HeliScopeCAGE signal within transcription initiation regions. (A) Width distribution of Tag Clusters. CpG and TATA association are shown as blue and red lines, respectively. (B) Fine level TSS preference differences between HeLa and THP-1 in the P2RY6 locus are revealed by HeliScopeCAGE.
To further test whether unamplified HeliScopeCAGE has an advantage over previous versions in determining the shape of TSS preference distributions, we repeated the above analysis on the THP-1 454 replicates generated with 28 cycles of PCR. For the 454CAGE libraries, the fraction of sharp peaks was much greater, ranging from 19% to 27% for the top 5000 tag clusters in three separate libraries. We hypothesized that this difference could be due to over-amplification of specific tags in the 454CAGE libraries, resulting in artifactual sharp peaks. To test this, we attempted a bias correction step in which CAGE tags with identical errors were counted only once and the cluster analysis repeated. This reduced the fraction of sharp peaks from 19%–27% down to 16%–23%. Another approach combined the signal for the three independent 454 libraries, and then the top 5000 clusters were annotated. Using this approach, the fractions observed for 454 and HeliScope were comparable (16% and 15%, respectively). Together this suggests that the PCR biases not only affect reproducibility but also affect TSS usage observations, overestimating the fraction of promoters that generate transcripts from sharp peak TSS regions by 4%–13% (Supplemental Table 6).
Exon painting signal observed in HeliScopeCAGE libraries
Previously we observed low levels of CAGE signal that originated from within internal exons rather than canonical 5′ ends of known transcripts. When mapped to the genome, these tags align to exons rather than introns, thereby “painting the exons” (as RNA-seq does). Recent work by Fejes-Toth et al. (2009) indicates that this signal may represent recapping of processed longer transcripts, supported by capped short RNAs that map along the length of processed transcripts including short RNAs that span splice junctions. Even more recently, using the HeliScope platform to sequence small RNAs, Kapranov et al. (2010) identified a set of transcripts antisense to the 3′ UTR of known transcripts that include a nongenomically encoded 5′ poly(U) sequence, indicative of an RNA-dependent RNA polymerase copying a poly(A) tail. This work also reanalyzed our previously published CAGE data and found evidence suggesting that these poly(U) transcripts were capped. With these observations in mind, we investigated whether our HeliScopeCAGE data sets showed evidence for exon painting and antisense signal. The highest amount of sense signal by far was associated at the 5′ end of transcripts (as is expected for CAGE), and for both known 5′ UTRs and the region 100 bases upstream of the transcript, we find enrichment on both the sense and antisense strands, indicative of bidirectional transcription (Fig. 5A). Interestingly, most of the antisense signal is observed in the 100 bases upstream of the 5′ end of transcripts, which is similar to a previous report of nonproductive promoter proximal antisense transcription for ∼30% of human genes using global run-on sequencing (GRO-seq) (Core et al. 2008).
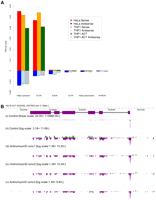
Sense-antisense HeliScopeCAGE signal. (A) Distribution of CAGE tag signal on the genome relative to 100 bp upstream, 5′ UTR, internal exons, 3′ UTR, and introns in HeliScope CAGE libraries from 5 μg of THP-1, 5 μg of HeLa, and 5 μg of THP-1 when the reverse transcription is carried out in the presence of actinomycin D. (B) Genomic view of the ACTB locus. (i) Linear scale 5 μg of THP-1; (ii) log scale 5 μg of THP-1; (iii–v) log scale 5 μg of THP-1 in the presence of 0.1, 0.2, or 0.4 mg/mL actinomycin D demonstrating sense and antisense painting of exons visually defining the gene boundaries (log scale).
Finally, we also observed HeliScopeCAGE signal painting both the sense and antisense of known exons, at levels significantly above what is seen for introns (Fig. 5A; Supplemental Fig. 12). As an example here, we show HeliScopeCAGE signal for the beta-actin locus (Fig. 5B). Note that we are able to visually identify gene boundaries by density of exon painting CAGE tags alone. The inclusion of actinomycin D effectively removes the majority of antisense signal on the exons, suggesting that at least 50% of this signal is due to second-strand synthesis by the reverse transcriptase. Inclusion of actinomycin D, however, severely reduces library yields (Supplemental Table 7); therefore, we currently omit actinomycin D from our standard production libraries.
Discussion
We have described the adaptation of the CAGE technique to the HeliScope true single-molecule sequencing platform. We demonstrate that this simplified technique is highly reproducible, has a dynamic range of more than 5 orders of magnitude, and that it outperforms microarrays in terms of sensitivity and isoform discrimination. It also outperforms previous CAGE protocols in terms of biases, depth, RNA requirements, and working time. Similar to RNA-seq, HeliScopeCAGE can identify novel regions of transcription and 5′ variant isoforms. Surprisingly, for many highly expressed loci, we are also able to visually identify exon boundaries from sense/antisense painting signal. This signal may be useful in the future for finding new genes and designing PCR primers for amplification of novel transcripts prior to full-length cloning and sequencing.
Within the ENCODE project, CAGE and RNA-seq are being used in combination to annotate transcribed regions of the genome and their expression levels. Both have strengths and weaknesses. We see these as complementary technologies. CAGE is clearly the method of choice for annotating 5′ ends of transcripts and RNA-seq the method for annotating transcript structure. When it comes to gene expression, however, they both have their advantages. In particular, for measurement of novel transcription, CAGE has the property of aggregating signal in a cluster, whereas RNA-seq is spread along the length of the transcript. The aggregation of CAGE signal may help to carry out differential gene expression analysis as we can simply cluster overlapping tags into de facto 5′ ends of gene models without any knowledge of the transcript structure, which simplifies the informatics. As we demonstrated in the section on novel transcripts, this works for DGE. However, this leaves us with no knowledge of the transcript structure (and whether multiple isoforms are generated); in this case, matching RNA-seq would be useful to elucidate the transcript structure. Finally, as an additional contrast, RNA-seq samples transcripts multiple times along their length, while CAGE is directed to the cap at the 5′ end present at only one copy per transcript. The advantage for CAGE is that it may allow for direct comparison of transcript abundance between loci, whereas RNA-seq requires length normalization to achieve this. The disadvantage for CAGE is that for low-quantity starting material, RNA-seq on fragmented RNA has more target molecules from which generate reads. Despite this, we still see these as complementary technologies that should be used in combination.
Using a triplicate design based on the highly reproducible HeliScopeCAGE protocol and edgeR, we were able to easily identify differentially expressed genomic regions between the two biological states, including known genes, alternative promoters, and novel transcripts. By manually examining the set of loci called as missed by the RefSeq analysis and detected by microarray, we found multiple cases in which the 5′ end of the annotated RefSeq transcript is either not used or is not the dominant 5′ isoform used in the cells. Based on our previous observations and the examples shown in the Supplemental Data of this manuscript, we estimate that up to 5% of loci use a different 5′ end from that of RefSeq transcripts. In a small fraction of cases in which the transcription initiation region is duplicated, we cannot uniquely assign expression to one position or the other, and although multimapping correction may help in some cases, an alternative technology that measures a unique region of the transcript is required for complete duplications (note that this is an issue for RNA-seq as well).
Finally, we have launched into the FANTOM5 project using HeliScopeCAGE data to build a global map of transcriptional regulation across the entire diversity of mammalian cellular states. To achieve this, reduction in sample requirements was needed because many primary cell types are not available in large quantities. Scaling down the RNA required from 50 μg to 5 μg has reduced this to the order of 1 million cells. We have also demonstrated that our low-quantity version with as little as 100 ng (about 20 to 100,000 cells) measures expression for about 8000 loci and is highly correlated with the 5-μg version (Fig. 2C; Supplemental Figs. 2, 13). Together this makes it possible to explore many subpopulations not previously profiled. The HeliScope platform is free of amplification biases, allows concurrent running of 48 samples, produces a depth of ∼20 million mappable reads per lane, and produces our highest-quality CAGE data to date. We expect that the increased quality and depth of the data will give great improvements to our attempts to map transcriptional regulatory networks. For these reasons, we have committed to this platform, and as of this date have already generated data for more than 1000 CAGE libraries for the FANTOM5 project. We openly invite researchers working on rare primary cell types to contact us if they would like to be involved in the project.
Methods
Cell culture and RNA/DNA preparation
THP-1 cells were cultured in RPMI1640 (Invitrogen) supplemented with 10% FBS, penicillin/streptomycin (Invitrogen), 10 mM HEPES (Invitrogen), 1 mM sodium pyruvate, and 50 μM 2-mercaptoethanol. HeLa cells were cultured in Eagle's MEM (Invitrogen) supplemented with 10% FBS, 1% NEAA (Invitrogen), and penicillin/streptomycin. Total cell lysates were harvested in TRIzol reagent (Invitrogen), total RNA was purified from TRIzol lysates according to the manufacturer's instructions, and we used RNase-free glycogen (Invitrogen) as carrier to the aqueous phase prior to precipitating the RNA with isopropyl alcohol. The quality of the extracted RNA was confirmed with the Agilent 2100 Bioanalyzer.
HeliScopeCAGE
First-strand cDNA synthesis
First-strand cDNA was synthesized from 5 μg of total RNA. RNA was mixed with 500 ng of random N15 primer in a volume of 6 μL, heat-denatured for 5 min at 65°C, and then chilled on ice/water and centrifuged briefly. Thirty-two microliters of an RT master mix (7.6 μL of 5× SuperScriptIII reaction buffer, 1 μL of 10 mM dNTP, 7.6 μL of 3.3 M sorbitol/27% trehalose solution, 1.9 μL of 0.1 mM DTT, 3.8 μL of 200 U/μL of SuperScriptIII [Invitrogen]) was then added and mixed gently. The reaction was then incubated at 25°C for 30 sec, 42°C for 30 min, 50°C for 10 min, 56°C for 10 min, and 60°C for 10 min, then chilled at 4°C. The cDNA/RNA hybrids were then purified with Agencourt RNACleanXP (Beckman Coulter) according to the manufacturer's instructions and eluted in 40 μL of water.
Oxidation and biotinylation
Forty microliters of purified cDNA/RNA hybrid was mixed with 2 μL of 1 M sodium acetate (pH 4.5) and 2 μL of freshly prepared 250 mM sodium periodate (aq) and chilled in ice/water for 45 min in the dark. The reaction was stopped by the addition of 2 μL of 40% glycerol and mixed with 14 μL of 1 M Tris-HCl (pH 8.5). The oxidized cDNA/RNA hybrid was then purified using Agencourt RNACleanXP. Four microliters of 1 M sodium acetate (pH 6.0) and 1 μL of freshly dissolved 150 mM biotin (long arm) hydrazide (VECTOR Lab) in DMSO were added, and the mixture was incubated for 2 h at 37°C in the dark. Twelve microliters of isopropanol was then added, and the biotinylated product was again purified with Agencourt RNACleanXP. Finally, single-stranded RNA regions not protected by a complementary first-strand cDNA strand were digested using RNase 1 (4.5 μL of 10× RNase I buffer and 0.5 μL of 10 U/μL RNase I [Promega]) for 30 min at 37°C.
Cap-trapping and release
One hundred fifty microliters of MPG streptavidin magnetic bead slurry (Takara Bio) was pre-blocked using 2 μL of 20 μg/μL tRNA (Sigma) on ice water for 30 min, two rounds of washing (buffer 1: 4.5 M sodium chloride, 50 mM EDTA at pH 8.0), and resuspended in 105 μL of buffer 1 containing 50 μg of tRNA. The biotinylated cDNA/RNA hybrids were then purified using 150 μL of blocked beads. Binding was carried out for 30 min at 50°C, then beads were purified using a magnetic stand and washed with 150 μL of the following buffers: once buffer 1(as above), once buffer 2 (0.3 M sodium chloride, 1 mM EDTA at pH 8.0), twice buffer 3 (1 mM EDTA, 0.4% sodium dodecylsulfate, 0.5 M sodium acetate, 20 mM Tris-HCl at pH 8.5), and twice buffer 4 (1 mM EDTA, 0.5 M sodium acetate, 10 mM Tris-HCl at pH 8.5).
Captured cDNA was released from the beads by heat shock and RNase I treatment. Beads were resuspended in 35 μL of 1× RNase I buffer, incubated for 5 min at 95°C, and transferred immediately to ice water. The supernatant containing cDNA was transferred to a fresh tube, the beads were washed with a further 30 μL of RNase I buffer, and the supernatant was pooled with the first elution and the volume adjusted to 65 μL. RNA was then removed by RNase treatment (3 μL of 2 U/μL RNase H [Invitrogen], 2 μL of 10 U/μL RNase I for 15 min at 37°C). Cap-trapped first-strand cDNA was then purified with Agencourt AMPure XP (Beckman Coulter) according to the manufacturer's instructions, and the cDNA quantity was determined with the OliGreen ssDNA Quantitation kit (Invitrogen). The typical yield for 5 μg of starting RNA is 10–20 ng of CAGE library.
Poly(dA) tailing and molar concentration measurement
The poly(dA) tailing reaction was done according to the manufacturer's recommendations as follows: A mix containing 10 ng of single-stranded CAGE library in 10.8 μL of H2O, 2 μL of 5 U/μL Terminal Transferase (NEB) 10× buffer, and 2 μL of 2.5 mM CoCl2 was denatured for 5 min at 95°C, followed by rapid cooling on a pre-chilled aluminum block kept in ice/water.
The mixture containing 1 μL of 5 U/μL Terminal Transferase, 50 μM dATP, and 0.2 μL of BSA (NEB) was added to the denatured 14.8-μL mixture. The reaction was incubated for 1 h at 37°C, followed by enzyme inactivation for 10 min at 70°C, then chilled 4°C. After the poly(A) tailing reaction, the 20-μL reaction was denatured for 5 min at 95°C followed by rapid cooling on a pre-chilled aluminum block kept in ice and water slurry.
Next, the following blocking mixture involving 1 μL of NEB Terminal Transferase 10× buffer, 1 μL of 2.5 mM CoCl2, 5 U of NEB terminal transferase, 0.5 μL of 200 μM biotin-ddATP, and 6.5 μL of nuclease-free water, was added to the denatured, poly-adenylated mixture to a total volume of 30 μL. The mixture was incubated for 1 h at 37°C, followed by enzyme inactivation for 20 min at 70°C, then chilled at 4°C. After the blocking reaction, 2 pM Carrier Oligonucleotide (5′-TCACTATTGTTGAGAACGTTGGCCTATAGTGAGTCGTATTACGCGCGGT[ddC]-3′) added to the heat-inactivated 30-μL terminal transferase reaction above. The sample was measured by Helicos OptiHyb assay for ChIP-Seq following the manufacturer's manual, LB-018_01.
Sequencing on HeliScope
Three nanograms (∼80 pM) of poly(dA)-tailed sequencing templates was loaded on a HeliScope flow cell according to the manufacturer's manual, LB-016_01 and LB-017_01. Sequencing on the HeliScope Single Molecule Sequencer was done according to the manufacturer's manual, LB-001_04.
Filtering and alignments of HeliScope reads
All raw reads were filtered with a method similar to Lipson et al. (2009), except for the approval read length (20 to 70 nt in this study) and alignment score (excluded if it is more than 3.5) to base-addition-order sequence. Reference sequences were based on the human (Homo sapiens) genome assembly hg18 (NCBI Build 36), downloaded from the UCSC Genome Browser database, and human ribosomal DNA complete repeating units (GenBank accession U13369). Each read was aligned using indexDPgenomic in the helisphere-0.14.a015 package, which is a pairwise-sequence aligner, as described in Lipson et al. (2009). We used some reads whose best alignment was to the human genome with score ≥3.5; those are “aligned” tags. Some reads, which had two or more alignments with the same highest score, were excluded.
HeliScopeCAGE for low-quantity total RNA
For low-quantity HeliScopeCAGE (1 μg or less of starting material), the protocol was essentially the same except for the following modifications: The biotin hydrazide amount was adjusted to 4 μL of 10 mM. DYNAL M270 streptavidin magnetic beads (Life Technologies) were used instead of MPG beads, and the added M270 beads slurry volumes were 20 μL for 1 μg of total RNA, or 15 μL for <1 μg of total RNA. In the subsequent purification step, we added 15 pmol of Carrier Oligonucleotide that was used in the “poly(A) tailing” step, prior to addition of AMPureXP beads slurry (in “capture and release”).
Expression quantification
Gene expression is measured with the number of reads aligned within 500-bp distance from RefSeq transcript 5′ ends (Maglott et al. 2000), where their genomic coordinates are downloaded from the UCSC Genome Browser database. The read counts are normalized to tag-per-million (tpm) based on the total number of aligned reads in the reference human genome except for the ribosomal unit. We grouped all CAGE tags overlapping with one or more base pairs on the same strand into a single tag cluster (TC) and normalized the reads to tpm in the analysis of alternative and novel promoters.
First-strand cDNA synthesis in the presence of actinomycin D
One milligram of actinomycin D (Sigma-Aldrich A1410-25MG) was dissolved in 90 μL of DMSO. The precise concentration was estimated with absorbance at 441 nm and absorption coefficient, 21,900, then adjusted at 1 mg/mL. Reverse transcription was done the same as above with the addition of actinomycin D at the final concentrations of 0.1, 0.2, and 0.4 μg/μL.
Microarray
Five hundred nanograms of total RNA was amplified using the Illumina TotalPrep RNA Amplification Kit (Ambion), according to the manufacturer's instructions. cRNA was hybridized to Illumina Human Sentrix-6 bead chips version 3, according to standard Illumina protocols. Chip scans were processed using Illumina BeadScan and BeadStudio software packages, and summarized data were generated in BeadStudio (version 3.4).
Acknowledgments
This work was funded by a Research Grant from the Japanese Ministry of Education, Culture, Sports, Science and Technology through the Cell Innovation Project and for the RIKEN Omics Science Center to Y.H. We thank the RIKEN Genome Network Analysis Service for data production and J. Severin for visualization. We also thank Kristen Kerouac, Phil Kapranov, Alix Kieu, Patrice Milos, and Chris Hart from the Helicos BioSciences Corporation for support of sequencing operations and analysis. Genomic visualization figures were produced using the Zenbu genome browser (http://fantom.gsc.riken.jp/zenbu/; Jessica Severin, in prep.).
Authors' contributions: M.K.-K., M.I., M.K., and A.K. developed the standard and low-quantity HeliScopeCAGE protocols. H.K., T.L., S.K., and N.B. analyzed the data. N.N. prepared RNA and carried out the microarray analysis. H.K. and A.R.R.F. designed the differential expression experiments and platform comparison. A.R.R.F., P.C., C.O.D., and Y.H. were involved in the planning of the project. A.F. drafted the manuscript with M.I., H.K., T.L., S.K., and N.B.
Footnotes
-
↵2 Corresponding authors.
E-mail alistair.forrest{at}gmail.com.
E-mail rgscerg{at}gsc.riken.jp.
-
[Supplemental material is available for this article. The sequencing data from this study have been submitted to the DDBJ Read Archive (http://trace.ddbj.nig.ac.jp/dra/index_e.shtml) under accession no. DRA000368. The gene expression profiles from this study have been submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accession no. GSE28148. Data for THP-1 and HeLa cells can also be viewed at the FANTOM web resource (http://fantom.gsc.riken.jp/5/suppl/Kanamori-Katayama_et_al_2011/).]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.115469.110.
- Received September 17, 2010.
- Accepted April 4, 2011.
- Copyright © 2011 by Cold Spring Harbor Laboratory Press








