
Deep-transcriptome and ribonome sequencing redefines the molecular networks of pluripotency and the extracellular space in human embryonic stem cells
- Gabriel Kolle1,
- Jill L. Shepherd1,
- Brooke Gardiner1,
- Karin S. Kassahn1,
- Nicole Cloonan1,
- David L.A. Wood1,
- Ehsan Nourbakhsh1,
- Darrin F. Taylor1,
- Shivangi Wani1,
- Hun S. Chy2,
- Qi Zhou2,
- Kevin McKernan3,
- Scott Kuersten3,
- Andrew L. Laslett2,4 and
- Sean M. Grimmond1,5
- 1Queensland Centre for Medical Genomics, Institute for Molecular Bioscience, The University of Queensland, Queensland 4072, Australia;
- 2CSIRO Materials Science and Engineering, Clayton, Victoria 3168, Australia;
- 3Life Technologies, Beverly, Massachusetts 01915, USA;
- 4Department of Anatomy and Developmental Biology, Monash University, Clayton, Victoria 3800, Australia
Abstract
Recent RNA-sequencing studies have shown remarkable complexity in the mammalian transcriptome. The ultimate impact of this complexity on the predicted proteomic output is less well defined. We have undertaken strand-specific RNA sequencing of multiple cellular RNA fractions (>20 Gb) to uncover the transcriptional complexity of human embryonic stem cells (hESCs). We have shown that human embryonic stem (ES) cells display a high degree of transcriptional diversity, with more than half of active genes generating RNAs that differ from conventional gene models. We found evidence that more than 1000 genes express long 5′ and/or extended 3′UTRs, which was confirmed by “virtual Northern” analysis. Exhaustive sequencing of the membrane-polysome and cytosolic/untranslated fractions of hESCs was used to identify RNAs encoding peptides destined for secretion and the extracellular space and to demonstrate preferential selection of transcription complexity for translation in vitro. The impact of this newly defined complexity on known gene-centric network models such as the Plurinet and the cell surface signaling machinery in human ES cells revealed a significant expansion of known transcript isoforms at play, many predicting possible alternative functions based on sequence alterations within key functional domains.
Human embryonic stem cells (hESCs) are an excellent model system for studying both pluripotency and directed differentiation. They also have the potential to provide an unlimited source of cells for therapeutic applications. The genes that maintain the undifferentiated stem cell state have been identified through a variety of array-based mRNA studies, proteomic profiling, functional genomic knockdown screening, and transcription factor ChIP approaches (Brandenberger et al. 2004a,b; Richards et al. 2004; Assou et al. 2007; Muller et al. 2008). These approaches have highlighted the importance of the “Plurinet,” a regulatory network of 299 protein-coding genes believed to interact and maintain the embryonic stem cell (ESC) state and the importance of the extracellular space in maintaining the stem-like state.
One challenge to studying pluripotency is that gene-centric models such as the Plurinet do not adequately reflect the true molecular complexity underlying these networks. This limitation arises from the “gene-centric” approach used to create these models, where each actively expressed gene is represented as a single node in the network that expresses a single transcript, which, in turn, encodes a single canonical peptide (Brandenberger et al. 2004a,b; Richards et al. 2004; Assou et al. 2007; Muller et al. 2008). Such models neglect the true complexity of transcriptional output in higher eukaryotes that is created by alternative splicing, multiple promoter usage, 3′-UTR switching, expression of non-coding RNAs, and RNA-mediated control via the likes of miRNAs (Carninci et al. 2005; The ENCODE Project Consortium 2007; Chekulaeva and Filipowicz 2009).
Until recently, defining transcriptional complexity in single biological states has proven challenging. Massive-scale shotgun next-generation sequencing of cDNAs (also known as RNA-seq) has been shown to provide the means for simultaneous monitoring gene activity and canonical and variant mRNA expression arising from alternate splicing, and the means to detect alternative promoter or 3′-UTR usage (Cloonan et al. 2008; Mortazavi et al. 2008; Wang et al. 2008; Wilhelm et al. 2008) in any biological state. Indeed, both mouse and human stem cells have recently been analyzed in this fashion (Cloonan et al. 2008; Wu et al. 2010). One major limitation arising in these studies has been the inability to confidently assign biological activity to the RNA detected by RNA-seq. While an mRNA may be actively transcribed, it may not be translated due to transcriptional pausing, illegitimate transcription, poor splicing, and nonsense-mediated decay (Chang et al. 2007; Maier et al. 2009). Other challenges in assigning biological activity to RNA detected by RNA-seq include the difficulty in distinguishing overlapping transcripts from a single locus and in accurately delineating novel expression when this expression is closely associated with known genes.
In this study, we report on the overall transcriptional output of human embryonic stem cells by deep sequencing of cytoplasmic RNA. We perform a systematic review of novel expression and show that sequence-based “virtual Northern blot” analysis improves variant detection and reveals a dramatic expansion of the hESC exome through long 3′-UTR usage. To help illuminate the biological activity of expressed mRNA, we have examined which actively transcribed mRNAs are also actively translated in hESCs. We describe the use of free and membrane polysome-associated RNA fractions to perform sequence-based translation state analysis and to identify which actively translated RNA transcripts encode peptides destined for the hESC extracellular space. We have identified hESC-specific non-canonical splice junctions and 3′-UTR extension events by comparing RNA-seq data from hES cells with that of three human tissues, namely, brain, heart, and liver. Finally, we have used this information to reannotate the classical models of key pluripotency regulatory networks so that they are now transcript-specific, uncovering a substantial increase in the number and complexity of components active in the hESC Plurinet.
Results
Sequencing the human ESC transcriptome
A thorough survey of the human ESC transcriptome (HES-2 cell line) was generated using strand-specific shotgun sequencing (Tuch et al. 2010) of the following ribosomal RNA-depleted RNA populations: (1) cytoplasmic, poly-adenylated RNA (known hereafter as the “mRNA” fraction); (2) cytoplasmic polyadenylated RNA size-fractionated into five subpopulations (<0.5 kb, 0.5–2 kb, 2–3.5 kb, 3.5–6.5 kb, >6.5 kb); and (3) membrane-bound (“MPR” fraction) and free/cytosolic (“CPR” fraction) polysome-associated RNAs isolated using cyclohexamide treatment and sucrose gradient ultracentrifugation (Supplemental Figs. S1, S2; Diehn et al. 2000, 2006; Stitziel et al. 2004). In total, 674 million mappable tags were generated across all libraries (Table 1) using a combination of single fragment or paired-end reads at lengths ranging from 25 to 75 bp. Tags were recursively mapped to both the human genome (GRCh37) and a curated library of known exon–exon junctions using BioScope (Life Technologies). All mapped data can be viewed as IGV-accessible BAM and wig/tdf files at http://grimmond.imb.uq.edu.au/hES_transcriptome.
Mapping statistics for hESC libraries
Quantifying human ESC locus activity
As a first step, the overall locus activity for hESCs was calculated. The majority (61%–91%) of unique reads mapped to exonic sequences of known or predicted genes (Table 1), which was consistent with other studies (Cloonan et al. 2008; Mortazavi et al. 2008). The directionality of the library preparative method was confirmed by a >10,000-fold enrichment of tags matching to exon junctions in a sense versus antisense orientation (Supplemental Fig. S3). A total of 14,081 unique genes were found to be active at a level of 1 mRNA transcript per cell or greater in the mRNA fraction (based on the estimate that one read per kilobase per million tags, or RPKM, equates to 1 transcript per cell) (Mortazavi et al. 2008).
To confirm that this active gene profile was accurate, the relative expression was compared with previously published strand-non-specific RNA sequencing of hESCs (Pearson correlation = 0.94) (Supplemental Fig. S4B; Wu et al. 2010). We also used these data to investigate expression of genes (n = 2990 genes) previously described as being inactive in hESCs based on microarray and massively parallel signature sequencing (MPSS) data (Boyer et al. 2005; Guenther et al. 2007). Despite the stringent criteria by which the inactive gene set was originally defined, we found that ∼20% of the genes (n = 535 genes) were detected by RNA-seq (RPKM ≥ 1). Furthermore, 115 of these genes were expressed at high levels (RPKM > 5) (Table 2; Supplemental Table S1). To increase confidence in expression of these genes, RNA-seq data for these genes were aligned with human ES cell chromatin immunoprecipitation sequencing (ChIP-seq) data for three histone H3 lysine trimethylation modifications indicative of transcriptional initiation (H3K4me3), elongation (H3K36me3), and repression (H3K27me3) (Ku et al. 2008). In the majority of such cases (77/115) expression was supported by the presence H3K4me3 and H3K36me3 as well as the absence of H3K27me3.
Analysis of cellular and membrane-polysome-enriched (MPR-enriched) expression in human ESCs
Alternative splicing in hESCs
Alternative splicing is a powerful method for expanding the transcriptomic and proteomic output of mammalian cells (Forrest et al. 2006). Alternative splicing events are readily identified in RNA-seq experiments using sequence tags that span exon–exon junctions. A total of 121,728 unique high-confidence exon junctions (supported by >8 tags) were identified within hESCs. Of these alternatively splicing events, 5698 were for non-canonical exon combinations, with 65% of these events independently supported by both fragment and paired-end sequencing (Supplemental Table S2A,B). In 250 cases, the non-canonical junction was expressed at least fivefold higher than the canonical (Supplemental Table S2). We also identified a further 252 genes that expressed at least two distinct 3′-terminal exons based on the distinct expression of two 3′-UTR sequences (Table 2; Supplemental Table S3).
Once variant junctions were defined, they were mapped against all well-defined full-length Ensembl transcripts to infer isoform-specific transcript expression (Supplemental Methods). Of the 5698 alternative variants, at least 11.2% (n = 639) resulted in a predicted protein domain architecture that differed from that of the canonical transcript (Supplemental Table S2A). Comparing non-canonical junction expression in hES cells and paired-end RNA-seq data for three human tissues revealed 2999 non-canonical junctions expressed exclusively in hES cells (Fig. 1C; Supplemental Table S2A). Of particular interest was the hES cell-specific splicing event detected for DNA methyltransferase 3B (DNMT3B), which was predicted to encode a truncation of the C5 DNA methyltransferase domain by 163 amino acids (Ensembl; Pfam) (Fig. 1A).
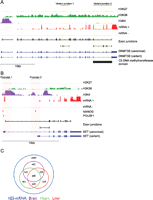
Alternative splicing in hES cells. (A) Integrated genome viewer (IGV) representation of the DNMT3B locus showing strand-specific RNA-seq data (mRNA±; scale: 0–5000 tags); exon junctions (black; detection threshold set at ≥8 tags); chromatin marks (H3K27, H3K36, and H3K4); two Ensembl transcripts (dark blue); and the C5 DNA methyltransferase functional domain (Pfam; bottom black). Detection of two non-canonical exon junctions (green shading) for DNMT3B indicated expression of at least two different transcript isoforms (dark blue), with expression of variant junction 2 predicted to result in truncation of the C5 DNA methyltransferase domain. (B) IGV view of the SET locus, including ChIP-seq data for POU5F1 (light blue) and NANOG (pink) binding sites, showing alternate promoter usage (red shading) and binding of both transcription factors at Promoter 1 and NANOG only at Promoter 2. (C) Venn diagram indicating tissue-specific expression of non-canonical alternative splicing events.
Alternative 5′-exon usage in hES cells
Alternative 5′-exon usage can be indicative of alternative promoter usage, and thus alternative mechanisms of transcriptional regulation. To identify discrete alternative 5′ usage, collated hESC transcriptome data were screened for regions sharing at least eight diagnostic exon–exon junction tags with 5′ exons of known alternative transcripts. A total of 8749 alternative 5′ exons were detected with high confidence (Supplemental Table S4), and 910 of these were non-canonical events. The genomic regions encoding these novel 5′ exons were then compared with hESC H3K4me3 ChIP-seq data (Ku et al. 2008), with the majority, 96% (877/910), overlapping a well-defined H3K4me3 region (i.e., SET) (Fig. 1B; Table 2; Supplemental Table S4). An additional class of variant 5′-UTR usage was also observed in which the exonic signal continued to extend 5′ of the canonical transcription start site (Supplemental Table S5, 44 genes). Taken together, these data provide a high-confidence set of transcripts expressed in hESCs for which alternative promoter usage may occur.
Transcriptome discovery
In addition to summarizing transcription from known loci, a detailed analysis was performed on expression arising outside known exons. Ninety-two percent of all novel expression clusters (>1 RPKM) mapped within the introns (sense strand) or flanking regions (within 10 kb) of well-defined genes (Fig. 2A). Examining this expression more closely, we found evidence for 45 genes containing intron retention events with high relative expression (Fig. 2C, OGT; Supplemental Table S6) and 10 genes that contained a putative extended internal exon (Fig. 2B, PHF10; Supplemental Table S6).
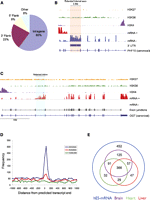
Novel sequence usage in hESCs. (A) Distribution of novel transcribed clusters compared with Ensembl gene models. (B) IGV view of the PHF10 locus showing expression (scale: 0–250 tags) of an extended internal exon (red shading). (C) IGV view of the OGT locus showing expression (scale: 0–500 tags) of a novel retained intron (blue shading) supported by junction tag evidence. (D) A consensus poly(A) sequence (blue) is highly enriched around the 3′ end of predicted long 3′ UTRs. Enrichment of a weaker poly(A) signal (red) is also shown, and a randomly chosen sequence (green) is not enriched. (E) Venn diagram indicating tissue-specific expression of 3′-UTR extension events detected in hES cell mRNA.
By far the most common novel expression event observed in hESCs was 3′-UTR extension of known genes (n = 1199) (Table 2; Supplemental Table S7). The average size of the extended 3′-UTR expression was 2.1 kb, with a range of 500 bp to 14 kb. The majority of long 3′ UTRs were found to possess a canonical poly-adenylation signal at their 3′ ends (733/1199). In almost all cases (92.4%), hES H3K4me3 ChIP-seq analysis failed to provide any epigenetic support for independent initiation of transcription driving these events. Comparing to heart, liver, and brain, we identified 452 long 3′ UTRs that were unique to ES cells (Fig. 2E).
To resolve the nature of these transcripts, a sequence-based “virtual Northern” analysis was used in which hES poly(A)+ RNA was separated into five size fractions (<500 bp, 0.5–2.0 kb, 2.0–3.5 kb, 3.5–6.5 kb, and >6.5 kb) and then subjected to sequencing. When gene expression was quantified across each fraction, a majority of genes (71%) displayed peak gene expression in the size fraction consistent with the canonical Ensembl annotation (Fig. 3A). In the case of genes predicted to express extended 3′UTRs that increased the transcript size by >2 kb, 71% (329/442) displayed peak levels of expression in a larger size fraction concordant with a 3′-UTR extension (Fig. 3B,C). The “virtual Northern” approach was subsequently used to validate other novel expression events (20/24 intron retention, 11/13 extended 5′ UTR) expected to substantially change the length of the expressed mRNA. In summary, most novel expression events that we identified are likely to be extensions of the surrounding gene, rather than independently transcribed events.
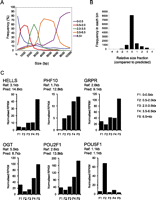
Transcriptome-sequencing based “virtual Northern” validation of long 3′-UTR expression. (A,B) Peak expression for most transcripts was detected within the size fraction corresponding to the canonical (Ensembl v55) transcript. (A) Graph of canonical transcript size against proportion of transcripts in each fraction. The percentage of transcripts in each bin is shown for each of the five size fractions (0–0.5 kb, 0.5–2 kb, 2–3.5 kb, 3.5–6.5 kb, and 6.5 kb+). (B) Distribution of peak gene expression relative to the expected (Ensembl canonical) size fraction. For example, if peak expression of a 2.5-kb gene was detected in the 6.5-kb+ size fraction, it would be assigned a relative value of 3. (C) Expression levels of individual genes across size fractions. Examples are shown of long 3′-UTR-containing genes (HELLS, GRPR, and POU2F1), a gene containing an extended internal exon (PHF10), and an intron-retaining gene (OGT), genes for which peak expression was detected in the fraction consistent with increased size. POU5F1 (no change expected) is shown for comparison.
Assessing subcellular enrichment and the preferential selection of transcriptional complexity for translation
The extracellular surface plays a vital role in the response of ES cells to external stimuli such as growth factors and differentiation cues. Previously, we have used membrane-polysome translation state analysis (MPTSA) and microarrays to separate membrane and secreted protein (known as M/S RNAs) encoding transcripts from all other free and cytosolic polysome-associated RNAs (C/N RNAs) based on their association with membrane-bound polysomes (Diehn et al. 2000; Diehn et al. 2006). To explore this subset of the transcriptome more closely and to begin to determine the relationship between transcriptional and translational complexity, we have performed MPTSA coupled with deep sequencing.
Sequence-based analysis of M/S RNAs versus C/N RNAs revealed a remarkable spatial separation of transcripts (Fig. 4A). At the locus activity level, 2903 genes were found to express RNAs significantly enriched in the M/S fraction, with a false discovery rate of <0.01 based on known gene classifications (Table 2; Supplemental Table S8; Stitziel et al. 2004; Kolle et al. 2009). Nine thousand one hundred seventy-three genes were significantly under-represented in the M/S fraction, suggesting cytoplasmic/nuclear localization (Supplemental Table S9). The observed ratio of M/S:C/N signal strongly correlated with previous published array-based studies on the same fractions (Pearson correlation = 0.95) (Supplemental Fig. S5; Kolle et al. 2009).
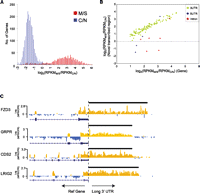
Enrichment of genes and alternative variants in the M/S enriched fraction. (A) Marked spatial separation of genes expressed in the membrane/secreted (M/S) and cytoplasmic/nuclear (C/N) fractions. The histogram shows frequency of genes expressed (RPKM > 1) plotted against the log2 ratio of M/S expression to C/N expression, log2(RPKMM/S/RPKMC/N). (B) Graph showing the correlation between canonical gene expression (x-axis) and expression of the novel annotated sequence (3′ UTR, 5′ UTR, intron retention; y-axis) for a subset of M/S genes. Notably, correlation with canonical gene expression is high for 3′ UTRs. (C) UCSC Genome Browser view of four genes for which novel long 3′ UTRs were detected by RNA-seq. Each track shows the log2 ratio of M/S to C/N expression calculated at 50-bp windows across the gene for the positive strand. (Orange) M/S-enriched regions; (blue) regions not enriched in M/S. Note the consistency of the M/S to C/S ratio across annotated 3′ UTRs, continuing through the length of the predicted novel 3′UTR.
Given the wealth of non-canonical transcripts we had observed to date, we next sought to determine what proportion of RNAs observed in the mRNA fraction enter the translational machinery. To address this, we looked specifically at all transcriptional events arising from the 2903 membrane-associated genes identified by MPTSA-Seq. Within these genes we identified 689 non-canonical variants and 229 genes that expressed two or more variants enriched in the M/S RNA fraction (Table 2; Supplemental Tables S10A,B, S11, S12). We subsequently validated 24 of these events by real-time PCR (Supplemental Fig. S6).
We were also interested in determining whether novel features such as extended 3′ UTRs or retained introns would be similarly enriched in the M/S fraction. To examine this, we looked at the enrichment ratio (M/S RNA: C/N RNA) of each novel feature and compared this with the canonical gene-enrichment ratio for each of the 2905 membrane-associated genes. We found strong evidence for membrane enrichment of the majority of 3′UTRs (Fig. 4 B,C), reinforcing their presence as a single long transcript. This was also confirmed for a panel of candidate genes by real-time PCR (Supplemental Fig. S7). In contrast, several 5′-UTR and intron retention events in M/S genes were not enriched in the M/S fraction (Fig. 4B).
Overlaying novel complexity on the gene-network controlling pluripotency
The impact of novel transcriptional complexity on pluripotency was examined in the context of the “Plurinet” gene network, a gene-centric model consisting of 299 nodes under the control of the master regulators of the stem cell state (transcription factors POU5F1, SOX2, KLF4, and NANOG). This model was originally created using extensive gene-expression array profiling of stem cells and progenitor populations; ChIP-chip analysis of the master regulators SOX2, POU5F1, and NANOG; plus functional studies (Muller et al. 2008). Overlaying hESC RNA-seq data confirmed that 96% (287/299) of the Plurinet loci were active in hES2 cells (Supplemental Table S14, RPKM ≥ 1, mRNA). In addition, we found 152 alternative splicing events for 98 genes, as well as alternate 5′-UTR expression (n = 23), 3′-UTR extensions (n = 27), and one instance of internal exon extension (Fig. 2B; Table 3, PHF10; Supplemental Table S14). These results are summarized in Table 3 and can be visualized within the context of the Plurinet network in Supplemental Figure S8. Alternative splicing events for 25 Plurinet genes were found to alter the canonical domain architecture (Supplemental Table S14), including DNMT3B as discussed above.
Summary of alternative splicing in Plurinet genes including non-canonical (NC) splicing events, alternative 5′UTRs, internal exon extension, and long 3′UTRs detected
A significant number of genes in the Plurinet have been predicted to be regulated by the pluripotency transcription factors POU5F1, NANOG, and SOX2. These annotations are based on overlaying canonical proximal promoters for each gene with data from ChIP-chip experiments (Boyer et al. 2005). We looked to redefine these models based on our RNA-seq data and recent ChIP-seq data for POU5F1 and NANOG (Kunarso et al. 2010). We found an additional 52 and 147 Plurinet genes that were targeted by POU5F1 and NANOG, respectively, based on canonical gene usage (Supplemental Fig. S8; Supplemental Table S14). Ten active alternate 5′ exons expressed by Plurinet genes show evidence for targeting by POU5F1 and 24 by NANOG (based on alignment with ChIP-seq data) (Supplemental Methods).
Impact of transcriptional complexity on the hES cell surface and signaling networks
Pluripotency is regulated by a balance of pathways and interactions (Vallier and Pedersen 2005; Vallier et al. 2009). Growth factors and other molecules present in the extracellular space bind to receptors expressed on the hES cell surface, triggering a cascade of reactions that, under appropriate conditions, favors hESC self-renewal and thus the maintenance of pluripotency. When cultured in suspension or as adherent single cells, hES cells rapidly differentiate (Draper et al. 2004), thus cell–cell and cell–matrix interactions also play important roles in the maintenance of pluripotent hES cells. By regulating the key growth factor receptors and mediating important cell–cell and cell–matrix interactions, the cell-surface proteome of hESCs is critical in governing hESC pluripotency and differentiation. We found evidence for the expression, alternative splicing, and membrane-polysome association of members of these pathways. Major signaling pathways involved in hESC growth factor signaling, including members of the TGFβ/activin/nodal, WNT, and FGF signaling pathways (Fig. 5) showed considerable variation both of ligands as well as cell-surface receptors. In addition, sequence data for several genes involved with cell-surface–extracellular-matrix interaction (Supplemental Fig. S9A) and cell–cell contact (Supplemental Fig. S9B) pathways also showed evidence for expression of non-canonical variant transcripts. Several M/S gene variants (12%) expressed in the mRNA fraction were not associated with membrane polysomes, indicating that variation at the mRNA level did not always predict complexity at the translational level.
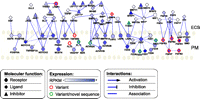
Transcriptional complexity for growth factor interaction model for pathways active on the hES cell surface. The model is based on KEGG pathway and IPA (Ingenuity Pathway Analysis, Ingenuity Systems) curated protein–protein interactions. Only genes enriched in the M/S fraction are shown because these are most likely to be actively translated. Nodes are colored according to M/S expression levels (RPKM). Node shape is dependent on molecular function according to IPA designation. Node outline color denotes the presence of at least two M/S-enriched alternatively spliced variants (red) or the presence of novel sequence feature(s) (green) for the gene in the M/S fraction. Interactions between genes (edges) are displayed for ligand–receptor (directed arrow), inhibitory action (directed inhibitory line), gene family member (dotted line), or non-directional interaction (solid line).
Discussion
We have used a strand-specific whole-transcriptome shotgun sequencing approach to discern the entire cytoplasmic transcriptome of human ES cells and the complement of genes and variants that is translated on membrane-bound polysomes. These data provide the largest resource to date of transcriptional output of the pluripotent state of human ES cells.
hESCs express a diverse array of transcript variants that have an impact on the proteome
Consistent with previous studies, we have identified a wide range of alternative splicing events that increase the transcriptional complexity expressed by mammalian cells (Cloonan et al. 2008; Mortazavi et al. 2008; Wang et al. 2008; Wilhelm et al. 2008). In particular, we have identified several alternate variant transcripts expressed by human ES cells within genes that are necessary for maintenance of pluripotency. For one of these, DNMT3B, at least two alternate transcripts are expressed, one of which is predicted to encode a protein product with a truncated C5 DNA methyltransferase domain relative to the canonical isoform based on Pfam domain annotation of Ensembl transcripts. Truncated DNMT3B isoforms are associated with altered patterns of DNA methylation in cancer cells (Ostler et al. 2007).
In some cases, we have found key pluripotency-related genes that predominantly express a non-canonical junction. An example of this is the Plurinet gene BIRC5 (also known as survivin) for which 359 junction tags support expression of a non-canonical junction, whereas expression of the canonical junction was supported by only 14 tags. In this case, the non-canonical transcript isoform encodes a functional survivin protein (Caldas et al. 2005), a 142-amino-acid component of the chromosome passenger complex (CPC) and a key regulator of chromosome segregation (Jeyaprakash et al. 2007). The canonical transcript isoform corresponds to survivin2b, a 165-amino-acid protein containing an alternate exon, 2b. Structural evidence suggests that survivin2b is unlikely to be capable of forming a functional CPC (Jeyaprakash et al. 2007). Thus, while RNA-seq evidence suggests that survivin2b is expressed, its role in hES cells is unclear.
Transcript complexity associated with translating ribosomes
Our study is the first to show at a full genome scale the number of transcripts that can be actively associated with translating membrane-bound ribosomes. We have used a method originally developed for use with microarrays (Diehn et al. 2000), which is based on the separation of RNA associated with membrane-polysomes from cytoplasmic polysomes and non-coding RNAs. Although polysome association using this method can only be accurately assessed for the M/S-encoding subset of the transcriptome, this method has some advantages over other polysome isolation techniques. First, separation between the membrane-polysome RNA fraction and the cytosolic-polysome/free fraction is substantial, with this method classifying more than two-thirds of expressed genes (Fig. 4A). Second, a vast majority of RNA transcripts that are enriched in the membrane-polysome fraction can be annotated by the existence of signal peptides, transmembrane anchors, or annotations consistent with membrane localization (Supplemental Table S8), allowing accurate thresholding for the discovery of novel transcripts and transcript variants.
We found that a small percentage (12%) of expressed transcript variants (measured by junction usage) within M/S genes were not actively translated on membrane-bound polysomes. We also observed (as in the case of AGPAT4) that most expressed introns are not enriched within membrane-bound polysomes. It is likely that a proportion of these transcript variants undergo nonsense-mediated decay (NMD) and are degraded prior to translation (Chang et al. 2007). NMD can occur when a transcript expresses a premature stop codon that is not in the last exon (Chang et al. 2007). These variants may also be inhibited at the level of translation, for example, by miRNAs. Together, these results also add to the disparity between transcriptional activity and translational output, which can differ by up to three orders of magnitude (Chang and Stanford 2008).
Despite the lower level of complexity associated with membrane-bound polysomes, there were clear examples of loci encoding two or more distinct transcripts predicted to output significantly different protein isoforms. Two pertinent examples are FLT1, encoding a receptor for the developmental regulator VEGF, and the orphan receptor-encoding gene ROR1 (Dormeyer et al. 2008). Both of these genes encode tyrosine kinase receptors (Forrest et al. 2006). In each case, there is sequence-based evidence for expression of a longer membrane-spanning receptor isoform and a shorter secreted isoform. Previous studies have shown that the secreted isoform of FLT1 can act as a decoy receptor inhibiting the response of the membrane-bound receptor to activation from the ligand VEGF (Kendall and Thomas 1993). The action of VEGF is important for directing hES cell differentiation to endothelial lineages (Nourse et al. 2010), but VEGF is not necessary for maintenance of hES cell pluripotency. Expression of secreted decoy receptors may be a way for hES cells to inhibit the function of certain receptors under conditions favoring pluripotency, while ensuring that the cells can rapidly respond in the presence of differentiation signals, such as increased concentrations of growth factors like VEGF, which is used in some directed differentiation protocols.
Abundance of long 3′ UTRs
One of the striking aspects of our study was the high frequency of long 5′ and 3′UTRs and retained introns detected by sequencing of the complete transcriptome of hESC. In particular, 3′ UTRs up to 14 kb were easily identified in our study, and long 3′UTRS of >500 bp accounted for at least 9% of transcripts expressed by hESCs. Many transcripts with long 3′ UTRs would have remained unknown to date because their size makes them difficult to clone, and, as a result, such transcripts are likely under-represented in current gene models such as Ensembl. Indeed, using a recent version of Ensembl (Ensembl v62, June 2011), we find upwards of 600 UTRs that are longer by 500 bases or more than the longest corresponding annotated UTR (Supplemental Table S7). Previous work has suggested that ES cells preferentially express shorter 3′ UTRs (Ji et al. 2009) compared with differentiated cells. While we do not object to that idea, our study does suggest that there are very long 3′-UTR-containing transcripts expressed in hESCs (Fig. 3C), and we have found evidence that these transcripts can be translated.
Several recent studies have predicted the presence of long 3′ UTRs based on overlapping ESTs, RNA-seq, and/or predictive methods (Mortazavi et al. 2008; Thorrez et al. 2010). However, in most cases, the majority of these transcripts have not been independently validated. We have shown here, using size fractionation/transcriptome sequencing, that a majority of those transcripts with an extended 3′ UTR predicted by analysis of sequencing data are true extensions of the canonical transcript rather than independently transcribed RNAs. Interestingly, several of these 3′ UTRs overlap clusters of ESTs and predicted full-length non-coding RNAs. It is likely that some of these predicted non-coding transcripts are cloning artifacts that underestimate the full-length transcript. However, as highlighted in several recent reports, these may be 3′-UTR-associated transcripts (TASRs) produced by cleavage (Fejes-Toth et al. 2009; Kolle et al. 2009; Mercer et al. 2010). Although we do not object to this idea, we have shown that a number of these long 3′-UTR-containing transcripts are the primary transcripts associated with translating polysomes, because there is no decay in either the signal or the polysome association ratio along the length of the long 3′ UTR.
Defining independent transcripts by cellular fractionation and sequencing
RNA sequencing technology has provided the means with which to examine the hESC transcriptome at unprecedented resolution. Using short-read high-throughput sequencing, combined with two independent methods of subfractionating RNA populations, we have produced a detailed characterization of the hESC transcriptome. Based on mRNA sequencing, we uncovered a vast array of alternative and novel expression events in hESCs, including alternative splicing of exons, alternate UTR usage, intronic expression, and UTR extension events. We have demonstrated the power of combining size fractionation and cellular subfractionation with RNA-sequencing by using these methods to resolve cellular transcriptional complexity within hESCs. Through systematic integration of our RNA-seq data with other data sets, we have redefined existing hESC regulatory networks such as the Plurinet and cell-surface interaction models. In doing so, we have produced compelling evidence that the alternative and novel expression events we have identified impact significantly on the regulation of the pluripotent state. We envisage that this work will provide a comprehensive resource to guide further research into the transcriptomic and epigenomic regulation of pluripotency.
Methods
Culturing of hESCs
HES2 hESCs were cultured as previously described (Laslett et al. 2007; Kolle et al. 2009) on mouse embryonic fibroblast (MEF) feeder cells with media containing 20% knockout serum replacer (KOSR)/Dulbecco's modified Eagle's medium (DMEM) F12, 1% L-glutamine, 1% nonessential amino acids, 90 μM 2B-β-mercaptoethanol (GIBCO Invitrogen; http://www.invitrogen.com), and fibroblast growth factor 2 (FGF-2, 10 ng/mL; Millipore; http://www.millipore.com).
Isolation of RNA populations
Approximately 1 × 108 HES-2 hESCs were resuspended in 500 μL of cold isotonic lysis buffer (10 mmol/L KCl, 1.5 mmol/L MgCl2, and 10 mmol/L Tris-Cl at pH 7.4) and lysed by multiple passage through a 21-gauge needle. Nuclei and cellular debris were removed by centrifugation at 2000g for 2.5 min at 4°C. Cytoplasmic poly-adenylated mRNA was extracted using the Oligotex Direct mRNA kit (QIAGEN). Ribosomal RNA was depleted from poly-adenylated mRNA fractions using the RiboClear kit (Ambion).
Membrane-polysome fractionation
Membrane-associated polysomes were fractionated from cytosolic and other RNAs by sucrose density gradient centrifugation as described previously (Kolle et al. 2009).
Size fractionation
Fifteen micrograms of mRNA (isolated as above) was size-fractionated as follows: mRNA was incubated for 5 min at 55°C in a 25-μL premix solution containing 5% formaldehyde, 40% formamide, and 1× MOPS buffer. Formamide loading buffer (5 μL of 80% formamide, 10 mM EDTA, 1 mg/mL xylene cyanol, and 1 mg/mL bromophenol blue) was added to the RNA/premix, and the sample was immediately loaded onto a 1.2% agarose gel containing 0.6% formaldehyde in 1× MOPS buffer. The gel was run at 75 mA for 1.5 h, and gel slices were taken corresponding to the following sizes: 0–0.5 kb; 0.5–2 kb; 2–3.5 kb; 3.5–6.5 kb, and 6.5–20+ kb. Gel pieces were dissolved in RLT buffer (QIAGEN) for 30 min at 37°C, purified using the RNeasy Mini Kit (QIAGEN) according to the total RNA preparation protocol, and eluted in a final volume of 100 μL. One one-hundredth of the RNA from each fraction was run on the Agilent Bioanalyzer (Agilent) to confirm the correct size distribution (Supplemental Fig. S2) and approximate yield.
Production of complex cDNA libraries for transcriptome sequencing
Libraries were generated following the Whole Transcriptome Analysis Kit (Ambion) protocol with some modifications. One hundred to 150 ng of RNA from whole-transcriptome and polysome-fractionated samples and 15 ng of size-fractionated mRNA were first fragmented into 50–100-–bp fragments by digestion with RNase III (Ambion) for 10 min at 37°C followed by heat inactivation for 20 min at 65°C. Fragmented RNA was purified on a Microcon YM30 column (Microcon) and concentrated to a final volume of 8 μL. Adapter sequences incorporating a single-stranded random hexamer overhang to allow directional capture of RNA molecules were hybridized to the RNA fragments and then incubated with RNA ligase overnight at 16°C. The ligation products were reverse-transcribed and used as templates for library PCR amplification. Libraries were amplified for 15 cycles (mRNA and polysome fractionated) or 25 cycles (size fractionated). Transcriptome library products were fractionated on 3% TAE/agarose gels (Bio-Rad), and gel slices corresponding to 125–150-nt and 150–175-nt fragments were excised and purified using the QIAquick Gel Extraction Kit (QIAGEN) according to the manufacturer's guidelines, except that the gel slices were dissolved by mixing at room temperature rather than at 55°C.
Solid sequencing
Library molecules were clonally amplified onto 1-μm magnetic beads according to the SOLiD Template Bead Preparation protocol and sequenced using a SOLiD Analyzer as per the manufacturer's instructions (Applied Biosystems).
Mapping SOLiD sequence data to the human genome and exon–exon junctions
All SOLiD data were mapped to human genome (build hg19 [GRCh37]) and exon–exon junctions (generated from human Ensembl v55 gene model) using the whole-transcriptome pipeline in BioScope 1.2.1 (Life Technologies).
Quantitative real-time PCR
cDNA was produced from 20–100 ng of RNA using SuperScript III (Invitrogen). Real-time PCR was performed using Sybr Green (Applied Biosystems) on an ABI 7900HT (Applied Biosystems) using the following conditions: 2 min at 50°C, 10 min at 95°C, and 40 cycles of 15 sec at 95°C, 1 min at 60°C. For isoform-specific real-time PCR, at least one primer was designed across the isoform-specific splice junction. Real-time PCR primers are detailed in Supplemental Table S15.
Determination of Ensembl canonical transcript length and long 3′-UTR size validation
Sequence reads from size-fractionated libraries were mapped to the human genome release hg19 (GHRC37) as above. The expression of each transcript in each fraction was denoted in reads per kilobase per million mappable tags. Values were offset by addition of 0.1 RPKM and normalized by quantile using the Bioconductor function “quantile” in R. A transcript was designated into a size fraction based on the peak normalized value. If a transcript had a predicted long 3′ UTR, it was considered validated if the peak expression was in a size range that corresponded with the increase in the length of the transcript (for transcripts with long 3′ UTRs of >2 kb).
Membrane-associated enrichment analysis
We used a Bayes Theorem–based classification as described previously (Stitziel et al. 2004) to determine a threshold ratio r (MPR/CPR), where the probability of membrane-associated and secreted genes in a test set above r is >99% (Stitziel et al. 2004). To determine the test set, all Ensembl transcripts were functionally annotated using subcellular localization information from manually curated literature mining (Ingenuity IPA; http://www.ingenuity.com), and in silico consensus prediction of signal peptides (SP) and/or transmembrane domains (TM) as described previously (Kolle et al. 2009). From these, a high-confidence classification set of membrane-associated/secreted (M/S) and cytoplasmic/nuclear (C/N) genes was generated. The expression data were floored at 0.1 RPKM, and r was calculated for each gene in the test set where the expression level in either the MPR or CPR is above 2 RPKM.
Bioinformatics analysis of sequencing data
All bioinformatics analysis of sequencing data, including gene expression counts, alternative splicing prediction, and prediction of long 5′ and 3′UTRs, was performed using customized scripts in Perl or R. Details of individual analysis pipelines are supplied in the Supplemental Methods.
Comparison with existing data sets
ES data were compared to published microarray and RNA-seq samples, and details of individual comparisons are supplied in the Supplemental Methods.
Data access
Raw and processed data (.wig) have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession nos. GSE25842 and GSE24355; and tdf (coverage), BAM (read alignment), and Ensembl RPKM files are available at http://grimmond.imb.uq.edu.au/hES_transcriptome.
Acknowledgments
We acknowledge the Australian Stem Centre for supporting this project. S.M.G. is an NHMRC Senior Research Fellow, and N.C. is an ARC Postdoctoral Fellow. We acknowledge the ARC Centre for Functional and Applied Genomics Array Facility for expression profiling.
Footnotes
-
↵5 Corresponding author.
E-mail s.grimmond{at}imb.uq.edu.au.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.119321.110.
- Received December 21, 2010.
- Accepted August 23, 2011.
- Copyright © 2011 by Cold Spring Harbor Laboratory Press
Freely available online through the Genome Research Open Access option.








