
Observation and prediction of recurrent human translocations mediated by NAHR between nonhomologous chromosomes
- Zhishuo Ou1,9,
- Paweł Stankiewicz1,9,
- Zhilian Xia1,
- Amy M. Breman1,
- Brian Dawson1,
- Joanna Wiszniewska1,
- Przemyslaw Szafranski1,
- M. Lance Cooper1,
- Mitchell Rao1,
- Lina Shao1,
- Sarah T. South2,
- Karlene Coleman3,
- Paul M. Fernhoff3,
- Marcel J. Deray4,
- Sally Rosengren5,
- Elizabeth R. Roeder6,
- Victoria B. Enciso6,
- A. Craig Chinault1,
- Ankita Patel1,
- Sung-Hae L. Kang1,
- Chad A. Shaw1,
- James R. Lupski1,7,8 and
- Sau W. Cheung1,10
- 1 Department of Molecular & Human Genetics, Baylor College of Medicine, Houston, Texas 77030, USA;
- 2 Departments of Pediatrics and Pathology, University of Utah, Salt Lake City, Utah 84112, USA;
- 3 Children's Healthcare of Atlanta, Atlanta, Georgia 30033, USA;
- 4 Department of Neurology, Miami Children's Hospital, Miami, Florida 33155, USA;
- 5 Hartford Hospital, Hartford, Connecticut 06102, USA;
- 6 Department of Pediatrics, UTHSCSA, San Antonio, Texas 78229, USA;
- 7 Department of Pediatrics, Baylor College of Medicine, Houston, Texas 77030, USA;
- 8 Texas Children's Hospital, Houston, Texas 77030, USA
Abstract
Four unrelated families with the same unbalanced translocation der(4)t(4;11)(p16.2;p15.4) were analyzed. Both of the breakpoint regions in 4p16.2 and 11p15.4 were narrowed to large ∼359-kb and ∼215-kb low-copy repeat (LCR) clusters, respectively, by aCGH and SNP array analyses. DNA sequencing enabled mapping the breakpoints of one translocation to 24 bp within interchromosomal paralogous LCRs of ∼130 kb in length and 94.7% DNA sequence identity located in olfactory receptor gene clusters, indicating nonallelic homologous recombination (NAHR) as the mechanism for translocation formation. To investigate the potential involvement of interchromosomal LCRs in recurrent chromosomal translocation formation, we performed computational genome-wide analyses and identified 1143 interchromosomal LCR substrate pairs, >5 kb in size and sharing >94% sequence identity that can potentially mediate chromosomal translocations. Additional evidence for interchromosomal NAHR mediated translocation formation was provided by sequencing the breakpoints of another recurrent translocation, der(8)t(8;12)(p23.1;p13.31). The NAHR sites were mapped within 55 bp in ∼7.8-kb paralogous subunits of 95.3% sequence identity located in the ∼579-kb (chr 8) and ∼287-kb (chr 12) LCR clusters. We demonstrate that NAHR mediates recurrent constitutional translocations t(4;11) and t(8;12) and potentially many other interchromosomal translocations throughout the human genome. Furthermore, we provide a computationally determined genome-wide “recurrent translocation map.”
Reciprocal (non-Robertsonian) translocations are one of the most frequently occurring human chromosomal aberrations. Balanced reciprocal translocations are found in one in approximately 600 individuals (Van Dyke et al. 1983); thus, one in approximately 300 couples are at risk for having chromosomally unbalanced offspring. In most cases, carriers of balanced reciprocal translocations do not have an abnormal phenotype but may experience reproductive issues such as infertility or multiple miscarriages. Interestingly, by empirical studies, ∼6% of de novo apparently balanced translocations are associated with clinical abnormalities (Warburton 1991). Recently, it has been shown by molecular analyses (e.g., array comparative genomic hybridization) that up to 40% of the apparently balanced reciprocal chromosome translocations in patients with an abnormal phenotype are accompanied by a chromosome imbalance, either at the translocation breakpoints or elsewhere in the genome (Gribble et al. 2005; De Gregori et al. 2007; Sismani et al. 2008). Little is known, however, about the mechanisms or genomic sequences involved in the formation of non-neoplastic reciprocal translocations (Abeysinghe et al. 2003; Higgins et al. 2008).
To date, only three recurrent constitutional non-Robertsonian translocations have been described in humans. The most frequent translocation, t(11;22)(q23;q11), is the result of a rearrangement between palindromic AT-rich cruciform structures in 11q23 and in low-copy repeat (LCR) LCR22-3a in 22q11.2 (Zackai and Emanuel 1980; Kurahashi et al. 2000; Edelmann et al. 2001; Kurahashi and Emanuel 2001; Ashley et al. 2006; Kato et al. 2006). Carriers of the balanced constitutional t(11;22) are phenotypically normal but are at risk of having progeny with the supernumerary der(22) syndrome (Emanuel syndrome; MIM 609029), resulting from asymmetric 3:1 meiotic segregation (Zackai and Emanuel 1980; Shaikh et al. 1999; McDermid and Morrow 2002). Recently, Sheridan et al. (2010) reported a second recurrent AT-rich palindrome-mediated translocation t(8;22)(q24.13;q11.21). The third recurrent translocation examined, t(4;8)(p16;p23), has been shown to result from a crossover between the olfactory receptor-gene cluster LCRs (Giglio et al. 2002; Maas et al. 2007). However, for this latter t(4;8), the precise breakpoint location or crossover was not determined at nucleotide sequence resolution.
We report the molecular and clinical data on four unrelated families with the same recurrent unbalanced chromosomal translocation der(4)t(4;11)(p16.2;p15.4), leading to monosomy 4p16.2-pter and trisomy 11p15.4-pter. When isolated, each genomic imbalance results in distinct, well-characterized syndromes. Deletion of 4p16.3 includes two proposed critical regions WHSCR1 and WHSCR2 (Zollino et al. 2008), and manifests clinically as Wolf-Hirschhorn syndrome (WHS; MIM 194190). Duplication of the imprinted 11p15.5 region results in Beckwith–Wiedemann syndrome (BWS; MIM 130650) when paternally inherited, or Russell–Silver syndrome (RSS; MIM 180860) when maternally inherited. However, when the imbalances are present together, the clinical manifestation reported in the literature represent a unique phenotype with overlapping features of WHS, BWS, or RSS, depending on the parental origin of the duplicated chromosome.
We provide evidence that this recurrent translocation between chromosomes 4 and 11 [t(4;11)] arises by nonallelic homologous recombination (NAHR) mediated by interchromosomal paralogous LCRs. To investigate the genomic potential for recurrent translocations to occur by NAHR, we analyzed the genome-wide distribution of interchromosomal LCRs with >94% sequence identity and between 5–10 kb, 10–20 kb, 20–30 kb, 30–40 kb, 40–50 kb, or >50 kb in length. Remarkably, we identified 295, 352, 184, 105, 45, and 162 pairs of interchromosomal LCRs, respectively, that may potentially act as NAHR substrate pairs. To demonstrate utility of this computationally generated “potential recurrent translocation map” we sequenced the NAHR crossover sites within paralogous LCRs in one of these predicted recurrent translocations, der(8)t(8;12)(p23.1;p13.31). We demonstrate that NAHR between interchromosomal LCRs on nonhomologous chromosomes mediate recurrent constitutional translocations potentially throughout the human genome and provide a computationally determined genome-wide “recurrent translocation map.”
Results
Genomic rearrangements identified by chromosomal microarray analysis and chromosomal studies
Chromosomal microarray analysis (CMA V5 BAC, V6 BAC, and V6 OLIGO) (Cheung et al. 2005), initially performed on patients 1, 2, and 3 (see Supplemental Notes) identified similar genomic imbalances: terminal deletion of 4p16.2-pter and duplication of 11p15.4-pter. Subsequent fluorescence in situ hybridization (FISH) analysis confirmed the CMA results and revealed an unbalanced translocation der(4)t(4;11)(p16.2;p15.4) in each patient. Two other related patients (patients 4 and 4U; referred to as patient 2 and patient 3 in South et al. 2008) with identical cytogenetic breakpoints were obtained for further molecular analyses on five subjects in total.
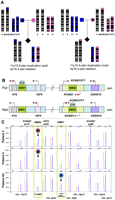
Methylation pattern and copy number analysis by MS-MLPA of patients with recurrent t(4;11). (A) Pedigrees illustrating the parental origin of the duplicated 11p material if the balanced translocation is present in the father (left), or the mother (right). (Blue) Paternal inheritance; (pink) maternal inheritance. (B) Schematic representation of the imprinted region at 11p15. The imprinted 11p15 region consists of two independent domains that are regulated by differentially methylated regions (DMR). The telomeric DMR1 is paternally methylated and regulates reciprocal expression of H19 and IGF2 genes. The centromeric DMR2 is maternally methylated and regulates expression of imprinted genes in this region including CDKN1C, KCNQ1OT1, and KCNQ1. CH3 represents the methylated allele. (C) Partial profile of MS-MLPA HhaI digestion/ligation products for patients 2, 4, and 4U (blue) compared to normal control (red). Increased peak intensities of IGF2 and KCNQ1 in patients 2, 4, and 4U indicate duplication of the 11p15 region. Gain of methylation in the HhaI sensitive DMR2 was detected in patients 2 and 4, whereas gain of methylation in the HhaI sensitive DMR1 was detected in patient 4U. The results demonstrated that maternal inheritance (M) of the duplicated 11p15 region in patients 2 and 4 and paternal inheritance (P) of the duplicated 11p15 region in patient 4U.
Translocation breakpoints map to LCR
Using a high-resolution SNP array, we fine-mapped the translocation breakpoints in patients 1, 2, 3, 4, and 4U to large LCRs, ∼359 kb in 4p16.2 and ∼215 kb in 11p15.4. As anticipated, patients 4 and 4U share the same breakpoints in both 4p16.2 and 11p15.4 (South et al. 2008). This high-resolution genome analysis also confirmed that the WHS critical regions WHSCR1 and WHSCR2 at 4p16.3 were deleted and the BWS and RSS genomic regions containing imprinted domains at 11p15.4 were duplicated (Table 1).
Summary of the parental origin, genomic rearrangement, and breakpoints of patients with recurrent t(4;11)
Parental origin of rearranged genomic sequences
FISH analysis using the same probes on the parental samples from patients 1 and 3 showed a balanced t(4;11) in their fathers. No parental samples were available for patient 2.
MS-MLPA analysis of DNA samples from patients 2, 4, and 4U showed increased peak intensities of IGF2 and KCNQ1, consistent with a duplication of the 11p15 region. The methylation studies revealed a difference in the methylation pattern in the 11p15 imprinted region in patients 2, 4, and 4U. Gain of differentially methylated imprinting centers DMR2 methylation was detected in patients 2 and 4, whereas gain of DMR1 methylation was detected in patient 4U (Fig. 1). These patterns are indicative of a maternal origin for the duplicated 11p15 region in patients 2 and 4, and paternal origin of the duplicated 11p15 region in patient 4U, consistent with the respective clinical findings.
Genomic architecture and sequence analyses
Bioinformatic analysis was performed comparing the breakpoint regions within the LCR blocks in 4p16.2 with that of the 11p15.4 region (genome build GRCh37/hg19). This analysis revealed that a 359-kb LCR cluster in 4p16.2 (genomic position 3.88–4.24 Mb) shares 204 kb of significant homology (DNA sequence identity >94%) with the 215-kb genomic segment on 11p15.4 (genomic position 3.41–3.62 Mb). Both paralogous regions harbor all the breakpoints of the five t(4;11) cases we studied (Fig. 2).
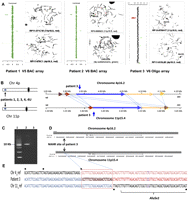
Identification of the LCR pairs acting as potential substrates for interchromosomal NAHR, resulting in the t(4;11) formation. (A) CMA profile of DNA from patient 1 (left). The mean normalized log2 (Cy3/Cy5) ratio of each BAC clone is plotted on the x-axis as dots with error bars, and arranged along the vertical axis from chromosome 1 at the top to chromosomes X and Y at the bottom. All 11 clones on the 4p subtelomeric region showed displacement to the left, indicating a deletion of 4p16.2-p16.3 material, whereas five clones on the 11p subtelomeric region are shifted to the right, indicating a duplication of 11p15.5 material in the patient versus the reference DNA. The results of FISH analysis of metaphase chromosomes prepared from the patient's peripheral blood lymphocytes with probe RP11-371C18 specific for chromosome region 11p15.5 (red) show the presence of 11p15.5 material on the derivative chromosome 4 [der(4)] (arrow), whereas the results of FISH analysis with probe RP11-478C1 specific for chromosome region 4p16.3 (red) show the deletion of 4p16.3 material (arrow). The CMA and FISH analyses revealed an unbalanced translocation between 4p16 and 11p15. CMA profile of DNA from patient 2 tested on V5 BAC array (middle). As in patient 1, the 4p deletion and 11p duplication were detected by displacement of 11 clones and five clones on the corresponding region, respectively. The results of FISH analysis with the PAC probe RP5-998N23 (red) specific for 11p15.5 indicate the presence of 11p15.5 material on der(4), whereas the results of FISH analysis in patient 2 with the 4p subtelomeric probe D4S3359 (green) show the deletion of 4pter material. CMA profile of DNA from patient 3 tested on BAC emulated Version 6 OLIGO array (right) revealed the same genomic aberrations as patients 1 and 2. The results of FISH analysis with RP13-870H17 (red) specific for 11p15.5 indicate the presence of 11p15.5 material on der(4), whereas FISH analysis for patient 2 with probe RP11-613L20 specific for chromosome region 4p16.3 (red) show the deletion of 4p16.3 material. (B) Five t(4;11) cases mapped this to NAHR substrate pair. Summary of the sequence similarity BLAST2 analysis of the 350-kb sequence surrounding the 4p16.2 (top) and 11p15.4 (bottom) breakpoint regions. The different color horizontal arrows depict the homologous LCR subunits. The numbers above and below the lines represent genomic distance (megabases) from 4p and 11p telomeres, according to NCBI human genome build 37 (GRCh37/hg19; Feb. 2009). The regions between 4p16.2 and 11p15.4 connected by dotted lines are >94% sequence identical. The translocation breakpoints in patient 3 are located in the homologous LCRs indicated by the vertical arrows, implying a NAHR-based recombination mechanism. (C) Ethidium bromide stained agarose gel image of the ∼9-kb t(4;11) patient 3-specific junction fragment amplified by long-range PCR with primers harboring trans-morphisms specific for each 4p16.2 and 11p15.4 LCR (lane 2). Lane 1 represents the DNA marker with the 10-kb band indicated to the left. Lane 3 represents a negative control. (D) The NAHR cross-over site for patient 3 is located in a 130-kb subunit with 94.7% DNA sequence identity. UCSC Genome Browser view of the homologous LCR blocks of the same orientation in the chromosome regions 4p16.2 (top) and 11p15.4 (bottom) indicated by the gray bars. The black arrows indicate the NAHR site for patient 3 determined by sequence analysis. (E) DNA sequence alignment of the PCR amplified translocation junction fragment in patient 3 (middle sequence). The NAHR site was narrowed to a 24-bp segment (red rectangle) with 100% DNA sequence identity between chromosomes 4 (top) and 11 (bottom). Blue nucleotides indicate alignment with the chromosome 11 sequence, red nucleotides indicates alignment with the chromosome 4 sequence, purple nucleotides indicate SNPs, and trans-morphic mismatches are indicated by black dots above or below the sequence.
To further delimit the exact crossover and the nature of the surrounding sequences in proximity to the strand exchange, we sequenced the recombinant NAHR site. In patient 3, sequencing of the long-range PCR products amplified from the patient's DNA enabled narrowing of the NAHR site. PCR amplification of the genomic DNA from patient 3 with forward primer GCCTAAACTATTTCTCAGCAAGGAGGAAGG and reverse primer CCCGAGTGGAGTTCTAGTATTTAAGGTGCTT revealed a patient-specific ∼9-kb product (Fig. 2C). Subsequent DNA sequencing analysis allowed us to narrow the NAHR sites to the 24-bp regions between chr4:3,940,888–3,940,911 and chr11:3,426,699–3,426,722 (Fig. 2E). The crossover occurred within interchromosomal, paralogous, directly oriented (centromere to telomere direction) LCRs of ∼130 kb in length and 94.7% DNA sequence identity located in olfactory receptor gene clusters; sequence analyses of a 10-kb flanking region (5 kb on each side of the breakpoints) revealed the recently proposed homologous recombination “hotspot” associated sequence motif CCNCCNTNNCCNC 1287 bp and 3209 bp telomeric and 636 bp centromeric to the chromosome 11p15.4 breakpoint and 642 bp centromeric to the chromosome 4p16.2 breakpoint. This 13-bp homologous recombination hotspot associated motif is purported to bind PRDM9; a zinc finger protein that causes histone H3 lysine 4 trimethylation (Baudat et al. 2010; Myers et al. 2010; Parvanov et al. 2010).
Clinical manifestations of chromosome imbalances
Clinical manifestations of WHS, BWS, and RSS along with the results of current (this work) and reported t(4;11) studies are summarized in Table 2. Our patients 1 and 3 with the der(4)t(4;11) of paternal origin demonstrate a unique phenotype. Their growth parameters are within the normal range, resembling neither WHS nor BWS. The facial features of patient 1 include arched eyebrows, short philtrum, micrognathia, and facial asymmetry, which partially resemble WHS. Features of patient 3 include micrognathia, seizures, and feeding difficulties consistent with WHS, whereas renal and cardiac anomalies occur in WHS and BWS. Macrosomia, macroglossia, and abdominal wall defect, which are defined as major features for BWS, were not detected in patients 1 and 3. Our data are consistent with previous reports, describing a unique phenotype with dominance of WHS features over the BWS features (Russo et al. 2006; Mikhail et al. 2007; South et al. 2008; Thomas et al. 2009).
Phenotypic features of WHS, BWS, and RSS compared to those found in the presented and reported patients
Patient 2, with a methylation pattern consistent with a maternal origin of t(4;11), has partial features of WHS and RSS, including prenatal onset of growth deficiency seen in both WHS and RSS. Hypertelorism, high arched eyebrows, cleft palate, and downturned mouth corners are seen in WHS, and the low nasal root, prominent forehead, and facial asymmetry are typical for RSS. Interestingly, most of these features were also observed in patient 4, in whom the derivative chromosome was also found to be maternally inherited (Table 2; Supplemental Notes).
A genome-wide recurrent translocation map
NAHR is a major mechanism for recurrent interstitial (i.e., within and between homologous chromosomes) rearrangements using either directly oriented (for deletions and duplications) or inversely oriented (for inversions) LCRs as homologous recombination substrates (Stankiewicz and Lupski 2002). We now sought to identify genome-wide interchromosomal LCRs that could potentially mediate rearrangements of nonhomologous chromosomes via NAHR.
The formation of a stable reciprocal translocation mediated by interchromosomal NAHR between LCRs is dependent upon the orientation of the LCRs and the chromosome arms involved. Interchromosomal NAHR between LCRs mapping in the same orientation and on the same chromosome arms (i.e., p-arm of one chromosome versus the p-arm of the other) or those in inverted orientation on opposite chromosome arms (i.e., p-arm from one chromosome versus q-arm of the other) are predicted to result in NAHR-mediated stable, monocentric reciprocal translocation chromosomes. In contrast, LCRs in opposite orientation on the same chromosome arms or those in the same orientation on opposite chromosome arms are predicted to result in either unstable dicentric or acentric chromosomes (Fig. 3).
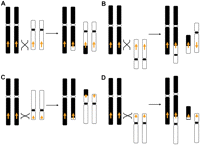
Potential outcomes of interchromosomal NAHR mediated by LCRs. Nonhomologous chromosomes are shown in black and white with the centromeres shown as circles. The arrows indicate the orientation of the LCRs. Only interchromosomal LCRs located in the same orientation on the same chromosomal arms (i.e., q-arm to q-arm) (A), or those in opposite orientation on different chromosomal arms (i.e., q-arm to p-arm) (C) are predicted to result in stable, monocentric reciprocal translocations. In contrast, LCRs located on the same chromosomal arm in opposite orientation (B) or on different chromosomal arms in the same orientation (D) would lead to unstable dicentric or acentric chromosomes, resulting in chromosome breakage or loss, respectively. Note: Both HR substrate orientation (direct versus inverted) and chromosomal arm location (p versus q), required for viable interchromosomal recombinant products.
We analyzed the human haploid genomic reference DNA sequence (NCBI36/hg18) for interchromosomal LCRs of >5 kb in length and >94% DNA sequence identity, and identified 1902 sequences that correspond to our inclusion criteria. We further segmented our genomic sequence analysis to include LCRs with 5–10 kb, 10–20 kb, 20–30 kb, 30–40 kb, 40–50 kb, and >50 kb in length, resulting in the identification of 295, 352, 184, 105, 45, and 162 pairs of interchromosomal LCRs, respectively. We also constructed a global view of potential translocations mediated by the interchromosomal NAHR between LCRs with >94% identity and >5 kb in size; parameters empirically shown to support recurrent translocation (Fig. 4). The global view was divided into 25% each, based on the size of the LCRs for easy visualization (Fig. 4A–D). Some of the potential interchromosomal NAHR pairs represent olfactory receptor gene repeats (Fig. 4, green lines). Importantly, both known recurrent translocations, t(4;8) (Giglio et al. 2002) and t(4;11), reported herein, are predicted by this translocation map (Fig. 4, red lines).
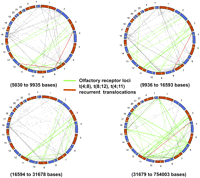
Recurrent translocation map. A global genomic view of interchromosomal LCR pairs with >5 kb in size and >94% DNA sequence identity represented by dotted lines and distribution divided into four groups based on the size of LCR. To create this plot we circularized the genome using polar coordinates. We then connected points between a pair of chromosomes linked by LCRs satisfying our size sequence identify criteria (see Supplemental Table 3). The midpoints of the LCRs were used to identify each segment with a single location on each chromosome. The red dotted lines indicate the translocations identified in our patient database, while the green dotted lines represent the olfactory receptor LCRs. (A) The size of LCR ranges from 5030 to 9935 bases in the first 25%. (B) The size of LCRs range from 9936 to 16,593 bases for the second 25% of LCRs. (C) The size of LCRs range from 16,594 to 31,678 bases for the third 25% of LCRs. (D) The size of LCRs range from 31,679 to 754,003 bases for the final 25% of LCRs.
Predicted recurrent translocations
To test the hypothesis that the identified 1902 candidate interchromosomal LCRs or 1143 LCR NAHR substrate pairs can potentially mediate different recurrent chromosomal translocations, we queried our patient database and found 105 patients with unbalanced translocations detected by array CGH analysis using CMA V6 OLIGO (44K), V7 OLIGO(105K), and V8 OLIGO (180K) arrays; custom whole genome arrays with varying densities of backbone interrogating oligonucleotides. In addition to the three cases with t(4;11), we found seven cases with t(4;8)(p16.2;p23.1), five with the derivative chromosome 4, and two with the derivative chromosome 8, and two cases with der(8)t(8;12)(p23.1;p13.31) (patients 5 and 6).
Bioinformatic analysis of the t(8;12) breakpoint regions revealed an ∼579-kb LCR cluster (genomic position 7.52-8.10 Mb) on chromosome 8p23.1 and an ∼287-kb LCR cluster on chromosome 12p13.31 (genomic position 8.31–8.60 Mb) that share 285 kb of significant homology (DNA sequence identity >94%). Both paralogous regions harbor all four breakpoints of the t(8;12) cases we studied (Fig. 5; Supplemental Table 1).
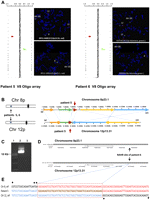
Identification of the LCR pairs acting as potential substrates for interchromosomal NAHR resulting in the t(8;12) formation. (A) CMA profiles of DNA from patients 5 (left) and 6 (right) tested on Version 8 OLIGO array (left) revealed a 7.9-Mb deletion of chromosome bands 8p23.1-pter and an 8.2-Mb duplication of chromosome bands 12p13.31-pter. The results of FISH analysis (right) with the probe RP11-440E12 (patient 5; red) or VIJTYAC14 (patient 6; green) specific for 12p33.33 indicate the presence of chromosome 12 material on the der(8), whereas the results of FISH analysis with probe RP11-1001A23 (patient 5; red) or D8S504 (patient 6; green) specific for the chromosome region 8p23 show the deletion on chromosome 8. (B) Two t(8;12) cases mapped this to NAHR substrate pair. Summary of the sequence similarity BLAST2 analysis of an ∼200-kb sequence surrounding the 8p23.1 and 12p13.31 breakpoint regions (bottom). (C) Ethidium bromide stained agarose gel image of the patient 5–specific ∼12-kb t(8;12) junction fragment amplified by long-range PCR with primers harboring trans-morphisms specific for each 8p23.1 and 12p13.31 LCR (lane 2). (Lane 1) The DNA marker with the 10-kb band indicated to the left. (Lane 3) A negative control. (D) The NAHR crossover site for patient 5 is located in a 7.7-kb subunit with 95.2% DNA sequence identity. UCSC Genome Browser view of the homologous LCR blocks in the 8p23.1 (top) and 12p13.31 (bottom) chromosome regions. (E) DNA sequence alignment of the PCR amplified translocation junction fragment for patient 6 (middle sequence). The NAHR site was narrowed to a 55-bp segment (red rectangle) with 100% sequence identity between chromosomes 8 (top, red) and 12 (bottom, blue).
DNA sequencing of the t(8;12) patient 5–specific ∼12-kb LR-PCR product amplified with forward primer TTCTTAATATCACTTTTCCCCACTCTAGTTC and reverse primer GTGTAAGACGTCGATACGATACGGCACTTC, enabled narrowing the NAHR sites to the 55-bp regions between chr8:7,884,979–7,885,033 and chr12:8,374,239–8,374,293 flanked by two paralogous sequence variants (Fig. 5). Sequence analyses of 10-kb flanking regions revealed a homologous recombination “hotspot” sequence motif CCNCCNTNNCCNC 2960 bp centromeric to the chromosome 8p23.1 breakpoint.
Locations of the LCR and breakpoints from empiric data
The chromosomal distribution of the identified 1902 LCR sequences is detailed in Figure 6 and Supplemental Table 4. We calculated that 33.64% of all 1902 interchromosomal LCRs map to the subtelomeric most distal 5 Mb regions of all chromosomes. From our clinical aCGH patient database, there were 105 patients with unbalanced constitutional chromosomal translocations; of these, 85 translocations had breakpoints that were resolved using custom whole genome clinical microarrays (105K or 180K). The line graph shows the density estimate for the distribution of LCRs on each chromosome. The red hash marks below the curve show the location of the LCR midpoints (Fig. 6). The 170 breakpoint coordinates from the aCGH-detected 85 translocations are depicted in green hash marks below the line plot.
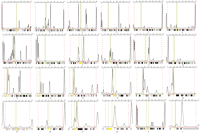
The distributions of 1143 LCR potential substrate pairs with relation to chromosome position. For each chromosome, the distribution of the LCR (x-axis) is plotted against the frequency of the LCR (y-axis) along the entire chromosome. The red dotted vertical lines on both ends of the chromosome represent the first and last 5 Mb of each chromosome; the yellow line represents the centromeric region as aligned with the karyogram on the bottom of each graphic. The red hash marks underneath the plots depict the density of the LCRs, while the green bar represents the distribution of the 170 breakpoint regions from our genome-wide unbalanced translocation data.
To determine whether the observed translocation breakpoints are more closely located to the LCR positions than expected by chance, we computed the minimum distance between the distal endpoint (furthest from telomere) of each translocation event and the LCRs located on that chromosome. We used the absolute value of the difference between the coordinate of the translocation distal breakpoint and the midpoints of the LCRs. In order to create a reference distribution for these values, we performed a Monte Carlo simulation, drawing 10,000 random coordinates along each chromosome and computing the minimum LCR absolute distance statistic for each draw. We standardized our observed translocation-LCR distance values by subtracting the simulation-derived mean and dividing by the simulation-derived standard deviations determined by the random draws for each chromosome. We then performed an analysis of the standardized distances to the LCRs using a Wilcoxon signed rank test with continuity correction, and this analysis determines a P-value of 1.410 × 10−7 for the observed translocation breakpoints against the null hypothesis that the breakpoints are located a random distance from the predicted LCRs against the alternative hypothesis that the breakpoints are closer to the LCRs than would be expected by chance. The median standardized distance between the observed and expected distance from a breakpoint to an LCR is −0.6285 standardized units, indicating together with the Wilcoxon P-value that the observed breakpoints are significantly closer to the LCRs than would be expected by chance.
Discussion
Balanced reciprocal translocations are one of the most commonly observed chromosomal abnormalities in humans. However, the molecular mechanisms of formation of these rearrangements remain elusive with the exception of the recurrent translocations, t(11;22)(q23;q11), t(8;22)(q24.13;q11.21), and t(4;8)(p16;p23) (Kurahashi et al. 2000; Edelmann et al. 2001; Kurahashi and Emanuel 2001; Giglio et al. 2002; Sheridan et al. 2010). NAHR between interchromosomal LCRs on different (i.e., nonhomologous) chromosomes has been suggested to result in chromosomal translocations (Lupski 1998; Stankiewicz and Lupski 2002). We now provide molecular evidence to support NAHR between interchromosomal LCRs as a potential major mechanism for recurrent reciprocal translocations.
For NAHR to occur, it has been proposed that 300–500 bp of perfect DNA sequence identity is the minimal efficient processing segment required to mediate meiotic NAHR between intrachromosomal LCRs (Reiter et al. 1998). LCRs of >10 kb in size and with >95%–97% DNA sequence identity have been shown empirically to result in the most common recurrent NAHR-mediated interstitial or intrachromosomal genomic rearrangements (Lupski 1998; Stankiewicz and Lupski 2002, 2006; Shaw and Lupski 2004; Lupski and Stankiewicz 2005; Sharp et al. 2005). The distance between LCRs is another factor apparently influencing NAHR, since larger-sized genomic rearrangements utilizing LCRs located further apart often correlate with large LCRs (Lupski 1998; Stankiewicz and Lupski 2002).
The “rules” for NAHR mediated interstitial chromosomal rearrangements have enabled predictions of genomic instability regions prone to deletions/duplications causing genomic disorders (Sharp et al. 2005). Five novel genomic disorders have been elucidated by this predictive “interstitial rearrangement map” and informed design of human genomic microarrays for array CGH analysis; these include: 1q21.1 microdeletion/microduplication (Brunetti-Pierri et al. 2008; Mefford et al. 2008), 15q13.3 microdeletion (Sharp et al. 2008; Ben-Shachar et al. 2009; Miller et al. 2009; van Bon et al. 2009), 15q24 microdeletion/microduplication (Sharp et al. 2007; El-Hattab et al. 2009, 2010), 17q12 microdeletion/microduplication associated with renal disease, diabetes, and epilepsy (Mefford et al. 2007; Moreno-De-Luca et al. 2010; Nagamani et al. 2010), and 17q21.3 microdeletion/microduplication (Koolen et al. 2006; Sharp et al. 2006; Shaw-Smith et al. 2006).
The understanding of the NAHR mechanism combined with the availability of the human genome sequence (International Human Genome Sequencing Consortium 2004) enabled the use of bioinformatics as a tool to predict hotspots for genomic instability that may be prone to recurrent translocations. Genome-wide bioinformatic analyses revealed 1902 interchromosomal LCR substrates or 1143 pairs, >5 kb in size and sharing >94% sequence identity that can potentially mediate recurrent chromosomal translocations via NAHR.
From our clinical aCGH patient databases, there were 105 patients with unbalanced constitutional chromosomal translocations with 85 of these translocations identified using custom whole genome clinical microarrays (105K or 180K). We found three recurrent translocations matching our predictions; seven t(4;8)(p16.2;p23.1), three t(4;11)(p16.2;p15.4), and two t(8;12)(p23.1;p13.31) with a detection rate of 12/105 = ∼11%. Although NAHR-mediated reciprocal translocations appear to be rare events, we contend that the frequency of NAHR in reciprocal translocations at the level of the genomic sequence has not been systematically assessed. Prior to our analysis, only two translocations, t(11;22)(q23;q11) and t(8;22)(q24.13;q11.21), have had their breakpoints sequenced. The t(4;11) reported in the current study is only the third recurrent translocation, in which the breakpoint region is established at the nucleotide sequence level. This was accomplished using arrays to identify unbalanced translocations, which narrowed the translocation breakpoint regions to a genome resolution level high enough for PCR amplification of the junction fragments.
Although the frequency of reciprocal translocation is relatively common, most are detected by GTG banded chromosome analysis. The resolution of banding is usually between 5–10 Mb and the band assignment for the breakpoint can deviate 10–20 Mb. This poor genome resolution renders translocation breakpoint assignments by karyotype analysis reported in the literature inherently inaccurate and unreliable for precise breakpoint mapping. Historically, FISH analysis using tiling BAC clones within the predicted translocation junction chromosome bands has been used to identify the clone that spans the breakpoint region. Currently, the focus of most of the studies investigating reciprocal translocation breakpoints utilizes array technology to determine whether an apparent balanced translocation by GTG banding analysis is indeed balanced (De Gregori et al. 2007; Fantes et al. 2008; Schluth-Bolard et al. 2009) rather than breakpoint identification per se.
Of the published 31 balanced translocations in which breakpoints have been mapped, none had homologous LCRs at the breakpoint regions (Baptista et al. 2008). These balanced rearrangements would not be detected by array CGH analysis. The fine mapping of balanced translocation breakpoint regions performed was focused predominately in segments containing genes, thus less likely to be associated with LCRs (Baptista et al. 2008). We believe the apparent discrepancy between our analysis and that published by Baptista et al. (2008) is due to the fact that the translocations in our database were unbalanced with breakpoints mapping in the distal portions of the chromosome arms that are enriched with interchromosomal LCRs (Linardopoulou et al. 2005). Telomeric imbalances that are smaller in size are less likely to be embryonically lethal and therefore may be viable. Translocations resulting in large imbalances are likely to be embryonically lethal, whereas translocations with small imbalances can be viable. Potentially, several thousands of translocation breakpoints may need to be mapped at high-resolution to assess reliable representative frequencies.
Recently, seven t(4;11)(p16.2;p15.4) cases with clustered breakpoints from six unrelated families have been reported (Russo et al. 2006; Mikhail et al. 2007; South et al. 2008; Thomas et al. 2009) (Table 1). The clinical features of the t(4;11) patients with 4p monosomy and 11p trisomy in these studies represent a unique combination of phenotypes with overlapping features of WHS and BWS or RSS, depending on the parental origin of the duplicated chromosome 11 (Table 2). The WHS phenotypic spectrum was observed more often than BWS (Russo et al. 2006; Mikhail et al. 2007; South et al. 2008; Thomas et al. 2009).
We describe the results of molecular cytogenetic and clinical analyses in three novel unrelated subjects and two published cases from one family (South et al. 2008) with an unbalanced translocation der(4)t(4;11)(p16.2;p15.4), resulting in segmental 4p monosomy and 11p trisomy with the translocation breakpoints mapping in the same LCR paralogues. Our high-resolution SNP array studies clearly demonstrated deletion of the WHS critical region and duplication of the BWS/RSS critical region in patients 1–4U. Family histories and/or MS-MLPA studies revealed paternal origin for the aberrations in patients 1, 3, and 4U and maternal origin in patients 2 and 4.
Although seven t(4;11)(p16.2;p15.4) cases with clustered breakpoints have been described, the specific breakpoints and DNA sequence of the junction on 4p16.2 and 11p15.4 have not been well characterized. Our genomic analysis of the breakpoint regions revealed the 204-kb homologous LCR portion of >94% interchromosomal DNA sequence identity. All analyzed translocation breakpoints mapped within the homologous subunits, suggesting that NAHR between the LCRs located on chromosome 4p16.2 and 11p15.4 is the likely mechanism for their formation. This hypothesis was further substantiated by breakpoint sequencing of two selected translocations, t(4;11) and t(8;12). As anticipated the breakpoints mapped to the “recurrent translocation map” identified LCR substrates.
Some of the other predicted recurrent translocations, however, may be underrepresented since derivative chromosomes with longer segments of imbalance are more likely to be incompatible with life. High-resolution genome analyses of additional balanced and unbalanced translocations will be required to further confirm the utility of our “recurrent translocation map.”
It is also likely that both balanced and unbalanced translocations are under-ascertained when studied by karyotype analysis alone. Because the subtelomeric regions of most chromosomes have a GTG-negative (light) banding pattern, the reciprocal exchange of chromosomal material at subtelomeres is likely to be cryptic. Human subtelomeric regions have been completely sequenced and it has been shown that the subtelomeric segmental duplicated region (also known as subtelomeric repeats) in humans make up 25% of the most distal 500 kb and 81% of the most distal 100 kb in human genome (Riethman 2008). These duplicated segments predispose to different types of genomic rearrangements (Linardopoulou et al. 2005). We find that 162 LCRs map to the most distal 100 kb on each chromosome, and 506 LCRs map to the most distal 500 kb. Interestingly, 22.97% of the breakpoints from the 85 unbalanced rearrangements are located within the first 5 Mb from each end of the chromosomes (Supplemental Table 2). These results support the hypothesis that segmental duplications in subtelomeric regions mediate translocations by interchromosomal NAHR mechanisms.
DNA sequencing analysis in one patient allowed us to narrow the NAHR sites to the 24-bp regions between chr4:3,940,888–3,940,911 and chr11:3,426,699–3,426,722 within interchromosomal paralogous directly oriented (centromere to telomere direction) LCRs of ∼130 kb in length and 94.7% DNA sequence identity located in olfactory receptor (OR) gene clusters. As much as half of the members of the OR gene family (∼852 genes) intercept copy number variation regions suggesting NAHR plays a major role in remodeling of the OR gene family (Young et al. 2008). In addition, the translocation map shows 25% of these OR genes intercept the first 5 Mb of the subtelomeric regions from each end of the chromosome.
The subtelomeric LCRs are very polymorphic and their structures differ between different individuals and populations, likely as a result of gene conversion events. The complexity of the genomic architecture for regions that are prone for rearrangements among different population is still largely unknown. For example, the 17q21.31 microdeletion apparently resulted from a meiotic recombination between the H1 and the inversion-bearing H2 haplotype, which is carried at a frequency of ∼20% in populations of European ancestry (Stefansson et al. 2005). However, the frequency of this inversion polymorphism has yet to be determined in other populations. Furthermore, both structural and nucleotide sequence diversity within LCRs (i.e., paralogous sequence and structural variations) were observed in the 24-kb-long Charcot-Marie-Tooth disease type 1A, CMT1A-REP, LCRs that sponsors deletion and duplication of this genomic region (Lindsay et al. 2006), high frequency of retroelement insertions, accelerated sequence evolution after duplication, and extensive paralogous gene conversion were observed. These findings were consistent with the recent observation that repetitive elements such as LINE-1 and Alu may also contribute significantly to structural variations (Beck et al. 2010; Ewing and Kazazian 2010; Huang et al. 2010; Iskow et al. 2010; Lupski 2010). Additionally, the observation of thousands of new structural variants with sizes ranging from kilobases to megabases using single molecule analysis only begins to reveal the magnitude of the structural variation complexity of the human genome (Conrad et al. 2010a,b; Pang et al. 2010; Park et al. 2010; Teague et al. 2010). The landscape and impact of these genomic variants that are individually rare but collectively common in the human population remains to be explored.
We show that the interchromosomal LCR harboring the olfactory receptor gene cluster in 11p15.4 is a novel genomic instability region that mediates the relatively common recurrent constitutional non-Robertsonian translocation t(4;11)(p16.2;p15.4) by NAHR. We identified the interchromosomal LCRs that can potentially mediate recurrent chromosomal translocations between nonhomologous chromosomes to construct a computationally derived “recurrent translocation map” and provide experimental evidence by virtue of t(8;12) breakpoint mapping to support the predictions. Our findings suggest interchromosomal LCR-mediated NAHR may be a major mechanism for recurrent constitutional translocation formation, in particular within the subtelomeric regions.
Methods
Informed consents approved by the Institutional Review Board for Human Subject Research at Baylor College of Medicine (BCM) were obtained for further delineation of the breakpoints and publication of photographs.
We obtained clinical information for three patients with the t(4;11) translocation, designated as patients 1 to 3. The Supplementary Notes contain detailed clinical information for these patients. We also obtained DNA from two other reported patients from a family with a similar t(4;11) translocation (South et al. 2008) (patient 2 and patient 3), designated here as patient 4 and patient 4U.
Chromosome microarray analysis
Blood samples were obtained from patients and their family members referred to the Medical Genetics Laboratories at Baylor College of Medicine for chromosomal microarray analysis (CMA). Samples from patients 1 and 2 were analyzed on the CMA V5 BAC array, and patient 3 on the CMA V6 OLIGO array.
Version 5 BAC array contained 853 BAC/PAC clones designed to cover genomic regions of 75 known genomic disorders, all 41 subtelomeric regions, and 43 pericentromeric regions. Version 6 BAC array consisted of 1472 BAC/PAC clones, covering ∼150 genomic disorders, all 41 subtelomeric regions up to 12 Mb, and 43 pericentromeric regions with backbone coverage of every chromosome at the 650-band level of cytogenetic resolution (http://www.bcm.edu/geneticlabs/?pmid=16207). The BAC microarrays were designed and manufactured at Medical Genetics Laboratories as previously described (Cheung et al. 2005). The procedures for DNA digestion, labeling, and hybridization as well as data analysis were performed as described (Lu et al. 2007). The BAC emulated Version 6 OLIGO array was comprised of ∼42,460 oligonucleotides representing 1400 BAC clones. The 42.46 K oligonucleotides (oligos) were selected from initial testing of 105,000 oligos derived from the Agilent eArray library with strict selection criteria and removal of repetitive sequences to ensure optimal performance with greater dynamic range (Ou et al. 2008). This targeted 42.46 K OLIGO array (V6 OLIGO) corresponds to genomic regions covered by the V6 BAC arrays and was manufactured in a 4 × 44 K format with an average of 28–30 oligos per region previously covered by a single BAC clone. The procedures for DNA digestion, labeling, and hybridization as well as data analysis were performed as previously described (Probst et al. 2007).
Affymetrix Genome-Wide SNP Array 6.0 arrays (Affymetrix, Inc.) were employed to define the breakpoints on chromosomes 4, 8, 11, and 12. Analysis was performed according to the Genome-Wide Human SNP Nsp/Sty Assay kit 5.0/6.0 protocol provided by the supplier. The arrays were scanned using a GeneChip Scanner 3000 7G (Affymetrix, Inc.) and results were analyzed using Genotyping Console version 2.1 software.
Cytogenetic and FISH analyses
GTG-banded chromosome analysis was performed using standard protocols. FISH was performed using standard procedures with BAC clones labeled by nick translation with SpectrumOrange or SpectrumGreen (Abbot). BAC clones specific for human chromosome regions 4p16.2 and 11p15.4, as well as 8p23.1 and 12p13.31 for confirmation of the CMA findings, were selected from UCSC Genome Browser (http://www.genome.ucsc.edu).
Long-range PCR and DNA sequencing
Long-range PCR primers were designed to harbor at least three nucleotides specific for one LCR on chromosome 4p16.2 or 8p23.1 and in the other primer for 11p15.4 or 12p13.31, respectively, to allow preferential amplification of the predicted chimeric fragment containing the junction between parts of these paralogous LCRs on nonhomologous chromosomes, but not the fragment of the original LCRs. The primers were designed using Primer 3 software (http://frodo.wi.mit.edu/primer3). Amplification of 8–15-kb fragments was performed using Takara LA Taq polymerase (Takara Bio), following the manufacturer's protocol. Briefly, we used 25-μL reaction mixtures containing 100 ng of genomic DNA, 0.4 mM dNTP (each), 0.2 μM primers (each), and 1.25 U of Taq polymerase. PCR conditions were: 94°C for 1 min, followed by 30 cycles at 94°C for 30 sec, 68°C for 12 min, and 72°C for 10 min. The PCR products were treated with ExoSAP-IT (USB) to remove unconsumed dNTPs and primers, and bidirectionally sequenced using the dye-terminator method (Lone Star Labs) with the primers used to amplify these DNA fragments and primers specific for both paralogous LCR copies to map the NAHR sites within the PCR products.
The genomic sequences defined by coordinates identified in the aCGH experiments, were downloaded from the UCSC Genome Browser (genome build GRCh37/hg19) and assembled and compared to the sequence from the junction fragments using the Sequencher V4.8 software (Gene Codes). Interspersed repeat sequences were identified using RepeatMasker (http://www.repeatmasker.org).
Methylation-specific multiplex ligation-dependent probe amplification
Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) analysis (Nygren et al. 2005; Scott et al. 2008) was performed in patients 2, 4, and 4U using a commercially available SALSA kit ME030 B1 (MRC-Holland). The ME030 B1 kit contains five HhaI sensitive probes in the DMR1 (differentially methylated region 1) and four in the DMR2 (differentially methylated region 2) imprinted 11p15 regions. In addition, it includes 17 probes that cover H19, IGF2, KCNQ1, and CDKN1C, and 19 reference probes located in other parts of the genome for a total of 45 probes. Analyses were performed according to the manufacturer's protocol. Briefly, 200 ng of DNA was denatured and hybridized to MLPA probes. The reaction was split into two aliquots. One aliquot was processed as a standard MLPA reaction for the copy number analysis. The restriction enzyme HhaI was added to the ligation reaction of the second aliquot. HhaI recognizes unmethylated DNA-probe hybrids, therefore only methylated DNA is PCR amplified. The amplification products of both aliquots were separated by capillary electrophoresis using an ABI 3730xl genetic analyzer (Applied Biosystems). Data were visually inspected and analyzed using GeneMarker software (SoftGenetics) for copy number alteration and methylation pattern.
Bioinformatics and in silico sequence analysis
We used the segmental duplications database from the University of Washington (Eichler laboratory, http://humanparalogy.gs.washington.edu/build36/oo.weild10kb.join.all.cull.xwparse) based on human genome build 36 (NCBI36/hg18), to obtain the coordinates and sequence identities of the known LCRs (Bailey et al. 2001). There are a total of 15,605 computationally determined interchromosomal LCRs with >1 kb in size and with >90% sequence identity occurring in ∼3%–4% of the human genome (Eichler Laboratory, http://eichlerlab.gs.washington.edu/evan.html) (Bailey et al. 2001). A subset of these LCRs with characteristics consisting of: (1) location on the same chromosomal arm in the same orientation, (2) location on different chromosomal arms in opposite orientation, (3) >5 kb in size, and (4) >94% sequence identity were computationally identified to derive a circle shaped global view genomic map of potential NAHR mediated recurrent, nonhomologous, interchromosomal translocations.
Genomic sequences of the breakpoint regions were downloaded from the NCBI (http://www.ncbi.nlm.nih.gov) and UCSC websites. The alignment of two given sequences was performed and assembled using the NCBI BLAST2 (http://www.ncbi.nlm.nih.gov/blast/bl2seq/wblast2.cgi).
Acknowledgments
We thank the patients and their families for participation in these studies. This work was supported in part by NINDS grant R01 NS058529 to J.R.L.
Footnotes
-
↵9 These authors contributed equally to this work.
-
↵10 Corresponding author.
E-mail scheung{at}bcm.edu; fax (713) 798-8937.
-
[Supplemental material is available online at http://www.genome.org. The sequence data from this study have been submitted to GenBank (http://www.ncbi.nlm.nih.gov/genbank/) under accession nos. HM989976 and HM989977 and to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accession nos. GSM578535–GSM578540, GSM578936–GSM578940, GSM578965, and GSM579028.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.111609.110.
- Received June 14, 2010.
- Accepted October 6, 2010.
- Copyright © 2011 by Cold Spring Harbor Laboratory Press
Freely available online through the Genome Research Open Access option.








