
Genome-wide identification of TAL1's functional targets: Insights into its mechanisms of action in primary erythroid cells
- Mira T. Kassouf1,
- Jim R. Hughes1,
- Stephen Taylor2,
- Simon J. McGowan2,
- Shamit Soneji1,
- Angela L. Green1,
- Paresh Vyas1 and
- Catherine Porcher1,3
- 1MRC Molecular Haematology Unit, Weatherall Institute of Molecular Medicine, John Radcliffe Hospital, Oxford University, Oxford OX3 9DS, United Kingdom;
- 2Computational Biology Research Group (CBRG), Weatherall Institute of Molecular Medicine, John Radcliffe Hospital, Oxford University, Oxford OX3 9DS, United Kingdom
Abstract
Coordination of cellular processes through the establishment of tissue-specific gene expression programs is essential for lineage maturation. The basic helix-loop-helix hemopoietic transcriptional regulator TAL1 (formerly SCL) is required for terminal differentiation of red blood cells. To gain insight into TAL1 function and mechanisms of action in erythropoiesis, we performed ChIP-sequencing and gene expression analyses from primary fetal liver erythroid cells. We show that TAL1 coordinates expression of genes in most known red cell–specific processes. The majority of TAL1's genomic targets require direct DNA-binding activity. However, one-fifth of TAL1's target sequences, mainly among those showing high affinity for TAL1, can recruit the factor independently of its DNA binding activity. An unbiased DNA motif search of sequences bound by TAL1 identified CAGNTG as TAL1-preferred E-box motif in erythroid cells. Novel motifs were also characterized that may help distinguish activated from repressed genes and suggest a new mechanism by which TAL1 may be recruited to DNA. Finally, analysis of recruitment of GATA1, a protein partner of TAL1, to sequences occupied by TAL1 suggests that TAL1's binding is necessary prior or simultaneous to that of GATA1. This work provides the framework to study regulatory networks leading to erythroid terminal maturation and to model mechanisms of action of tissue-specific transcription factors.
Understanding the mechanisms by which stem and multipotential progenitor cells progressively commit to uni-lineage programs of gene expression are key biological questions. Red blood cell production (erythropoiesis) has been extensively characterized, providing an ideal model to study cell differentiation. The red blood cell lineage is characterized by the maturation of erythroid precursors into terminally differentiated, enucleated erythrocytes. Progressive erythroid cellular maturation stages have been defined by morphological criteria (for review, see Klinken 2002) and expression of cell surface markers (Socolovsky et al. 2001). Early erythroid precursors express increasing levels of erythropoietin receptor (EPOR), which is required for terminal erythroid maturation (Lin et al. 1995; Wu et al. 1995). Cells of the next erythroid maturation stage, proerythroblasts, express high levels of the transferrin receptor TFRC (also known as CD71) and start to produce hemoglobin. The red cell–specific antigen Ter119 is expressed on all subsequent differentiating murine erythroid cells, while expression of TFRC decreases as maturation progresses. An important characteristic of erythrocytes is the unique composition of their membrane and cytoskeleton, which is required to confer high flexibility to mature red blood cells while maintaining their transport and mechanical properties (Mohandas and Gallagher 2008). In mammals, definitive red cell production initially occurs in the fetal liver and then shifts to the bone marrow in adult life.
Tissue-specific transcriptional regulators play essential roles in establishing red cell–specific gene expression programs (Cantor and Orkin 2002). One example is TAL1 (formerly SCL), a basic helix-loop-helix (bHLH) transcription factor (Lecuyer and Hoang 2004). TAL1 is initially required for specification of hemopoietic cells during embryonic development (Shivdasani et al. 1995; Porcher et al. 1996; D'Souza et al. 2005; Patterson et al. 2005). Later in hemopoietic differentiation, continued TAL1 expression is critically required for erythroid maturation as lack of TAL1 leads to a block in erythropoiesis (Hall et al. 2003, 2005; Mikkola et al. 2003; Schlaeger et al. 2005; McCormack et al. 2006).
TAL1 functions as an obligate heterodimer. It interacts with the ubiquitously expressed bHLH E-proteins (or TCF3 [also known as E2A]) to bind to its DNA recognition motif, an E-box (CANNTG). In red cells, TAL1 is part of multiprotein complexes that include the LIM-only domain protein LMO2 and the LIM domain-binding protein LDB1. The TAL1/TCF3/LMO2/LDB1 complex recruits cofactors with activator or repressor functions, such as EP300, GFI1B, CBFA2T3 (ETO2), and KDM1A (Huang et al. 1999, 2000; Schuh et al. 2005; Goardon et al. 2006; Hu et al. 2009). It can also bind other DNA-bound transcription factors such as the hemopoietic regulator GATA1 (to form the “pentameric complex”; Wadman et al. 1997) or the ubiquitously expressed protein SP1 (Lecuyer et al. 2002). Evolutionary conserved association of E-box and GATA motifs separated by 9 to 12 nucleotides [GATA(n9–12)CANNTG] has been reported in regulatory regions of many erythroid-specific genes (Anderson et al. 1998; Vyas et al. 1999; Lahlil et al. 2004).
To date, only a handful of red cell–specific genes have been reported as direct, functional TAL1 target genes. These include genes coding for the transcription factors erythroid Kruppel-like factor 1 (KLF1) and GATA1, the membrane proteins Band 4.2 (EPB4.2) and glycophorin A (GYPA), the cytokine receptor Kit as well as the alpha- and beta-globin gene clusters (Hba and Hbb, respectively) (Vyas et al. 1999; Anderson et al. 2000; Lecuyer et al. 2002; Xu et al. 2003; Anguita et al. 2004; Lahlil et al. 2004; Song et al. 2007; Kassouf et al. 2008; Manwani and Bieker 2008). TAL1 can be recruited to DNA either directly by its basic DNA-binding domain or independently of its DNA-binding activity. Extending our initial in vitro observations (Porcher et al. 1999), we recently reported on the DNA-binding independent functions of TAL1 in a knock-in mouse model expressing a DNA-binding mutant form of TAL1 (TAL1RER) (Kassouf et al. 2008). We showed that direct DNA binding is dispensable for hemopoietic specification in early development. In contrast to conventional Tal1-null embryos that die from complete absence of embryonic blood at embryonic day (E)9.5 (Shivdasani et al. 1995), Tal1RER/RER embryos survived at that stage. They die from day E14.5 onward with anemia at a time when massive expansion of erythroid cells occurs in the fetal liver to satisfy the oxygen transport requirements in the growing embryo (Kassouf et al. 2008). Though Tal1RER/RER erythroid precursors are specified, terminal maturation of Tal1RER/RER erythroid cells is grossly perturbed, with decreased numbers of TFRC+/Ter119+ red cells that fail to fully hemoglobinize and mature. Therefore, direct DNA binding by TAL1 is required for terminal erythroid maturation. At a molecular level, expression of many known TAL1 erythroid target genes was deregulated. We also reported that the DNA-binding mutation did not always fully prevent binding of TAL1 to its target loci in red cells (Kassouf et al. 2008), thereby supporting previous observations that TAL1 can be recruited to gene loci in absence of functional E-boxes (Vyas et al. 1999; Lecuyer et al. 2002).
Here, using chromatin immunoprecipitation followed by massive parallel sequencing (ChIP-seq), we present a genome-wide characterization of the sequences bound by TAL1 in wild-type and Tal1RER/RER early Ter119− fetal liver erythroid precursors, coupled with gene expression analyses. The aims of the work are to characterize the breadth of red cell processes regulated by TAL1 that execute terminal differentiation, distinguish TAL1 direct and indirect DNA binding functions, and define DNA motifs underlying the sequences bound by TAL1. More generally, we hoped this would characterize networks and processes coordinated during lineage maturation and model the mechanisms of action of bHLH tissue-specific transcription factors.
Results and Discussion
Genome-wide mapping of sequences bound by TAL1
We performed anti-TAL1 ChIP assays from material isolated from immature, Ter119− erythroid cell populations derived from day E12.5 wild-type (Tal1WT/WT) fetal livers followed by high-throughput sequencing (ChIP-seq) (Fig. 1A). To compare TAL1's direct versus indirect DNA-binding activities, we also analyzed material isolated from Tal1RER/RER fetal livers; importantly, expression levels of the TAL1 protein in Ter119- Tal1RER/RER cells remained similar to those observed in wild-type cells (Kassouf et al. 2008). Approximately 4 million uniquely mapped reads were aligned for each sample (Fig. 1A) and displayed on the Generic Genome Browser (GBrowse). Using Cisgenome (Ji et al. 2008), 4364 peaks were identified from the Tal1WT/WT sample and 694 peaks from the Tal1RER/RER sample. The peaks were ranked by the number of reads mapped in a region, with peak 1 having the greatest number of reads. After appropriate quality filtering, peaks 1–2994 (Tal1WT/WT sample) and 1–594 (Tal1RER/RER sample) were retained for further analyses.
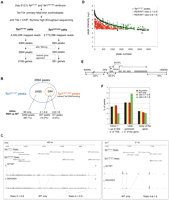
Detection of ChIP-seq peaks in Tal1WT/WT and Tal1RER/RER samples. (A) Outline of the experimental strategy. (B) Venn diagram showing that the peaks identified in material isolated from Tal1RER/RER Ter119− fetal liver cells (594 peaks, in orange circle) are a subset of the peaks identified in material isolated from Tal1WT/WT cells (2994 peaks, in blue circle). Below, the peaks are divided into three categories: “WT only” when not detected in the Tal1RER/RER sample; “0.1–0.8” or “0.8–1.8” according to the ratio of intensity between Tal1RER/RER and the corresponding Tal1WT/WT peaks. (C) TAL1 ChIP-seq peaks are displayed on two genomic loci (on chromosomes 8 and 7, top track) on GBrowse. For both sets of samples (Tal1WT/WT and Tal1RER/RER), the sequencing reads, identified as peaks, are mapped onto the chromosome view along with their coordinates and visualized along the sequence in GBrowse. The peaks exclusively detected from the wild-type sample (Tal1WT/WT Peaks) are labeled “WT only.” All the peaks detected from the mutant population (Tal1RER/RER Peaks) correspond to genomic locations also identified as peaks in the wild-type population. For those peaks, the ratio of intensity between wild-type and mutant samples is shown (RER/WT ratios 0.1–0.8 or 0.8–1.8). (D) The distribution of the 594 peaks detected in the Tal1RER/RER sample (RER/WT ratios 0.1–0.8 and 0.8–1.8) is compared with that of their corresponding peaks (i.e., detected at the same position) in the Tal1WT/WT sample, according to their intensities. The “WT only” peaks are not shown. (E) Genomic distribution in percentages of the Tal1WT/WT peaks with respect to gene loci. In gray, exons; position of intron 1 is shown; thin lines on either side of the locus represent upstream and downstream flanking sequences; the arrow shows position of the transcription start site (TSS). (F) Distribution in percentages of the Tal1WT/WT peaks as a whole (All peaks) and after fractionation according to their requirement for direct DNA-binding activity (WT only, ratios 0.1–0.8 or 0.8–1.8), with respect to the three main genomic locations, as indicated on the graph.
We then compared the genomic coordinates of the peaks detected in the Tal1WT/WT and Tal1RER/RER samples. Out of the 2994 peaks from Tal1WT/WT cells, 2400 were not detected in the Tal1RER/RER sample; this group was termed “WT only” (80.1% of total peaks; Fig. 1B, Supplemental Table 1). The remaining 594 peaks were identified in both samples. Thus, peaks detected in the mutant samples are a subset of the peaks present in the wild-type sample. A quantitative analysis revealed that the intensity of the peaks shared between Tal1WT/WT and Tal1RER/RER samples varied. This allowed us to divide the shared peaks into two categories, based on the ratio of their intensity in mutant (RER) versus wild-type (WT) samples (Fig. 1B). The first category represented peaks partially affected by the DNA-binding mutation (suggesting the occurrence of both direct and indirect TAL1 binding) and defined by RER/WT ratios between 0.1 and 0.8 (457 peaks representing 15.2% of total peaks; Fig. 1B; Supplemental Table 2). The second category represented peaks minimally affected by the DNA-binding mutation (suggesting that recruitment to DNA may occur mainly independently of direct TAL1 DNA-binding activity) and defined by RER/WT ratios between 0.8 and 1.8 (137 peaks representing 4.6% of total peaks; Fig. 1B; Supplemental Table 3). As an example, Figure 1C shows the peaks in the three different categories (WT only, 0.1–0.8, and 0.8–1.8) over two genomic regions in GBrowse.
The distribution of the peaks detected in the Tal1RER/RER sample was compared with that of their corresponding wild-type peaks, according to their intensities. In Figure 1D, the same peak number is associated to a peak in the Tal1RER/RER sample and its corresponding wild-type peak. All the peaks belonging to the category RER/WT 0.1–0.8 were found within the peak number range 1–1705. The category RER/WT 0.8–1.8 shows fewer peaks of high intensity and more peaks of medium to low intensity (peak numbers ∼1500–2985). This suggested that, among the sequences with the strongest affinity for TAL1 (peaks one to ∼1700), some might contain specific arrangements of cis-acting elements allowing for recruitment of TAL1 independently of its DNA-binding activity, albeit with less affinity than through direct binding. Sequences likely to recruit TAL1 through both direct and indirect mechanisms with similar efficiency show a lower affinity for TAL1.
This initial analysis provides genome-wide molecular evidence that direct DNA-binding activity is required to recruit TAL1 to ∼80% of its red cell target sequences. Thus, it is not surprising that loss of direct TAL1 DNA binding causes a dramatic erythroid phenotype leading to embryonic lethality (Kassouf et al. 2008). As the peaks detected from the Tal1RER/RER sample are a subset of the wild-type peaks, mutation of the DNA-binding domain of TAL1 did not create novel DNA-binding specificities but, instead, suggests that, at a subset of target sequences, TAL1 is likely to be normally recruited to DNA indirectly, as previously suggested by our group and others (Porcher et al. 1999; Lecuyer et al. 2002; Kassouf et al. 2008).
Detection of known TAL1 binding sites and validation of new binding sites
Using the nearest gene approach, we identified 2195 genes associated with the 2994 peaks in the Tal1WT/WT sample (Fig. 1A). TAL1 binding peaks were distributed between the proximal promoter and intragenic and distal binding sites (representing 33.5%, 24.9%, and 41% of total peaks, respectively; Fig. 1E; Supplemental Table 4), suggesting both short and long-range transcriptional control. Although peaks at proximal promoters are more likely to be functionally attributed to the relevant gene, our data identify potential active distal sites for investigation. Of note, we noticed an enrichment of peaks that are bound by the DNA-binding mutant form of TAL1 (RER/WT ratios 0.8–1.8) at distal cis-elements, at the expense of proximal regions (Fig. 1F; Supplemental Table 4). This suggested that, when TAL1 recruitment is largely independent of its direct DNA-binding activity, this is more likely to occur at distant enhancers, rather than promoters.
The list of TAL1 candidate target genes identified by ChIP-seq contains most, if not all, previously reported functional targets of TAL1 in red cells. For example, TAL1 was bound to the promoter region and the +40 downstream element in the Tal1 locus itself and to the promoter and enhancer regions of Epb4.2, Hba and Hbb loci, Klf1, and Gypa (Fig. 2A, top panels, black tracks) (for review, see Ogilvy et al. 2007; Kassouf et al. 2008). New, previously unreported TAL1 binding sites were identified in the loci of known TAL1 target genes, such as the intragenic peaks detected in the Gypa gene (Fig. 2A). ChIP-seq also revealed binding to genomic sequences associated with numerous genes previously not reported as TAL1 targets. We selected six of these elements, two located in promoter regions (Aqp9 and Trim10), three within intragenic sequences (Alad, Prdx2, and Mmel1), and one upstream of the transcriptional unit (Ssr1), for validation purposes (Fig. 2A, bottom panels, black tracks).
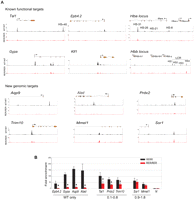
Profile of TAL1 binding on chosen loci. (A) Selected known functional or novel genomic targets of TAL1 are represented. For each locus are shown (from top to bottom): the RefSeq annotation of the gene or part of the gene (orange, exons; thin lines, introns; arrow, position of the TSS); the ChIP-seq profiles in Ter119- populations from Tal1WT/WT (black tracks) and Tal1RER/RER (red tracks) fetal liver cells. (B) Real-time PCR analysis of anti-TAL1 ChIP on selected loci. Chromatin derived from Ter119- populations from Tal1WT/WT and Tal1RER/RER fetal liver culture cells was immunoprecipitated using anti-TAL1 antibodies and the loci indicated on the graph analyzed by real-time PCR. The y-axis represents the enrichment over input DNA, normalized to a control sequence in the Gapdh gene. N, negative control. Error bars, ±1 SD, from at least three independent experiments (*P < 0.01). Below the graph are shown the categories the peaks belong to, as detected by ChIP-seq.
ChIP-seq analysis of these same loci from material derived from the Tal1RER/RER sample revealed either absence of the corresponding wild-type peaks (Epb4.2, Gypa, Hba HS-26, HS-21, HS-8 and Hbb HS1–3, Aqp9, and Alad), decreased binding (Tal1, Prdx2, Trim10, Hba HS-31), or unperturbed binding (Hba HS4, Mmel1, and Ssr1) (Fig. 2A, red tracks).
Real-time PCR (qPCR) analysis of TAL1-ChIP material confirmed binding of TAL1 on known and newly discovered genomic targets (Fig. 2B, black bars; data not shown). It also confirmed binding of the DNA-binding mutant form of TAL1 (Fig. 2B, red bars; data not shown). Importantly, this provided us with a means to validate the measurements of peak intensity, as RER/WT ratios calculated from ChIP-seq data correlated with those obtained from qPCR analyses. The “WT only” category represented by Epb4.2, Gypa, Aqp9, and Alad shows minimal TAL1 binding in Tal1RER/RER samples. Tal1, Prdx2, and Trim10 from the RER/WT “0.1–0.8 ratio” category show a decrease in TAL1 enrichment by qPCR in Tal1RER/RER samples. Finally, there is no statistically significant difference in enrichment on the Mmel1 and Ssr1 loci when comparing Tal1WT/WT and Tal1RER/RER samples, thereby validating the category RER/WT 0.8–1.8.
In summary, all known erythroid TAL1-bound sequences interrogated in our analysis were identified in our ChIP-seq experiment. This ChIP-seq analysis has yielded a high-resolution map of possibly all genomic sequences bound by TAL1 in erythroid precursors and comparison between Tal1WT/WT and Tal1RER/RER peaks gives a sense as to whether TAL1 is recruited directly or indirectly to any one binding site.
TAL1 candidate target genes are involved in regulatory and red cell–associated processes
Two-thousand-thirty-seven out of 2195 genes associated with sequences bound by wild-type TAL1 had Gene Ontology (GO) (Fig. 3A; Supplemental Tables 5,6). One-third of the genes are involved in transcription and signaling (16.8% and 15.8%, respectively). Close examination of the next set of GO categories (metabolism, transport, adhesion/migration, cytoskeleton, and redox processes, altogether accounting for 35.8% of genes associated with TAL1-occupied DNA segments) highlighted genes coding for proteins involved, among other tissues, in red cell structures and functions, such as the heme pathway enzymes, ion/water channel proteins and solute carriers, proteins involved in membrane integrity, cell–cell interaction, and oxidative processes. The remaining categories comprised other general cellular processes, accounting for 15.8% of genes. This overview revealed the breadth of the transcriptional control exerted by TAL1 on red cell–specific processes.
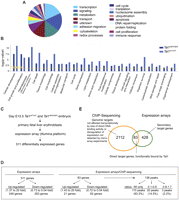
Combining ChIP-seq data with gene expression analyses. (A) Pie chart showing the distribution of the genes identified as candidate targets of TAL1 in Tal1WT/WT fetal liver cells according to their GO. (B) Highly enriched functional categories (1.1 × 10−11 < P-values < 0.05) were identified in the gene sets characterized from Tal1WT/WT and Tal1RER/RER samples using Ingenuity software. The Ingenuity Knowledge Base served as background population and the Fisher's exact test was used. The threshold corresponds to a P-value of 0.05. (C) Microarray analysis: outline of the experimental strategy. (D) Characteristics of genes revealed by expression arrays (511) and those identified in the intersection with ChIP-seq data (83). See text for details. (E) Venn diagram showing the overlap between the genes detected by ChIP-seq and those revealed by expression array.
To check whether the sequences that can be bound by the TAL1RER protein were associated with a functional subset of TAL1's target genes, we have identified and compared high-level functional categories in both sets of genes using the Ingenuity Pathway Analysis software (Fig. 3B). The same significantly enriched categories were found in wild-type and Tal1RER/RER samples (1.1 × 10−11 < P-values < 0.05), indicating that TAL1 indirect DNA binding does not preferentially occur on specific subsets of targets. Cellular development, cell growth and proliferation, hematopoiesis, and cell death were among the most enriched categories.
ChIP-seq data combined with gene expression analyses reveal functional locus occupancy by TAL1
The impact of TAL1 direct DNA binding on gene expression was assessed in the same populations as those interrogated by ChIP-seq (Tal1WT/WT and Tal1RER/RER Ter119− fetal liver erythroid populations) using microarray analysis (Fig. 3C). Five-hundred-eleven differentially expressed genes were identified (Supplemental Table 7), of which 248 (49%) and 263 (51%) were up- and down-regulated in mutant cells, respectively (Fig. 3D).
To identify direct targets of TAL1, we focused on the differentially expressed genes (511 genes) whose genomic loci contained sequences present in the ChIP-seq data set of 2195 genes. Eighty-three genes were in this intersection and likely represent direct TAL1 binding target genes (Fig. 3E; Supplemental Table 8). The remaining 428 differentially expressed genes could either represent secondary targets or be bound by TAL1 below the level of detection or at distal elements not identified by the nearest gene approach. Expression of the remaining TAL1-bound 2112 genes is not altered by loss of direct TAL1 DNA-binding activity (Fig. 3E). This may occur for a number of reasons. First, 19.1% of TAL1's genomic targets are bound by the DNA-binding mutant form of TAL1 (peak categories RER/WT 0.1–0.8 and 0.8–1.8) and may not be transcriptionally sensitive to the DNA-binding mutation. Second, redundancy between TAL1 and another hematopoietic-specific bHLH protein, such as LYL1, could explain the lack of perturbation of expression of some targets. Lyl1 and Tal1 show very similar patterns of expression in hematopoietic tissues, including erythroid cells (Visvader et al. 1991; Giroux et al. 2007); they were also recently found to have redundant functions in hematopoietic stem cells (Souroullas et al. 2009). Third, TAL1 might not exert a function at all the sites it interacts with, or might bind genomic sites to prepare them for activation at later stages of differentiation, upon subsequent recruitment of transcriptional regulators. Finally, technical limitations associated with expression microarrays (such as probe design and hybridization conditions) could also lead to failure to detect differentially expressed genes and contribute to the underrepresentation of genes transcriptionally affected.
Further characterization of these 83 putative direct TAL1 target genes revealed that they were associated with 138 peaks of TAL1 binding in the ChIP-seq database (Fig. 3D; Supplemental Table 8). As expected, the large majority of these peaks (135/138) were affected in the Tal1RER/RER sample (Fig. 3D); they were either absent (115/138, 83%, WT only category), or reduced (20/138, 14.5%, 0.1–0.8 RER/WT ratios). The genomic distribution of these peaks was very similar to the whole set of TAL1 genomic targets (Supplemental Table 4, last column). Finally, the distribution of these 83 differentially expressed direct TAL1 target genes in the GO categories was similar to the whole TAL1 gene list (Supplemental Table 9). In summary, the 83 genes presented features very similar to those of the overall set of genes bound by TAL1.
Strikingly, 62 out of 83 (75%) of the differentially expressed genes were down-regulated in Tal1RER/RER erythroid populations compared with control cells, whereas 25% (21 out of 83) were up-regulated (Fig. 3D). The high proportion of down-regulated genes suggested that direct DNA binding is required for TAL1 to preferentially exert activator function in red cells. The fold change in expression did not exceed 2.5 for the majority of the genes (63 out of 83 genes; Supplemental Table 8). Only a few genes were dramatically down-regulated (Lpl, Txnl1, Aqp9, Prdx2, Slc4a1) or up-regulated (Trib2, Dapp1, Hbb-b2) (by 3.5–20-fold). There was no difference in the genomic localization of the peaks associated with activated or repressed genes, when all peaks (138) or only the nearest peak associated to each gene (83) were considered (data not shown).
An integrated approach identifies TAL1's core transcriptional red cell network
We set out to identify targets of TAL1 with functions in erythropoiesis that may contribute to the erythroid phenotype of the Tal1RER/RER mouse when their expression is perturbed. From the list of genes identified through ChIP-seq, 80 loci were selected that (1) contained sequences bound by wild-type TAL1, (2) showed reduced or absence of binding by the DNA-binding mutant form of TAL1, and (3) have functions relevant or potentially relevant to red cell biology, based on previously reported functional in vitro and in vivo studies (Table 1). Importantly, when mouse models of these genes are available, the reported phenotype has similarities with the TAL1 DNA-binding mutant mice.
Genes bound by TAL1 with known erythroid functions and with functions potentially relevant in erythropoiesis
To this first level of analysis, we have incorporated our gene expression data. Out of these 80 genes, 23 showed deregulated expression (Table 1). Among them are some of the most transcriptionally affected genes (such as Aqp9, Slc4a1, Prdx2, Lpl, Hbb-b2, Dapp1). Confirming our hypothesis that microarray analyses might only detect a fraction of the genes transcriptionally affected, we also found, by qPCR, that expression levels of selected red cell–specific targets of TAL1, not present in the microarray list, were deregulated in Tal1RER/RER cells. Three of these are included in Table 1 (Tspan33, Trim10, and Ank1).
These 80 genes can be divided into six main groups: organization of the cell membrane and cytoskeleton, signaling, redox processes, the heme biosynthetic pathway, as well as transcription and lipid metabolism. Figure 4 gives a schematic representation of the genes that, we believe, form the core of the TAL1 transcriptional red cell network and their cellular localization. Below, we describe some of these genes and associated processes (see Table 1 for references).
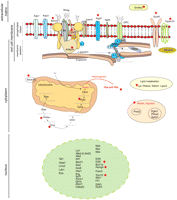
Schematic representation of selected pathways and molecules identified in this study as functional, direct targets of TAL1 in red cells. The red star indicates genes whose expression is perturbed in Tal1RER/RER Ter119- fetal liver cells, when compared with wild-type controls.
Membrane proteins and cytoskeleton organization
The red cell membrane is a highly specific structure characterized by a lipid bilayer anchored to a spectrin-based filamentous network of skeletal proteins. Numerous transmembrane proteins serve diverse functions such as transport and adhesion/migration (for review, see Mohandas and Gallagher 2008). Genes encoding 26 membrane-associated proteins are candidate targets of TAL1 (Table 1; Fig. 5; data not shown). They are involved in processes including transport, formation, and function of the erythroblastic islands (Manwani and Bieker 2008) and cytoskeleton organization.

Profile of TAL1 binding on gene loci involved in red cell–specific processes or with functions potentially relevant in erythropoiesis. For each locus are shown (from top to bottom): the RefSeq annotation of the gene or part of the gene (orange, exons; thin lines, introns; arrow, position of the TSS); the ChIP-seq profiles in Tal1WT/WT (black tracks) and Tal1RER/RER (red tracks) cells. GO biological processes are indicated at the left of the figure.
Heme pathway, heme complex: Redox processes, hypoxia
TAL1's target genes control the complex series of enzymatic steps involved in the heme biosynthetic pathway. TAL1 binding was detected at the proximal promoter or intronic regions of all the genes encoding enzymes in the pathway (Supplemental Fig. S1). TAL1 could also be involved in the transport of heme through the mitochondria and in the control of oxygen homeostasis (Abcb10 and Egln3; Table 1).
Signaling: Transcription, cell cycle, proliferation
TAL1 may also control expression of proteins involved in key signaling and regulatory pathways in red cell survival, proliferation, and terminal maturation (Table 1). Examples are TSPAN33, a newly described erythrocyte trans-membrane protein that belongs to a protein family believed to function as organizers of membrane microdomains and supramolecular signaling complexes, and the main red cell–specific cytokine receptor, EPOR, as well as a number of molecules involved in this pathway (FOXO3, PIM1, DYRK3, and DAPK2) (Fig. 5; Table 1).
Key transcriptional regulators of erythroid development are among the candidate targets of TAL1. To cite but a few, all members of the pentameric complex (including TAL1 itself), LYL1, NFE2, ZFPM1 (FOG1), FLI1, SFPI1 (PU.1), CBFA2T3 (ETO2), E2F2, E2F4, TRIM10, and HMGN are some of the critical regulators that orchestrate erythroid cell commitment, proliferation, and differentiation (Table 1; Supplemental Fig. 2). Interestingly, some of these transcription factors are also known partners of TAL1, such as E2A, LMO2, LDB1, GATA1, and CBFA2T3 (Wadman et al. 1997; Schuh et al. 2005), or proteins interacting with partners of TAL1, such as ZFPM1 (partner of GATA1) (Tsang et al. 1997). These interactions underlie the complexity of genetic regulatory networks and emphasize the importance of feed forward and autoregulatory transcriptional loops (Swiers et al. 2006; Fujiwara et al. 2009).
A recent study identified 139 putative targets of TAL1 in a hemopoietic progenitor cell line (Wilson et al. 2009). More than half of the genes (80/139) are also targets of TAL1 in erythroid cells, thereby highlighting a conserved role for TAL1 at certain loci across hematopoietic development. These include genes coding for cytoskeleton proteins (Epb4.1), for proteins located in the plasma membrane (Flt1), and for nuclear proteins (Cbfa2t3, Cebpe, E2f2, Gata2, Gfi1b, Fli1, Hhex, Nfe2, Runx1). In addition to binding events common to the progenitor cell line and the fetal liver Ter119− erythroid cells, additional TAL1-bound genomic segments can be detected in some of these loci in red cells (Cbfa2t3, Nfe2, and Tal1 loci; Supplemental Fig. 2, black track, stars; data not shown) suggesting that TAL1 exerts distinct transcriptional control over the same targets in distinct biological contexts.
Novel potential players in erythropoiesis
Finally, potentially new players in erythroid biology have been identified, thereby demonstrating the strength of our comprehensive approach (Table 1; Fig. 5). MICALL2 is a protein associated with the cytoskeleton that has been implicated in adhesion and repulsion mechanisms. In light of a possible involvement of TAL1 in the regulation of the function of the erythroblastic islands, investigation of the functional significance of this molecule and its regulation by TAL1 in erythropoiesis is of interest. The lipoprotein lipase Lpl is a functional target of TAL1 (its expression is 20-fold down-regulated in Tal1RER/RER cells), suggesting that this protein is likely to play a major role in maintaining the specific phospholipid composition of the red cell membrane bilayer. Further supporting this observation, TAL1 may also regulate expression of Lipin2, Ptdss2, and Ddhd1, all involved in phospholipid metabolism.
Additional examples are P4ha2 and Sphk1, which encode proteins involved in response to hypoxia; Dapp1 (or Bam32), which codes for a hematopoietic adaptor protein thought to control proliferation; finally, Emilin2, encoding an extracellular matrix protein that activates the extrinsic apoptosis pathway, is one of a number of apoptotic genes that are putative targets of TAL1 (see Supplemental Tables 5, 6). Our data would suggest that Emilin2 is normally repressed by TAL1 (Table 1), in agreement with TAL1's proposed role in forestalling apoptosis (Zeuner et al. 2003; Martin et al. 2004; Souroullas et al. 2009).
In conclusion, these few examples emphasize the strength of ChIP-seq in characterizing the whole repertoire of cis-elements bound by TAL1 and, therefore, candidate target genes. The comprehensive nature of this study provides a platform for assembling and testing networks of processes coordinated by TAL1 in red cells. It has also unveiled putative novel players in normal erythropoiesis and opened up new investigative routes for understanding inherited and acquired anemias.
De novo search for motifs underlying TAL1 peaks reveal E-boxes, GATA, and CACC sequences, as well as two novel motifs
To better understand the molecular mechanisms underlying TAL1's recruitment to its genomic targets in red cells, de novo motif finding was performed on the genomic sequences bound by TAL1. We first used the algorithm Weeder to search sequences underlying the 2994 peaks bound by wild-type TAL1 (Fig. 6A). GATA motifs were the most prevalent consensus sequences. Our data highlighted A/TGATAA (and its extension to C/GA/TGATAAG) as the in vivo preferred GATA sequence associated with TAL1 peaks, a subset of the WGATAR consensus motif initially identified in vitro (Orkin 1992). The same consensus site (WGATAA) was recently identified in two independent reports as the preferred sequence for occupancy by GATA1 (Yu et al. 2009; Zhang et al. 2009). WGATAA is therefore the dominant motif for GATA1.
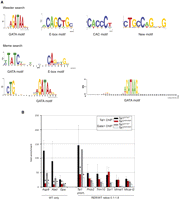
DNA motifs underlying the TAL1 peaks. (A) Logos representing the motifs identified in the sequences underlying the TAL1 peaks using de novo Weeder and Meme searches. (B) Real-time PCR analysis of anti-TAL1 and anti-GATA1 ChIP on selected loci. Chromatin derived from Ter119- populations from Tal1WT/WT and Tal1RER/RER fetal liver culture cells was immunoprecipitated using anti-TAL1 and anti-GATA1 antibodies and the loci indicated on the graph analyzed by real-time PCR. The y-axis represents the enrichment over input DNA, normalized to a control sequence in the Gapdh gene. Error bars, ±1 SD from at least three independent experiments (*P < 0.01). Below the graph are shown the categories the genes belong to, as detected by ChIP-seq.
After masking the GATA sites, E-box motifs were the next most prominent motif detected. CAGCTG was the TAL1 preferred consensus sequence, rather than the in vitro described variations of CAGATG (Hsu et al. 1994) or CAGGTG (Wadman et al. 1997).
After masking both GATA and E-box sequences, CAC motifs (consensus CACCC) were identified, as well as a novel motif CTGCCA/TGNNG. This motif presents similarities with the first five positions of the recently described motif GCCAGC that is significantly enriched in GATA1-occupied DNA segments in erythroid cells (Zhang et al. 2009). It also overlaps in a similar way with a consensus sequence present in occupied composite E-box/GATA sequences in red cells (T/CC/TC/TTGG/TG/CC/GA/TGT/G, the sequence common to all three motifs, GCCAG, is underlined) (Wozniak et al. 2008). Altogether, this further supports a strong correspondence between TAL1 and GATA1 occupancy in erythroid cells, as recently observed by Cheng et al. (2009).
No other motifs were identified, even when the three categories (WT only and RER/WT ratios 0.1–0.8 and 0.8–1.8) were interrogated individually (data not shown).
In a parallel analysis using the MEME algorithm (Fig. 6A), the enrichment of GATA and E-box motifs was confirmed. In addition, two longer motifs were characterized: the well-described E-box-GATA element [TATC(n9)CAGCTG], and a new, related, composite motif consisting of a GATA sequence and the trinucleotide CTG, separated by 9 bp [CTG(n9)GATA]. To confirm the existence of this composite motif, the 2994 TAL1-bound peaks were searched for WGATAA and sequences 5′ and 3′ of this motif examined. A significant proportion of the sequences were found to be associated to a CTG motif (Fig. 6A, bottom). The trinucleotide is half an E-box, inviting the hypothesis that such a sequence may allow less stringent binding of a TAL1 dimer with bound GATA factors stabilizing TAL1 recruitment. Supporting a biological meaning to this finding, a similar motif [CTG(n7–8)WGATA] was recently described in peaks co-occupied by GATA1 and TAL1 (Soler et al. 2010).
To measure the frequency of the motifs generated by Weeder and MEME, an in-house Perl script calculated the mean frequency of a motif per sequence over a region encompassing the peaks and flanking 100 bp and against a background distribution (see Methods; Table 2). Motif occurrence (number of peaks with the motifs) was also calculated (Table 2).
Motif frequency and motif occurrence
Although CAGCTG only appeared at a frequency of 0.18 motif/sequence, when relaxed to CAGNTG, E-boxes were the most frequent motifs (0.68 motif/sequence, P = 0; present in 39% of all peaks); the degenerated motif CANNTG was not found to be significantly enriched over the background distribution (frequency 0.71, P = 0.97). We therefore propose that the relaxed CAGNTG motif is TAL1-preferred E-box sequence in erythroid cells. The A/TGATAA motif was found at a frequency of 0.53 motif/sequence (46% of all peaks) and confirmed to be dominant over A/TGATAG (frequency 0.19, occurrence 17%). CAC motifs were also frequent (0.42 motif/sequence, occurrence 34%). The new motif, simplified to CTGCCA/TG, was present at a frequency of 0.16 (occurrence 14%). As for composite motifs, the novel sequence [CTG(n9)GATA] dominated with a frequency of 0.19 motif/sequence (occurrence 13%) whereas, surprisingly, the E-box-GATA motif was associated with a frequency of only 0.03 motif/sequence (occurrence 4.5%).
Enrichment in novel motifs in a subset of peaks
We repeated the motif analysis on subsets of peaks that had been grouped by various parameters: peak localization, whether the peaks were associated with activated or repressed genes and according to the mechanisms of recruitment of TAL1 to DNA (Table 2).
The frequency or nature of DNA motifs did not vary according to peak localization (as defined in Fig. 1E). The enrichment in recruitment of the DNA-binding mutant protein on distal cis-elements as opposed to proximal promoter regions (Fig. 1F) is not, therefore, due to specific arrangements of the motifs identified de novo.
We then looked at peak composition according to the transcriptional state of the gene. There was no substantial change in the overall frequency of E-boxes or A/TGATAA motifs between active and repressed genes. However, our search of motifs in sequences underlying TAL1 peaks pointed to a fourfold enrichment of composite E-box/GATA sequences in the peaks associated with activated genes when compared with repressed genes (12% versus 3% of peaks, respectively). This is in agreement with recent data suggesting that TAL1 cooperates with GATA1 in red cells mainly to activate target loci (Cheng et al. 2009; Tripic et al. 2009; Yu et al. 2009). In addition, the novel motif CTGCCA/TG, shows a 2.3-fold increase in occurrence in TAL1 peaks associated with activated genes when compared with repressed genes (21% versus 9%), highlighting its potential role in regulatory regions of activated genes.
Finally, to better define the mechanisms of recruitment of TAL1 to its genomic targets, the sequences underlying the peaks in the three categories reflecting the ratios of direct versus indirect DNA binding were investigated (Table 2). As expected, the percentage of peaks containing the E-box motif CAGNTG was observed at the highest occurrence (42%) in the sequences underlying the ”WT only” peak category. This decreased by 1.5-fold to 30% and 28% in the sequences where the RER/WT ratios were 0.1–0.8 and 0.8–1.8, respectively. In contrast, the A/TGATAA motif occurrence increased by ∼1.5-fold in the ”WT only” sequences when compared with sequences with RER/WT ratios 0.1–0.8 and 0.8–1.8 (42%, 62%, and 55%, respectively). In agreement with this, the occurrence of the composite motif CTG(n9)GATA was increased by two- to threefold in the sequences bound by TAL1RER (10%, 27%, and 22%, in sequences associated to “WT only” and RER/WT ratios 0.1–0.8 and 0.8–1.8, respectively). Although of moderate amplitude, these changes reflect the two mechanisms of recruitment of TAL1 to DNA: The frequency of E-boxes is the highest when TAL1 direct DNA-binding activity is exclusively employed (“WT only” category) and the presence of GATA motifs alone or in the new composite element becomes more frequent in the categories where TAL1 recruitment requires other DNA-binding proteins to be tethered to DNA. Investigating what binds the sequence CTG(n9)GATA in addition to GATA proteins would give insight into what makes this composite motif more tolerant of a defective DNA-binding form of TAL1. Finally, motif overrepresentation analysis was performed on all peak categories using consensus sites associated to blood development-specific factors including ETS factors (SFPI1, SPI1, FLI1), NFE2, RUN1, GFI1B, and SP1. No specific enrichment was detected for any of the motifs in any of the categories studied (data not shown).
TAL1's binding is required prior or simultaneously to that of GATA1 at sites of co-occupancy
As the frequency of GATA1 binding site motifs increases when TAL1 recruitment requires other DNA-binding proteins to be tethered to DNA, we hypothesized that GATA1 might be the main protein to recruit TAL1 to its target sequences in Tal1RER/RER cells. To test this, we performed GATA1 ChIP on selected loci, from material isolated from Tal1WT/WT and Tal1RER/RER samples (Fig. 6B). In agreement with our hypothesis, we observed binding of GATA1 on all the sequences able to recruit the TAL1RER protein (peaks belonging to the RER/WT ratios 0.1–1.8) both in wild-type and Tal1RER/RER cells (with the exception of one locus, Mmel1). However, the levels of enrichment of GATA1 on these loci in wild-type and Tal1RER/RER cells paralleled that of TAL1; for example, decreased levels in TAL1 enrichment at the Tal1 promoter (Tal1 prom.) in Tal1RER/RER cells correlated with a proportional decrease in enrichment of GATA1 binding; little or no variation in TAL1 binding on sequences such as Prdx2, Trim10, Ssr1, and Mical-l2 in Tal1RER/RER cells correlated with similar changes in GATA1 enrichments. As Gata1 expression is not affected in Tal1RER/RER cells (Kassouf et al. 2008), these data suggested that recruitment or stabilization of GATA1 on DNA relies on the presence of TAL1. Supporting these observations, GATA1 binding was severely affected at sequences not able to recruit TAL1 in Tal1RER/RER cells (WT only, Aqp9, Alad, Gpa loci).
Therefore, our analysis of transcription factor recruitment in Tal1RER/RER cells shows that, when co-localizing on target sequences, TAL1 and GATA1 might cooperate and stabilize each other's binding. This does not exclude the possibility that TAL1 is recruited to DNA by other, yet unidentified mechanisms. Analyses of the recruitment of these factors on “WT only” sequences suggests that TAL1's binding is necessary prior or simultaneous to that of GATA1. This agrees with earlier studies performed on the Hba locus, showing that TAL1 is present on the locus in early hematopoietic progenitors, before binding of GATA1 is firmly established in erythroid precursors (Anguita et al. 2004). Whether TAL1 can open compacted chromatin to allow subsequent recruitment of additional transcriptional regulators, thereby acting as a “pioneer” transcription factor (Lupien et al. 2008; Sekiya et al. 2009), remains to be investigated.
Conclusion
In conclusion, our attempt to elucidate the mechanisms of recruitment of TAL1 to DNA emphasized the robust and well-characterized observation of an E-box/GATA interplay at the genome-wide scale. The functional relationship between TAL1 and GATA1 is, however, likely to be quite complex and will need to be further dissected mechanistically. Novel motifs [CTGCCA/TG and CTG(n9)GATA] were described that remain to be studied functionally. This will help distinguish activated from repressed sequences and define which elements, in addition to GATA and CACC sites, help recruit the TAL1 DNA-binding mutant. However, as previously reported (Wozniak et al. 2008; Steiner et al. 2009), these studies have limitations; chromosomal environment and chromatin structure, dynamic recruitment of additional trans-acting factors, DNA looping, and influences of long-range regulation are elements to take into account when analyzing DNA/protein interactions that cannot be appreciated solely through ChIP-seq and motif prediction analyses. The biological intricacies of tissue development and underlying molecular dynamics are a challenge to capture at the genome-wide scale.
Methods
Cell culture
Fetal liver cells from day E12.5 Tal1WT/WT and Tal1RER/RER embryos were expanded for 3 d, and Ter119- erythroid progenitors were purified as previously described (von Lindern et al. 2001; Schuh et al. 2005).
ChIP assay
Anti-TAL1 and anti-GATA1 ChIP assays were performed on chromatin prepared from Tal1WT/WT and Tal1RER/RER Ter119− erythroid purified progenitor populations as described (Schuh et al. 2005). Anti-TAL1 antibody has been described previously (Porcher et al. 1999). Anti-GATA1 antibody was from Abcam.
ChIP-seq sample preparation, library construction, and data processing
Tal1WT/WT and Tal1RER/RER anti-TAL1 and “no antibody” (input controls from both Tal1WT/WT and Tal1RER/RER cells) ChIP DNA were processed for Illumina high-throughput sequencing at the Center for Biomics, Erasmus MC, Rotterdam. Linker annealing, amplification, and gel purification were performed according to Illumina protocol. Data analysis was undertaken by the Computational Biology Research Group CBRG (Oxford University). ChIP-seq data sets have been submitted to the Gene Expression Omnibus (GEO) database under accession number GSE18720.
Approximately 12 million reads of 35 bp each were produced from Tal1WT/WT and Tal1RER/RER anti-TAL1 and “no antibody” ChIP DNA. Sequences were mapped to the repeat-masked reference mouse (build m37) genome using MAQ (Li and Durbin 2009). Repeat masking included simple, complex, and ribosomal repeats using data from the UCSC Table Browser “rmsk,” and was done to avoid complications of full or partial multiple mapping of sequences when using the MAQ program which usually randomly maps such sequences to a single position in the genome. Approximately 4 million uniquely mapped reads were counted for every nucleotide position. The data were displayed on the Generic Genome Browser (Gbrowse, http://tinyurl.com/SCLtargets) (Stein et al. 2002). Peak height (as seen in GBrowse) reflected the number of sequences that map to the genomic region. Cisgenome (Ji et al. 2008), using the two-sample method (which allows comparison of antibody versus no antibody data) was used to call and quantify the peaks with an FDR cutoff of 0.1, positive/negative background ration of 0.5, and minimum read number per window of 3 over a window size of 100. Quality filtering was performed by visual inspection of the peaks in GBrowse. An arbitrary cutoff was set up to reduce the noise level. To confirm that no potentially relevant peaks had been removed, a motif search was run on these peaks (as described below); no significant enrichments were observed. The identified peaks were associated to the nearest RefSeq genes to aid investigation of the data.
The genomic sequences of the Cisgenome-determined peaks were extracted using a custom in-house PERL scripts and analyzed for overrepresented sequence motifs using Weeder version 1.3 (Pavesi et al. 2004) and MEME (Bailey and Elkan 1994). Sequence logos of the motifs were generated using WebLogo (Crooks et al. 2004).
The frequency of motif occurrence (mf) was tested for significance against a background distribution which was generated by
drawing 1000 regions S times from a repeat masked version of the mm9 genome. In each case, the random peaks were twice the size of the real TAL1
peaks (200 bp versus an average of 107 bp) and matched for repeat content. For each round of sampling (s), the motif frequency mf(s) was calculated, and P was calculated by: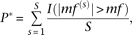 where I is 1, if the condition inside the parentheses is true. In this case S was set to 1000.
where I is 1, if the condition inside the parentheses is true. In this case S was set to 1000.
To test the applicability of the background model, sequences of 200 bp were isolated 500 bp and 1 kb upstream of and downstream from the TAL1 peaks, again controlling for useable sequence content. The frequency of the motifs within these data sets is now not significantly different from the background model.
All scripts for the analysis and Makefiles that were used to run the peak calling pipeline are available on request.
Quantitative real-time PCR analysis
For expression analysis, RNA was extracted from Tal1WT/WT and Tal1RER/RER Ter119−-purified erythroid progenitors using the RNAeasy Micro RNA isolation kit (Qiagen), DNase-treated (Qiagen), and cDNA was synthesized using the Sensiscript kit (Qiagen). Primers for Ank1, Tspan33, and Trim10 were designed using MacVector (MacVector, Inc.). SYBR Green-based quantitative qPCR (ABI SYBR Green PCR master mix, Applied Biosystems Inc.) was perfomed on three independent populations. Samples were analyzed in duplicates using an ABI Prism 7000 sequence detection system (Applied Biosystems Inc.). Data were normalized relative to Gapdh.
For ChIP experiments, primers and 5′-6-carboxyfluorescein-3′-6-carboxy tetramethylrhodamine-labeled probes were selected from unique sequences in the Epb4.2, Gypa, Hba, and Hbb loci using Primer Express (Kassouf et al. 2008). Primers for Aqp9, Alad, Tal1, Prdx2, Trim10, Ssr1, and Mmel1, designed using MacVector, were used with SYBR Green PCR master mix (Applied Biosystems Inc.) for ChIP quantitation. Input and immunoprecipitated material were analyzed in duplicates relative to a sequence in the Gapdh locus as previously described (Anguita et al. 2004) on three independent Tal1WT/WT and Tal1RER/RER samples. Primers for negative points were selected on all the loci, and “no antibody” ChIP reactions were used as controls. Primer sequences are available upon request.
Expression microarray
Expression profiling was performed using Sentrix Mouse-6 Expression BeadChip arrays from Illumina (Illumina Inc.) on three independent Tal1WT/WT and Tal1RER/RER Ter119−-purified erythroid progenitor populations. RNA was extracted using RNAqueous (Ambion) and assessed for integrity using the Agilent Bioanalyzer 2100 (Agilent Technologies). All samples presented RNA integrity (RIN) scores above 9.5. Samples were then processed for array hybridization and data accumulation at the Wellcome Trust Center for Human Genetics, Genomics Group. In brief, amplification was performed using the Illumina TotalPrep RNA Amplification kit (Ambion) according to the manufacturer's instructions. Amplified cRNA was hybridized to the BeadChip arrays according to the manufacturer's guidelines and detected with Fluorolink Streptavidin-Cy3 (Amersham Biosciences). The raw intensity values obtained for the scanned array images were compiled using Illumina BeadStudio. The data were filtered so that any probe with a detection score of <0.95 across all samples was removed from the analysis prior to log transformation (base2) and quantile normalization. Differentially expressed genes were identified using limma (Smyth 2004) for R. Expression array data sets have been submitted to the Gene Expression Omnibus (GEO) database under accession number GSE21877.
Acknowledgments
We thank Wilfred van Ijcken from Erasmus MC Centre for Biomics for the Illumina data. We thank the Genomic Service (The Wellcome Trust Centre of Human Genetics, Oxford) for the Illumina gene expression analyses. We thank D. Higgs for critical reading of the manuscript. M.T.K. was funded by the Leukaemia Research Fund. P.V. acknowledges funding from the MRC Disease Team Award, the MRC Molecular Haematology Unit, and the Oxford Partnership Comprehensive Biomedical Research Centre with funding from the Department of Health's NIHR Biomedical Research Centres. This work was supported by the Medical Research Council.
Footnotes
-
↵3 Corresponding author.
E-mail catherine.porcher{at}imm.ox.ac.uk; fax 44-1865-222-500.
-
[Supplemental material is available online at http://www.genome.org. The ChIP-seq and expression array data from this study have been submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under accession nos. GSE18720 and GSE21877, respectively. Processed data is also available at https://gbrowse.molbiol.ox.ac.uk/cgi-bin/gbrowse/SCLtargets/.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.104935.110.
- Received January 6, 2010.
- Accepted May 19, 2010.
- Copyright © 2010 by Cold Spring Harbor Laboratory Press








