
Sequence features that drive human promoter function and tissue specificity
- Jane M. Landolin1,5,
- David S. Johnson2,5,6,
- Nathan D. Trinklein3,
- Shelly F. Aldred3,
- Catherine Medina2,6,
- Hennady Shulha4,
- Zhiping Weng4,8 and
- Richard M. Myers2,3,7,8
- 1 Division of Life Sciences, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA;
- 2 Department of Genetics, Stanford University, Stanford, California 94305-5120, USA;
- 3 SwitchGear Genomics, Menlo Park, California 94025, USA;
- 4 Program in Bioinformatics and Integrative Biology, Department of Biochemistry and Molecular Pharmacology, University of Massachusetts, Worcester, Massachusetts 01655, USA
-
↵5 These authors contributed equally to this work.
Abstract
Promoters are important regulatory elements that contain the necessary sequence features for cells to initiate transcription. To functionally characterize a large set of human promoters, we measured the transcriptional activities of 4575 putative promoters across eight cell lines using transient transfection reporter assays. In parallel, we measured gene expression in the same cell lines and observed a significant correlation between promoter activity and endogenous gene expression (r = 0.43). As transient transfection assays directly measure the promoting effect of a defined fragment of DNA sequence, decoupled from epigenetic, chromatin, or long-range regulatory effects, we sought to predict whether a promoter was active using sequence features alone. CG dinucleotide content was highly predictive of ubiquitous promoter activity, necessitating the separation of promoters into two groups: high CG promoters, mostly ubiquitously active, and low CG promoters, mostly cell line–specific. Computational models trained on the binding potential of transcriptional factor (TF) binding motifs could predict promoter activities in both high and low CG groups: average area under the receiver operating characteristic curve (AUC) of the models was 91% and exceeded the AUC of CG content by an average of 23%. Known relationships, for example, between HNF4A and hepatocytes, were recapitulated in the corresponding cell lines, in this case the liver-derived cell line HepG2. Half of the associations between tissue-specific TFs and cell line–specific promoters were new. Our study underscores the importance of collecting functional information from complementary assays and conditions to understand biology in a systematic framework.
Regulation of transcription is critical in every biological process, from embryonic development to stress response (Kadonaga 1998). Sequence elements and nuclear proteins, including core promoter elements, enhancers, repressors, chromatin factors, and epigenetic modifications, interact to regulate the expression of genes. These interactions have been revealed through a large variety of methodologies, including global measurements of transcripts in different cell types (Carninci et al. 2005; Harrow et al. 2006; Wakaguri et al. 2007), studies of proteins binding to DNA (The ENCODE Project Consortium 2007; Johnson et al. 2007), measurements of enhancers in reporter genes in mice (Pennacchio et al. 2006), measurements of DNA methylation (Weber et al. 2007; Brunner et al. 2009), and others (Trinklein et al. 2003; Cooper et al. 2006; The ENCODE Project Consortium 2007). Of the sequence elements that affect gene expression, transcriptional promoters have been the most widely studied in both prokaryotes and eukaryotes (Myers et al. 1986). Although the lengths and sequence contents of extended promoters vary, core sequence elements are usually contained in a short sequence, from ∼100 bp upstream to ∼100 bp downstream of the transcription start site (TSS).
Promoters contain short sequence features, or motifs, to which transcription factors (TFs) bind and regulate transcription (Myers et al. 1986; Johnson et al. 2005; Cooper et al. 2006; Brown et al. 2007; The ENCODE Project Consortium 2007; Johnson et al. 2007; Lin et al. 2007). Large-scale efforts to identify and characterize human promoters are typically transcript-based, including aligning full-length cDNA sequences to the genome (Imanishi et al. 2004), mapping 5′ ends by CAGE tags (Shiraki et al. 2003), and performing gene expression microarray analyses (Su et al. 2002, 2004). These methods measure transcript abundance in steady state and can be performed in a highly parallel fashion, but do not determine the activity of the cis-acting motifs directly.
A direct method for characterizing functional promoter sequences is the transient transfection promoter activity assay. In this method (Myers et al. 1986; Trinklein et al. 2003; Cooper et al. 2006), a putative promoter sequence is fused in a plasmid construct with a reporter gene, such as luciferase, and the recombinant plasmid is transfected into mammalian tissue culture cells. This method is advantageous in that it measures the function of a specified DNA fragment and thus directly connects sequence features with transcription output, albeit only the cloned portion of the promoter is assayed. This method removes the promoter from its native genomic context, such that long-range regulatory elements are mostly missing, and although plasmids develop some chromatin structure when transfected, the chromatin structures of promoters on such constructs are clearly not in the same form or context, nor at the same amount, as the chromatin structures of endogenous genes and promoters in the chromosome. Nevertheless, transient transfection reporter assays often yield tissue-specific information (Cooper et al. 2006), and the promoter activity assay remains a useful method for promoter identification and characterization.
Here, our goal was to build a comprehensive model of the sequence features that drive human promoter function and tissue specificity by combining extensive experimental and computational analyses. First, we used the transient transfection reporter assay to measure the activities of 4575 putative human promoters, comprising ∼5% of the promoters in the genome, in eight immortalized human tissue culture cell lines. In parallel, we measured endogenous transcripts from about 20,000 genes from each of these cell lines. We analyzed the sequence features that drive cell line–specific gene expression by examining the effects of all known TF motifs. Because we are interested in TFs that contribute to promoter activity, our modeling strategy is formulated to identify motifs that best correlate with promoter activity or the expression of the downstream gene. For many of the cell lines tested, we found motifs associated with expression specific to only that cell line. Half of the motifs that we identified in our computational screen are recognized by TFs known to function in the corresponding cell type. Thus, our approach can identify key components of transcriptional regulatory networks.
Results
Promoter activity and gene expression data
To predict putative TSSs in silico, we first used published data that describe human transcribed sequences, such as full-length cDNAs and 5′-end cDNA sequence tags (Carninci et al. 2005; Ruan et al. 2007; Wakaguri et al. 2007), which specify TSSs when aligned to the genome. We aligned a database of more than 250,000 human cDNAs and predicted about 37,000 gene models, with about 22,000 gene models represented by two or more cDNAs. We then used the 5′ ends defined by these data sets to formulate a confidence score for the TSS of each gene model. Generally, lower scores produced fewer positives, but these potentially contain novel uncharacterized promoter types. We selected 4575 of these TSS predictions (2083 had low TSS scores defined as below 20) and built plasmids containing the promoters driving a luciferase reporter gene for transient transfection experiments. These plasmids contained, on average, 1000 bp of DNA spanning the putative promoter, from ∼900 bp upstream of the TSS to ∼100 bp downstream of the TSS, but lacking the translation start site, so that the luciferase protein would be translated from its own AUG. These plasmids contain nearly all of the putative promoters on human chromosome 7, as well as 2266 promoters from various genes across the genome. We then performed transient transfection experiments in triplicate for each promoter construct in eight immortalized human cell lines (HT1080, G402, T98G, HCT116, HeLa, HepG2, AGS, and U87MG). The averaged promoter activities are included in Supplemental Data S2. Cell lines were chosen to represent a variety of parent tissue types, from hepatocyte (HepG2) to neuroblastoma (T98G). For detailed descriptions of each cell line, see Supplemental Table T1.
To complement the transient transfection data, we measured the expression of 20,589 genes in each of the eight cell lines (Supplemental Data S2). This provides a molecular phenotype and measures the steady-state transcript levels in each of the cell lines under endogenous conditions. The distributions of both promoter activity and endogenous gene expression scores were bimodal, and we defined thresholds for each assay at the trough of their respective distributions indicated by vertical dashed lines in Figure 1. The threshold for active promoters was set at log2(promoter activity score) = 0 (Fig. 1A), corresponding to a promoter activity score of 1, the point at which the luciferase signal exceeds the signal of negative controls in the same experiment. Likewise, the threshold for endogenous gene expression was set at log2(gene expression score) = 7 (Fig. 1B), roughly corresponding to the maximum intensities of the internal negative probes built into the expression microarrays (i.e., probes that should not hybridize to any sequence in the human genome).
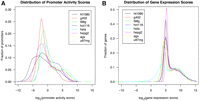
Distribution of transient transfection promoter activities and endogenous gene expression scores in eight cell lines. (A) The threshold for active promoters in the transfection assay is set at log2(promoter activity score) = 0, corresponding to the point where promoter activity scores exceed the scores of negative control sequences. (B) The threshold for expressed genes is set at log2(gene expression score) = 7, corresponding to the trough of the bimodal distributions displayed in all eight cell lines.
The TSS confidence score was a strong predictor of the average promoter activity in the eight cell lines (Pearson correlation coefficient r = 0.51, P-value < 2.2 × 10−16). Because low scoring putative promoters have scarce cDNA evidence, we termed the 2083 promoters with scores of less than 20 as putatively novel. Transient transfection assays revealed that 1082 (67%) of these novel promoters were active in at least one cell line, contributing ∼30% more promoters to the current repertoire of human transcriptional promoters (3067) in chromosome 7 and a few other regions from which we isolated these fragments (Fig. 2).
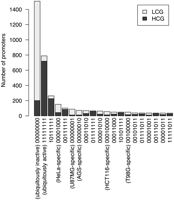
Binary encoding of promoter activity patterns. Active promoters were encoded with the number 1, and inactive promoters were encoded with the number 0.
DNA sequence and epigenetic contributions to endogenous gene expression
To quantify the relationship between promoter activity and endogenous gene expression, we identified all genes that lacked a known alternative TSS in RefSeq annotation and could be unambiguously matched with the promoters in our data set (Supplemental Fig. F1). The Pearson correlation coefficient (r) between the averaged promoter activity and averaged endogenous expression levels across cell lines for these 1188 promoter–gene pairs is 0.43 (Supplemental Fig. F2; Supplemental Table T2), compared with 6.5 × 10−4 for randomly matched promoters and genes (P-value < 2.2 × 10−16). This result is in agreement with our previous study with a smaller number of promoters, where the correlation between promoter activity and endogenous RNA transcript levels (as determined by quantitative RT-PCR) was 0.53 (Cooper et al. 2006).
To investigate the possibility that DNA methylation might partially explain the discrepancy between promoter activity and endogenous expression, we integrated Methyl-seq data in HCT116 cells (Brunner et al. 2009) with the data on matched promoters and genes. The Methyl-seq technique combines DNA digestion by a methyl-sensitive enzyme with next-generation DNA sequencing to identify regions that lack DNA methylation throughout the human genome. Overall, the Pearson correlation coefficient between endogenous expression and promoter activity in HCT116 cells is 0.32 (P-value < 2.2 × 10−16), and the correlation between endogenous expression and lack of methylation in the promoter region is 0.38 (P-value < 2.2 × 10−16). The partial correlation coefficient between the lack of DNA methylation and endogenous expression, given promoter activity as measured by transfection experiments, was 0.23, indicating that lack of DNA methylation may provide additional information beyond promoter activity toward predicting endogenous gene expression.
Sequence features and the effect of CG dinucleotides
As the transient transfection assay directly measures the ability of specific DNA segments to drive transcription, it is especially suitable for identifying active TF motifs and specific sequence features that are encoded in the assayed promoters. We found that CG content alone is highly predictive of ubiquitous promoter activity, with r = 0.75 and area under the receiver operating characteristic (ROC) curve (AUC = 94%), surpassing the contribution of any single TF motif. Note that an AUC of 100% represents the ideal discriminator, and an AUC of 50% represents a random discriminator. This statistic characterizes the trade-off between the sensitivity and specificity of a discriminant model.
Normalized CG content (defined in Methods) of the 4575 promoters displayed a bimodal distribution (Fig. 3), similar to the distribution of a previously reported genome-wide set of human promoters (Saxonov et al. 2006). We separated the promoters into high CG (HCG) and low CG (LCG) classes at the trough of the distribution of normalized CG content (Fig. 3) and found that grouping promoters into two classes substantially decreased the predictive ability of CG content on ubiquitous promoter activity. For HCG promoters, r = 0.22 and AUC = 60.8%; for LCG promoters, r = 0.5 and AUC = 77.5%. In all subsequent analyses, we considered the performance of CG content as the baseline against which our motif predictions are compared.
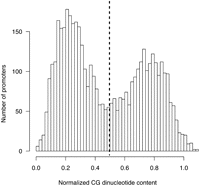
Distribution of normalized CG dinucleotide content among 4575 promoters. The normalized CG dinucleotide content is defined as the ratio of observed over expected number of CG dinucleotides (see Methods). LCG promoters have normalized CG content <0.5 and HCG promoters have normalized CG content > 0.5.
HCG promoters tend to be ubiquitously active, whereas LCG promoters tend to be cell line–specific and contain a variant of the TATA box motif (consensus: TATAAA). Promoters that were active in the transfection assay in all eight cell lines were dominated by HCG promoters (91%; 719/789), whereas only 9% (70/789) of the LCG promoters were active in all cell lines. For those promoters that were active in only one cell line, only 21.7% (105/483) were HCG and 78.3% (378/483) were LCG. Similar results were observed among matched promoters and genes. These findings are consistent with previous reports associating LCG promoters with tissue-specific genes and HCG promoters with housekeeping genes that are constitutively active across all tissues (Saxonov et al. 2006).
Many novel promoters were cell line–specific and served as important training examples for predicting motif modules that regulate cell line–specific promoter activity. Among the novel promoters confirmed by the promoter activity data, 66.8% (723/1082) had low CG dinucleotide content, compared with only 24.7% (491/1985) in known promoters (Supplemental Fig. F3). Novel promoters tended to be active in fewer cell lines, with 26.7% (289/1082) being specifically active in only one cell line. Only 11% (114/1082) of novel promoters were active in all eight cell lines. This trend was most pronounced among novel LCG promoters (P-value < 2.2 × 10−16) and also significant among novel HCG promoters (P-value = 1.85 × 10−5).
Predicting ubiquitous and cell line–specific active promoters
Using support vector machines (SVMs), we designed discriminative models to predict whether a promoter would be active or inactive based on the set of TF motifs. We designated promoters that were active in eight cell lines as ubiquitous, and promoters that were active in only one cell line as cell line–specific. The promoter activities of cell line–specific promoters are plotted in Figure 4. TF motifs, were represented by position specific scoring matrices (PSSMs), which we collected from three sources: the TRANSFAC database (Matys et al. 2006), our previous analyses on the human genome (Wei et al. 2006; Lin et al. 2007; Xi et al. 2007), and a catalog of mammalian motifs detected by evolutionary conservation (Xie et al. 2005), in total 691 motifs. Because multiple instances of the same motif can be present in a promoter, we used the algorithm Clover (Frith et al. 2004) to obtain a composite score for each motif in each promoter, quantifying the equilibrium binding potential of a TF to a promoter according to a thermodynamic model (for details, see Methods).
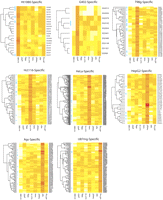
Promoter activities of cell line–specific promoters. Cell line–specific promoters are mainly active at medium levels (orange) and rarely active at high (red) levels in the cell line of interest, but the tissue specificities of all promoters are clearly distinguishable by eye. High CpG promoters are indicated by blue bars in the left margin.
We assumed that HCG and LCG promoters as well as ubiquitous and cell line–specific promoters were regulated by different cis-regulatory codes, and designed separate SVM models for each promoter set. We did not analyze promoter sets that had fewer than 20 promoters because there would be insufficient instances to produce a robust model evaluation criterion by fivefold cross-validation. Most cell line–specific models originated from the LCG promoter class. In total, we considered nine promoter sets: two sets for HCG promoters (Ubiquitous and HeLa-specific) and seven sets for LCG promoters (Ubiquitous, U87MG-, AGS-, HepG2-, HeLa-, HCT116-, and T98G-specific).
As the combinatorial examination of all 691 TF motifs would be computationally intractable, we first ranked motifs by their individual AUCs and then added them sequentially until there was no improvement on the AUC. We also attempted more complex feature selection strategies such as recursive feature elimination (Guyon et al. 2002) and RSVM (Zhang et al. 2006), but they produced overall lower AUC scores than those produced by simply adding motifs according to their ranks. Cumulative AUC and individual motif ranks are provided in Supplemental Data S3. All nine models performed well, with an average AUC of 92.8% for the ubiquitous models and 90.4% for the cell line–specific models. The average AUC was also significantly higher compared with the average AUC using CG content alone (Table 1).
AUCs of best performing models using the promoter activity data set
Ubiquitous models required fewer TF motifs to achieve a high AUC than did cell line–specific models. Only 27 motifs were required in the HCG ubiquitous model, and only six motifs were required in the LCG ubiquitous model. In contrast, the number of motifs required in the cell line–specific models ranged from 23–163 motifs, with an average of 89 motifs per model. This suggests that the complex regulation of cell line–specific promoter activity may require many TFs, whereas a few key TFs can adequately regulate ubiquitous promoters. Cell line–specific promoters likely contain a variety of activators, repressors, insulators, and other motifs that provide contextual information for specific regulation.
Cis-regulatory module discovery
Next, we sought to identify groups of co-occurring motifs, or cis-regulatory modules that were most informative of cell line–specific activity. The TFs that bind to cis-regulatory motifs are thought to interact cooperatively to drive promoter function (Johnson et al. 2005). Therefore, a thorough characterization of these modules is a key step for understanding gene regulatory networks. We found that our models maintained high predictive ability with modules containing as few as four motifs. The performance of these modules exceeded that of CG dinucleotide counts by an average of 23.2% AUC (Table 2).
Cis-regulatory modules discovered using the promoter activity data set
Each model identified unique subsets of TFs that regulate cell line–specific promoter activity. Several TFs known to regulate activity in specific tissue types were recapitulated in the corresponding models. For example, HNF4A in liver (Watt et al. 2003) and CREB in brain (Mantamadiotis et al. 2002; Gass and Riva 2007; Han et al. 2007) were specific to the HepG2-specific (i.e., hepatocyte-derived) model and the T98G-specific (i.e., neuronal cell–derived) model, respectively. Half (13/26) of the motifs in the four-motif modules corresponded to TFs that are supported by published reports with regards to tissue specificity, and 19.1% (five of 26) of the motifs were potentially novel as they did not associate with a TF known to be specific for the corresponding tissue types.
Predicting ubiquitous and cell line–specific expressed genes
Computational strategies to discover TFs that regulate tissue-specific transcription have typically relied on gene expression data derived from microarray hybridization experiments (Smith et al. 2006, 2007; Davies et al. 2007). Likewise, we compared our results from promoter activity assays with the endogenous gene expression measurements we performed on the same eight cell lines (Supplemental Tables T3, T4). The models that were trained and tested on promoter activity performed better than models that were trained and tested on gene expression (each with cross-validation). The average AUC above CG content was 35.3% for the promoter activity models, compared with 26.1% for the gene expression models. This is most pronounced in ubiquitous models, where the performances of four-TF modules above and beyond CG content were 24% and 13.4% for the ubiquitous HCG and LCG promoter activity models, respectively (Table 2). In contrast, the AUC of the corresponding models trained and tested on endogenous gene expression data did not exceed 6% (Supplemental Table T3).
Comparing the literature support of specific TF motifs, we found that 28% (nine of 32) of the TFs identified in our endogenous gene expression analyses were supported by published reports, lower than the 50% (13/26) reported for the promoter activity analysis. The promoter activity assay is more appropriate for sequence-based modeling than endogenous expression, because the promoter activity assay directly measures the activity of a known fragment of DNA, whereas gene expression data can be additionally influenced by chromatin structure, regulation in trans, RNA stability, transcription elongation regulation, and epigenetic mechanisms. Motifs that are not included in the cloned promoter construct or act in trans to the motifs in the promoter, however, cannot be included in such analysis.
Discussion
In this study, we functionally validated the activities of putative human promoters, identified novel promoters, and characterized ubiquitous and cell line–specific transcriptional mechanisms by modeling how cis-regulatory motif enrichment could predict promoter activity. Transient transfection measures the promoting activity that corresponds to a defined DNA sequence, while endogenous gene expression is additionally influenced by chromatin states and long-range elements. Ubiquitously active promoters tend to have high CG content and are regulated by few TFs, while cell line–specific promoters tend to have low CG content and are regulated by many different TFs. A model demonstrating these regulatory modes is illustrated in Figure 5.
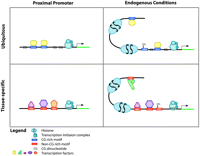
Elements of transcription regulation. Ubiquitous promoters have high CG content and are regulated by a few TFs. Tissue-specific promoters tend to have low CG content and a TATA box and are regulated by many TFs. Promoter activity of the proximal promoter is primarily determined by sequence content, while endogenous gene expression is additionally influenced by chromatin, DNA methylation, and long-range elements. Note that the molecules in this figure are not drawn to scale.
Interestingly, ubiquitous HCG promoters require more regulatory elements than ubiquitous LCG promoters. This difference could be explained by the notion of a default state for promoters, leading them to require different combinations of activating and repressive TFs. Low CG promoters may default to the ubiquitously off state and only require a few strong activators to become active across several cell types. In contrast, high CG promoters may default to the ubiquitously active state and require motifs for both activating and repressive TFs to discriminate between ubiquitously active and ubiquitously inactive states in our computational models. Indeed, there is a mixture of well-characterized activators (Ets, Sp1, Nrf-1, E2F) and repressors (Nf-μE1, ETV7, Ap4, NRSF) among the 27 motifs that are most discriminating in the high CG promoter models. In contrast, all six motifs that are most discriminating for the low CG promoter models have primarily activating functions (Sp1, EGR, CACC-BF, GABP, Churchill, and NF-κB). The complete list of motifs is provided in Supplemental Data 3.
The contribution of core promoters to ubiquitous or tissue-specific transcription is largely determined by the presence of a few key sequence elements, most of which are associated with known TFs. The list of TFs in Table 2 can be used to prioritize future chromatin immunoprecipitation (ChIP) experiments to be performed in the appropriate tissue types. Indeed, the promoters we predicted to be ubiquitously active in all eight cell lines overlap the ChIP hits of many general TFs for which ChIP data are available, such as SP1 and E2F1 (analysis not shown). The sequence features we used were composite scores characterizing TF binding potential along the entire promoter, and further studies would be necessary to map the exact locations of the functional TF binding sites. Mutagenesis experiments can then be performed to test and validate the predicted motif associations. As our promoters are large (∼1 kb) and contain multiple TF binding sites, mutagenesis of multiple binding sites and their combinations may be needed to detect significant changes in promoter activity. We are developing an approach to validate the regulatory modules presented in this study.
Genome-wide features such as promoter activity, endogenous expression, TF binding, and other genomic and epigenomic marks are being built for many tissue types and environmental contexts (The ENCODE Project Consortium 2007). Computational methods such as the one presented here will have to evolve to integrate increasing amounts of data from diverse lines of biological evidence. Integrative approaches will be crucial for unraveling the intricacies of transcriptional networks.
Methods
Promoter transient transfection reporter assay
We performed large-scale promoter transient transfection reporter assays as described previously (Trinklein et al. 2003; Cooper et al. 2006; Lin et al. 2007) with some modifications. We tested all promoter constructs in triplicate across each of the eight cell lines. Each plate contained four positive control promoters that have a range of activity levels, as well as four negative control DNA fragments. The 5′ reporter plasmids were constructed by SwitchGear Genomics, and DNA preparations for the transfections were also provided by SwitchGear. These promoter fragments in the plasmids are, on average, 1000 bp in length and contain ∼900 bp upstream and ∼100 bp downstream of the TSS. The AUG codon driving translation in all the plasmids is that of the luciferase gene and not that of the human genes from which the promoters were derived. The plasmids were constructed by cloning a PCR amplified human genomic DNA fragment corresponding to each promoter into the pGL4 reporter vector (Promega). The accurate representation of each promoter was confirmed by DNA sequencing.
Endogenous gene expression analysis
We purified total RNA from each of the eight immortalized human cell lines in three separate growths of cells. Cells were homogenized in TRIzol (Invitrogen) with a QIAshredder (Qiagen) according to the manufacturer's protocols. We assessed the purity and quantity of the RNA by using the NanoDrop (ThermoScientific). We then isolated total RNA from the homogenate by using RNeasy Mini Kits (Qiagen) and labeled and amplified this material (Illumina TotalPrep RNA amplification kit, Ambion).
We hybridized the labeled cRNA to Illumina HumanRef-8 v2 whole-genome Expression BeadChips to measure endogenous expression in each of the three biological replicates. We extracted and normalized the data with the rank-invariant method (Illumina BeadStudio software) and matched gene identifiers from all of the transcripts on the BeadArrays to promoter predictions from SwitchGear Genomics (http://www.switchdb.com; score > 20) to find transcripts that were not associated with alternative promoters.
Definition of high- CG and low CG promoter classes
The normalized CG dinucleotide content is defined as the ratio of observed over expected number of CG dinucleotides, where the expected number of CG dinucleotides is defined as [(G% + C%)/2]2. The normalized CG content for the 4575 assayed promoters follow a bimodal distribution (Fig. 3), and LCG and HCG promoters were separated at the trough of the distribution where normalized CG dinucleotide content was 0.5.
Thermodynamic model of TF binding potential
Each motifs was represented by a composite score computed by the algorithm Clover (Frith et al. 2004). A raw score is first computed representing the likelihood that a motif is present at a particular location on a given sequence.
This was defined as log-likelihood-ratio (LR1) in Frith et al. (2004), where p(L) is the background probability of observing nucleotide L at position k, and q is the foreground probability defined by a given PSSM: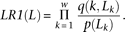
Multiplying over w, the width of the motif, LR1 has been shown to be proportional to the equilibrium occupancy of a binding site by the corresponding TF, according to statistical mechanical theory (Schneider et al. 1986; Berg and von Hippel 1987; Stormo 2000). The raw scores for all locations along a promoter are averaged to produce the composite score described in this article. This represents the likelihood of binding along the entire promoter.
We tested several metrics for how well they predicted promoter activity, and found that the metric for the positional preferences of motifs was far less predictive (r < 0.2) than other metrics, including the composite score above, that took into account the number and scores of binding sites along the promoter (r > 0.4). The composite score we used incorporated the likelihood of occurrences of multiple motifs and their combinations, but did not consider their positions relative to each other.
SVM implementation
SVMs are among the best available tools for classifying data with few examples in very large dimensional spaces. To avoid over-fitting, we used cross-validated trials where we partitioned the promoters for each model into five parts, reserving 80% of the data for training and the remaining 20% for testing. We used all the data available by rotating the training and testing sets five times. This procedure favors models that will generalize well to an independent test set. We considered nine promoter sets: two sets for HCG promoters (Ubiquitously inactive and HeLa-specific) and seven sets for LCG promoters (Ubiquitously active, U87MG-, AGS-, HepG2-, HeLa-, HCT116-, and T98G-specific). The most abundant promoter activity pattern was used as background promoter sets in our discriminative models. In LCG promoters, the most abundant pattern was the ubiquitously inactive pattern (labeled 00000000 in Fig. 2), whereas the most abundant pattern in HCG promoters was the ubiquitously active pattern (labeled 11111111 in Fig. 2). Some of the foreground promoter sets contained fewer than 20 promoters, which was not enough to produce robust AUC scores by fivefold cross-validation. These cases were discarded.
For each model, we estimated the cumulative distribution function of the test statistic using the AUCs tested on randomly sampled motif combinations across five cross-validated trials (80 motif combinations × 5 trials × 10 samples = 4000 data points). The P-value was then computed using this background distribution and represents the probability of obtaining a model with that AUC or better by chance. A higher AUC would result in a lower P-value. If we select the model that had the minimum P-value (instead of the maximum AUC), four models would have fewer numbers of motifs. The overall observation that tissue-specific models required more motifs than ubiquitous models would not change.
All models were implemented in R 2.5.0, using the e1071 package interface to the libsvm algorithm written by Chih-Chung Chang and Chih-Jen Lin (software available at http://www.csie.ntu.edu.tw/∼cjlin/libsvm). We used a linear kernel with default parameters. As the combinatorial space of all the TFs would be intractable to traverse, we first ranked the TFs by their individual AUCs and then added them sequentially to form a cumulative model. This resulted in motif combinations that had better AUCs than motif combinations discovered using other feature selection strategies. The cumulative AUCs, P-values, and ranks for individual motifs are provided in Supplemental Data S3.
Reducing redundancy in motif sets
After training and ranking all motifs, we clustered and pruned out redundant motifs in each model to eliminate biases caused by multiple testing of the same motif. This step was done after modeling because there was no a priori information as to which PSSM variant would perform best, when multiple PSSMs are available for the same TF. We used the PSSM with the best AUC and clustered redundant motifs that had pairwise Pearson correlation coefficients greater than 0.2. We used a program we developed previously (Haverty et al. 2004) to align locally and compute Pearson correlation coefficients between pairs of motifs. We then picked the motif with the best rank in each model to represent the group of redundant motifs, roughly halving the number of motifs in the final nonredundant list. All nonredundant motif ranks and plots of their predictive abilities are provided in Supplemental Data S3.
Footnotes
-
↵8 Corresponding authors.
E-mail Zhiping.Weng{at}umassmed.edu; fax (508) 856-2392.
E-mail rmyers{at}hudsonalpha.org; fax (256) 327-0978.
-
[Supplemental material is available online at http://www.genome.org. The gene expression data from this study have been submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under accession no. GSE21045.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.100370.109.
- Received September 5, 2009.
- Accepted April 12, 2010.
- Copyright © 2010 by Cold Spring Harbor Laboratory Press








