
Estrogen-mediated epigenetic repression of large chromosomal regions through DNA looping
- Pei-Yin Hsu1,
- Hang-Kai Hsu1,
- Gregory A.C. Singer1,
- Pearlly S. Yan1,
- Benjamin A.T. Rodriguez1,
- Joseph C. Liu1,
- Yu-I Weng1,
- Daniel E. Deatherage1,
- Zhong Chen2,
- Julia S. Pereira3,
- Ricardo Lopez3,
- Jose Russo3,
- Qianben Wang2,
- Coral A. Lamartiniere4,
- Kenneth P. Nephew5 and
- Tim H.-M. Huang1,6
- 1 Human Cancer Genetics Program, Department of Molecular Virology, Immunology, and Medical Genetics, The Ohio State University, Columbus, Ohio 43210, USA;
- 2 Department of Molecular and Cellular Biochemistry, The Ohio State University, Columbus, Ohio 43210, USA;
- 3 Breast Cancer Research Laboratory, Fox Chase Cancer Center, Philadelphia, Pennsylvania 19111, USA;
- 4 Department of Pharmacology and Toxicology, University of Alabama at Birmingham, Birmingham, Alabama 35294, USA;
- 5 Medical Sciences, Indiana University School of Medicine, Bloomington, Indiana 47405, USA
Abstract
The current concept of epigenetic repression is based on one repressor unit corresponding to one silent gene. This notion, however, cannot adequately explain concurrent silencing of multiple loci observed in large chromosome regions. The long-range epigenetic silencing (LRES) can be a frequent occurrence throughout the human genome. To comprehensively characterize the influence of estrogen signaling on LRES, we analyzed transcriptome, methylome, and estrogen receptor alpha (ESR1)-binding datasets from normal breast epithelia and breast cancer cells. This “omics” approach uncovered 11 large repressive zones (range, 0.35∼5.98 megabases), including a 14-gene cluster located on 16p11.2. In normal cells, estrogen signaling induced transient formation of multiple DNA loops in the 16p11.2 region by bringing 14 distant loci to focal ESR1-docking sites for coordinate repression. However, the plasticity of this free DNA movement was reduced in breast cancer cells. Together with the acquisition of DNA methylation and repressive chromatin modifications at the 16p11.2 loci, an inflexible DNA scaffold may be a novel determinant used by breast cancer cells to reinforce estrogen-mediated repression.
Epigenetic control of gene silencing plays a causal role in the development of human cancers. This form of transcriptional repression is commonly associated with loss of tumor-suppressor functions, DNA mismatch repair, cell cycle regulation, and drug resistance (Costello et al. 2000; Feinberg et al. 2006; Roberti et al. 2006; Jones and Baylin 2007). De novo DNA methylation is a frequent epigenetic hallmark observed in promoter CpG islands of repressed genes (Clark 2007; Jones and Baylin 2007). Unlike genetic mutations, this epigenetic change does not alter the DNA sequence itself, but can stably transmit the heritable information from progenitor cells to the progeny (Feinberg et al. 2006; Ho et al. 2006; Newbold et al. 2006). In addition, the silencing state is initiated and coordinately maintained by histone modifications (Clark 2007; Jones and Baylin 2007). For example, polycomb-modified trimethylation of histone H3 on lysine 27 (H3K27me3) may result in a closed chromatin configuration, rendering a promoter CpG island inaccessible to RNA polymerase II (Pol II) transcription complexes (Jones and Baylin 2007; Hansen et al. 2008). The process of acquired DNA methylation then occurs slowly. Increased methylation density in the CpG island may mark the entry of a gene into permanent silencing (Leu et al. 2004; Robertson 2005; Jones and Baylin 2007).
Using candidate gene approaches, molecular mechanisms underlying epigenetic gene silencing were generally thought to occur at the local chromatin level primarily in promoter regions of individual loci. However, recent genome-wide surveys uncovered contiguous loci frequently hypermethylated in cancer cells, suggesting that epigenetic repression can have a “geographic influence” on neighboring genes (Frigola et al. 2006; Novak et al. 2006; Clark 2007). Specifically, Frigola et al. (2006) found hypermethylation of 12 nearby CpG islands located within a ∼4 Mb cluster on chromosome 2q14.2 in colorectal cancer. Interestingly, neighboring genes spanning the region were also epigenetically suppressed, even though the involved CpG islands remain unmethylated. Moreover, despite the lack of promoter CpG island hypermethylation, the suppression of these genes could similarly be relieved by demethylating drug treatment (Frigola et al. 2006). Concurrent hypermethylation of adjacent CpG islands was also observed in smaller (<100 kb) regions of contiguous genes, including the HOXA@ on 7p15.2 in breast cancer (Novak et al. 2006), the TNFRSF10 cluster on 8p21 in neuroblastoma (van Noesel et al. 2003), the HLA class I genes on 6p21.2 in esophageal cancer (Nie et al. 2001), and the TCF21-related loci on 6q23-24 in lung and head and neck cancers (Smith et al. 2006). Taken together, these studies indicate that long-range epigenetic silencing (LRES) (Clark 2007) may be a more common phenomenon in cancer than previously recognized. However, the mechanism responsible for concurrent distal regulation is currently unclear.
Previously, our laboratory developed an in vitro model for initiating epigenetic alterations in breast progenitor cells (Cheng et al. 2008; Hsu et al. 2009). In this model, pre-exposure to estrogen and related endocrine disruptors during mammosphere stage permanently alters estrogen receptor alpha (ESR1)-mediated transcription programs in progenitors and subsequently triggers epigenetic repression of target genes later observed in the differentiated progeny (Cheng et al. 2008; Hsu et al. 2009). This epigenetic process likely disrupts tumor-suppressor functions and promotes neoplastic transformation of ESR1-positive epithelial cells (Cheng et al. 2008; Rodriguez et al. 2008; Hsu et al. 2009).
The objective of the current study was to determine whether LRES can occur in large chromosomal regions of the breast epithelial genome. We extended the study of our progenitor model, coupled with global analysis of gene transcription, DNA methylation, and ESR1 binding data. Our integrative analysis identified 11 large conserved chromosomal regions concurrently repressed by estrogen signaling. Functional analysis of normal breast epithelial cells showed that activation of estrogen signaling induces transient formation of DNA loops in a 16p11.2 gene cluster by bringing 14 distant promoters to two ESR1 binding sites for coordinate repression. This transient movement is lost in cancer cells, and the establishment of permanent loop structure is present in the epigenetically repressed region. The specific DNA loop structure described in this study may be a novel epigenetic mark used to reinforce LRES in cancer cells.
Results
Identification of large chromosomal regions subject to estrogen-mediated epigenetic repression
Breast progenitors were propagated as spherical mammospheres in suspension culture (Dontu et al. 2003) and pre-exposed to 17β-estradiol (E2, 70 nM) or vehicle for 3 wk (Fig. 1A). When transferred to collagen substratum, these progenitors attached to the floor of culture dishes and subsequently differentiated into epithelial cells, also called mammosphere-derived epithelial cells (MDECs) (Hsu et al. 2009). To determine heritable influence of this estrogenic exposure on gene repression, step-wise integration of global mapping data obtained from MDECs and breast cancer cells was conducted (Fig. 1B). First, differential profiles of Pol II binding were determined in E2-pre-exposed MDECs relative to control cells by chromatin immunoprecipitation coupled with massively parallel sequencing (ChIP-seq) (Fig. 1C; Table 1). Enriched (n = 1106) and depleted (n = 683) Pol II binding sites were identified in E2-pre-exposed MDECs (Fig. 1D). Second, to identify genomic regions that were subject to estrogen-mediated repression during neoplastic development, we compared profiles of depleted PoI II binding in MDECs with those of the ESR1-positive MCF-7 breast cancer cells stimulated with E2 for 4 h (Fig. 1E). The aim of this initial analysis was to identify chromosomal regions in E2-stimulated MCF-7 cells devoid of PoI II binding (for example, see Fig. 3A, below). Third, to confirm whether this repression is attributed to an epigenetic mechanism, we integrated the aforementioned PoI II data with global DNA methylation profiles of MCF-7 cells derived from a MeDIP-chip (methylated DNA immunoprecipitation microarray) experiment. Last, we examined the presence of ESR1 binding sites in these DNA methylation-enriched regions using a published ChIP-chip data set of MCF-7 cells (Carroll et al. 2005, 2006). With this step-wise integrated approach, 11 large chromosomal zones (0.35–5.98 Mb) were identified, including a total of 108 candidate genes that might undergo estrogen-mediated epigenetic repression in breast cancer cells (Fig. 2; Supplemental Fig. S1; Supplemental Table S1). In silico analysis of published expression data from 48 breast cancer cell lines (Neve et al. 2006) confirmed our initial finding that genes localized in these large chromosomal regions are preferentially down-regulated in ESR1-positive cancer cell lines (Fig. 3B; Supplemental Fig. S1A–J).
Total sequence reads of RNA polymerase II binding by ChIP-seq
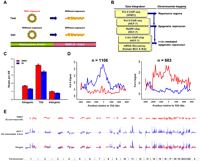
Integrative mapping of large genomic zones subject to estrogen-mediated epigenetic repression. (A) Experimental scheme. Progenitor cells, propagated into floating mammospheres, were exposed to different estrogenic compounds for 3 wk. Upon the removal of these compounds, cells were induced to epithelial differentiation on two-dimensional collagen substratum for 2∼3 wk. The differentiated progeny, called mammosphere-derived epithelial cells (MDECs), were used for ChIP-seq analysis. (B) Flowchart showing step-wide identifications of chromosomal regions associated with ESR1-mediated repression. Omics data, including ChIP-seq for differential RNA polymerase II (Pol II) binding sites and MeDIP-chip for DNA methylation profiling, were acquired for identifying the epigenetically repressed regions in the human genome. ChIP-chip and expression microarray data collected from published information were coupled with omics data to identify the ESR1-mediated repressive regions. (C) Genome-wide distribution of Pol II binding sites in the human genome. After 3 wk of E2 or DMSO pre-exposure, MDECs were subjected to ChIP-seq with Pol II antibody. The binding locations of Pol II were categorized into three groups, intragenic area, regions closed to transcription start sites (TSS), and intergenic region. Gene definitions were taken from the UCSC Genome Browser's RefGene table (Pruitt et al. 2007; Karolchik et al. 2008). For genes with more than one isoform, all TSSs were considered, but intragenic region measurements were taken from the longest isoform of the gene (less TSS regions). (D) Distribution of differential expressed genes regulated by E2 in MDECs. Cutoffs for Pol II activity were determined by examining levels of enrichment within known housekeeping genes and choosing the lower fifth percentile (Eisenberg and Levanon 2003). Similarly, cutoffs for lack of binding activity were established by studying enrichment values from gene desert regions. Comparing the E2-pretreated and control mammosphere populations, we found 1106 activated (left) and 683 repressed (right) genes upon E2 treatment. (E) Genome-wide sound-track of differential gene expression in response to E2 treatment in human normal and malignant cells. ChIP-seq with Pol II antibody was conducted on E2-treated MDECs (red track) and MCF-7 cells (blue track).

Genome-wide mapping of DNA methylation in MCF-7 breast cancer cells. MCF-7 cells were stimulated with E2 for 4 h and then subjected to MeDIP-chip. Combined with Pol II ChIP-seq data of MDECs and MCF-7 cells, 11 hypermethylated zones, as indicated by the blue open rectangles, are potential regions susceptible to estrogen-mediated epigenetic silencing.
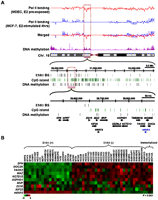
Concurrent silencing of a gene cluster on chromosome 16p11.2. (A) Representative example of a repressive gene cluster (black, n = 14) on chromosome 16p11.2 identified through integrative mapping. Within this gene cluster, HIRIP3 (blue) was found to be positively regulated by ESR1. The expression of this gene was used as a positive control for the study. (B) In silico expression analysis of the 16p11.2 cluster in 48 breast cancer cell lines. Microarray expression data (Neve et al. 2006) of nine of the 14 genes were available for the construction of a heat map as shown. Statistical analysis used to distinguish ESR1-positive from ESR1-negative breast cancer cell lines was calculated by Wilcoxon rank sum test.
Concurrent silencing of a 14-gene cluster on chromosome 16p11.2 is mediated through an ESR1-dependent pathway
To experimentally validate the aforementioned global findings, we focused on a cluster (∼0.4 Mb) of 14 repressed genes located on 16p11.2 (Fig. 3A). These genes are known to encode diverse cellular functions associated with stress response, mitogen-activated protein kinases, ion transport, and chromatin remodeling (Supplemental Table S2). RT-qPCR analysis of the gene cluster was conducted in unexposed MDECs transiently stimulated with E2 or a synthetic estrogen, diethylstilbestrol (DES, 70 nM). Progressive repression (1.2- to 2.0-fold decrease) of these genes was observed over a 24-h period (Fig. 4A; Supplemental Fig. S2). The expression levels of these 14 genes appeared to be down-regulated by 1.9- to 6.8-fold in unstimulated MCF-7 cells compared with unstimulated MDECs (Supplemental Figs. S2, S3). However, greater repressive effects (1.3- to 3.9-fold decrease) were observed when MCF-7 cells were further stimulated with E2 (Fig. 4B, lanes 1,5; Supplemental Fig. S3). To investigate whether this repression could be attributed to epigenetic mechanisms, MCF-7 cells were treated with a demethylating agent, 5-aza-2′-deoxycytidine (DAC, 1 μM), and/or a histone deacetylase inhibitor, trichostatin A (TSA, 1 μM). Treatment with either epigenetic drug resulted in partial gene reactivation, while synergistic re-expression of the 14 genes was observed following combined DAC plus TSA (Fig. 4B, lanes 1–4). Subsequent E2 treatment led to resilencing of these genes (lanes 5–8), suggesting a significant role of estrogen signaling in mediating this epigenetic repression. Although treatment of MCF-7 cells with an ESR1 antagonist, ICI182780, alone partially abrogated repression of these 14 genes, this derepression was more profound after further epigenetic drug treatment (i.e., DAC and/or TSA) (lanes 9–12). Since E2 treatment was not included in these treatments, we could not overlook the possibility that a nonestrogen-stimulated ESR1-dependent pathway, e.g., mitogen-activated kinase signaling transduction (Jensen and Jordan 2003), was responsible for repression of the 14 genes. Therefore, to further prove that this epigenetic repression was mediated through an estrogen-stimulated ESR1-dependent pathway, we treated MCF-7 cells with the combination of ICI182780 and E2 (lane 13). Gene reactivation by the combined treatment was greater than ICI182780 alone (lane 9). In the presence of epigenetic drugs, increased reactivation of the 14 genes was further observed (lanes 14–16).
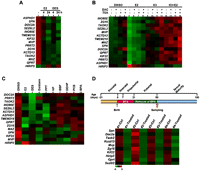
ESR1-mediated concurrent silencing of a 14-gene cluster on chromosome 16p11.2. (A) Progressive repression of the 14-gene unit in mammosphere-derived epithelial cells treated with 17β-estradiol (E2, 70 nM) or diethylstilbestrol (DES, 70 nM). Total RNA was isolated and subject to quantitative RT-PCR analysis from three independent sets of MDEC samples. Expression data are summarized in heat maps (see also detailed results in Supplemental Fig. S2). (B) ESR1-mediated epigenetic repression of the gene cluster in MCF-7 cells. Six hours before E2 stimulation, cells were pretreated with 5-aza-2′-deoxycytidine (DAC, 1 μM), trichostatin A (TSA, 1 μM), and/or ESR1 antagonist ICI182780 (ICI, 1 μM). Total RNA was subjected to RT-qPCR analysis, and expression data are summarized in heat maps (see also detailed results in Supplemental Fig. S3). (C) Coherent repression of the 16p11.2 gene cluster in differentiated cells pre-exposed to endocrine disruptors. Progenitors were exposed to E2 (70 nM), diethylstilbestrol (DES, 70 nM), daidzein (10 μM), 1,3,5-tris(4-hydroxyphenyl)- 4-propyl-1H-pyrazole (PPT, 0.1 nM), 4-nonylphenol (NP, 1 μM), N-butyl-benzyl phthalate (BBP, 10 μM), di(2-ethylhexyl)-phthalate (DEHP, 10 μM), 4,4′-dichloro-biphnyl (PCB, 0.1 nM), and bisphenol A (BPA, 4 nM) for 3 wk. After the pre-exposure, the differentiated cells in the absence of estrogenic exposure were subjected to RT-qPCR analysis. Data was calculated from three independent sets of MDEC samples and presented in heat maps (see also detailed results in Supplemental Fig. S4). (D) Concurrent silencing of 16p11.2 homologs (n = 10) in a rat model pre-exposed to bisphenol A (BPA). After birth, rats were exposed to sesame oil (Ctrl) or BPA (Treated; 250 μg/kg BW) during the prepubertal period (∼21 d). Then, total RNA of mammary gland obtained from 50-d-old animals prepubertally exposed to BPA were collected for expression analysis. Results of four independent sets of rat samples are shown in heat maps (see also detailed results in Supplemental Fig. S5)
While the results of these pharmacological experiments underscore that control of gene silencing in breast cancer is highly complex, our observations suggest that (1) estrogen signaling initiates the repression of the 16p11.2 loci in breast epithelial cells; (2) this repression is partly mediated through an ESR1-dependent pathway; and (3) persistent repression of the 16p11.2 loci in cancer cells may be further maintained by DNA methylation and histone modifications.
Concurrent silencing of the 16p11.2 gene cluster is initiated in estrogen-exposed progenitor cells and recapitulated in an animal exposure model
To determine whether long-term environmental exposure to estrogen-like chemicals can initiate this epigenetic repression (Fenton 2006; Ho et al. 2006; Schaffer et al. 2006; Dolinoy et al. 2007), progenitor-containing mammospheres were exposed to E2, DES, daidzein, 1,3,5-tris(4-hydroxyphenyl)-4-propyl-1H-pyrazole (PPT), 4-nonylphenol (NP), N-butyl-benzyl phthalate (BBP), di(2-ethylhexyl)-phthalate (DEHP), 4,4′-dichloro-biphnyl (PCB), and bisphenol A (BPA) for 3 wk (see the treatment dosages in Fig. 4 legend). Without further exposure, these progenitors were differentiated into epithelial cells (MDECs) in two-dimensional collagen stratum for 2∼3 wk. Expression profiles of the 16p11.2 cluster in the pre-exposed MDECs were then analyzed by RT-qPCR. Concurrent repression of these genes was confirmed (1.4- to 3.4-fold decrease) in E2-pre-exposed MDECs relative to the control (Fig. 4C; Supplemental Fig. S4). Suppressive effects varied for the different environmental exposures, indicating differential sensitivity of progenitors to these estrogenic ligands.
Since this 16p11.2 cluster is highly conserved in mammalian species (see the list of the sequence homology between human and rat in Supplemental Table S3), we extended the above study to a rat model with prepubertal BPA exposure (250 μg/kg BW/day; Fig. 4D, top). Expression profiling of mammary glands collected from pre-exposed 50-d-old rats (n = 4) revealed concurrent repression in 10 genes homologous to those of the 16p11.2 loci (bottom). Moreover, down-regulation was apparent for 75 rat genes located in the other 10 aforementioned regions (Supplemental Fig. S5). Taken together, these findings suggest that (1) this conserved cluster is susceptible to estrogen-mediated repression in both human and rat mammary epithelial cells, and (2) continuous exposure to estrogenic ligands may lead to permanent silencing of the 16p11.2 loci.
Concurrent silencing of the 16p11.2 gene cluster is associated with H3K27me3 and DNA hypermethylation in breast cancer cells
Based on the pharmacologic study described in Figure 4B, we proposed that repressive chromatin marks may be recruited to the 16p11.2 loci in cancer cells. One such mark is the polycomb-modified H3K27me3, which is frequently present in promoter regions of silent genes (Cao et al. 2002; Cao and Zhang 2004). ChIP-qPCR was used to determine enrichment levels of H3K27me3 in the 5′-end regions (−3 to 1 kb from each TSS) of the 14 genes in MDECs and MCF-7 cells (Fig. 5A). In E2-stimulated MDECs, a low level of enrichment was observed in these gene promoters for a 24-h period. Interestingly, H3K27me3 enrichment was greatly enhanced (two- to fourfold) in unstimulated MDECs with prior exposure to estrogen during the mammosphere stage. This polycomb-mediated repression was more dramatic when MCF-7 breast cancer cells were stimulated with E2; significant increases (eight- to 10-fold) in H3K27me3 recruitment were found in at least 12 of the 14 genes analyzed. This enrichment was not present in two other repressed genes, SPN and MVP, suggesting that other repressive histone marks may be involved in regulating these two gene repressions. Taken together, the results indicate that (1) prolonged exposure of breast epithelial cells to estrogen may establish a “semirepressive” chromatin state in the 16p11.2 area, and (2) upon activation of estrogen signaling, this heterochromatic environment further promotes incremental repression of the loci in breast cancer cells.
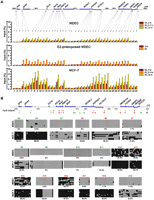
Epigenetic changes in the 16p11.2 region. (A) Distribution patterns of the repressive H3K27me3 mark in the 16p11.2 region. The diagram indicates the regions surveyed by ChIP-qPCR. The locations of interrogating regions (−3 to +1 kb of TSS) were labeled in red. DNA samples from E2-stimulated MDECs and MCF-7 cells and E2-pre-exposed MDECs were immunoprecipitated with anti-H3K27me3 antibody. Immunoprecipitated DNA was subjected to quantitative PCR to measure enrichment levels of H3K27me3. Mean ±SD (n = 3). (B) De novo DNA methylation analysis of CpG islands located on the 16p11.2 region. The diagram indicated the genomic locations of 20 CpG islands on the 16p11.2 region. The analyzed CpG islands (n = 9; nos. 5, 6, 7, 13, 14, 15, 16, 17, 18) located at the promoter and first exon regions of genes were labeled in red; other nonpromoter CpG islands (n = 9; nos. 2, 3, 4, 8, 9, 10, 11, 12, 19) were labeled in green. Two unanalyzed CpG islands were labeled in black. DNA methylation of 18 of 20 CpG islands located on the 16p11.2 region was surveyed by bisulfite sequencing. Primers flanking the cores of these CpG islands were designed for PCR amplification of bisulfite-treated DNAs from normal (MDEC) and cancer (MCF-7) cells. Ten clones per CpG island were selected for sequencing. (Black dot) methylated CpG dinucleotide; (white dot) unmethylated CpG nucleotide. Methylation percentage is calculated from the number of black dots to total dots in each sample.
To confirm the global methylation profiling of the gene cluster, we conducted bisulfite sequencing in 18 of 20 CpG islands located within this region in breast normal and cancer cells (Fig. 5B; see also Supplemental Fig. S6 for the sequenced regions). High to moderate (90%–30%) levels of DNA methylation were found in 12 (67%) of the 18 CpG islands in MCF-7 cells relative to MDECs (Fig. 5B). Among these, hypermethylation was observed in eight of nine promoter CpG islands; however, existing methylation of the ASPHD1, KCTD13, and TMEM219 CpG islands (n = 4) was already present in MDECs. The remaining six CpG islands (numbers 4, 5, 9, 10, 11, and 19) exhibited low (<30%) or no methylation in this epigenetically repressed cluster in cancer cells. Furthermore, five other genes, SPN, QPRT, ZG16, SEZ6L2, and DOC2A do not have CpG islands localized near their 5′-end promoter regions; as shown in Figure 4, we found that the expression of these non-CpG island containing genes was similarly repressed through estrogen signaling in breast cancer cells. While hypermethylation of local CpG islands may not be a dominant epigenetic feature in this estrogen-mediated repression, our data suggest that both DNA-methylated and neighboring unmethylated genes could be coordinately regulated by a common mechanism in a large chromosomal region.
Concurrent silencing of the 16p11.2 gene cluster is mediated through ESR1-modulated DNA looping
Based on the above observation, we speculated that repression of this 14-gene cluster may be remotely controlled by two functional ESR1 binding sites, termed ESR1-1 and ESR1-2, located 13-kb apart near the ZG16 locus (see the ChIP-qPCR results in Supplemental Fig. S7). Long-range interactions, through DNA looping, have been shown to regulate gene transcription (West and Fraser 2005; Kleinjan and Lettice 2008). To investigate whether such spatial networking is operating on the 16p11.2 cluster, we conducted chromosome conformation capture (3C)-qPCR assay (Hagege et al. 2007), a technique for detecting cross-linking frequencies between gene promoters and distant binding sites. In E2-stimulated MDECs, the frequency of DNA looping between these promoters and ESR1-1 was gradually increased (4.3- to 9.6-fold) within 24 h (Fig. 6B, brown labels). DNA loop formation was not detected in control regions distal from the transcription start sites (#11 and #12) and in the 3′-end (#14 and #15) or intragenic (#27 and #28) regions of selected genes. When these cells were pretreated with the ESR1 inhibitor ICI182780, we observed reduced frequencies (1.2- to 2.6-fold) of DNA loop formation in E2-stimulated MDECs, suggesting a direct role of ESR1-1 in mediating this loop formation (Fig. 6B, pumpkin labels). However, this ESR1-mediated loop formation was not apparent in the MCF-7 cancer line when these cells were stimulated with E2 (Fig. 6C).
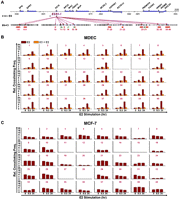
Generation of ESR1-1-mediated DNA loops in the 16p11.2 gene cluster. (A) Diagram of a genomic region surveyed by chromatin conformation capture (3C)-qPCR assays. Forty reactions (number in violet) of 3C-qPCR were performed in BbvCI-digested DNA samples. Areas (red) selected for 3C-qPCR were frequently localized at the 5′-ends of genes. Inter- and intragenic regions (green) were also selected as negative controls for the assay. (B) Formation of DNA loops at ESR1-1 upon E2 stimulation in normal cells. In the presence of E2, MDECs were treated without (E2) or with (E2+ICI) the ESR1 antagonist, ICI182780 (ICI, 1 μM) for two time-periods (0.5 and 24 h). BbvCI-digested DNA fragments were subjected to 3C-qPCR. Data are shown in relative cross-linking frequencies compared with that of GAPDH as an internal control. Mean ±SD (n = 3). (C) Formation of DNA loops at ESR1-1 in breast cancer cells. BbvCI-digested DNA fragments from E2-stimulated MCF-7 cells (0.5 and 24 h) were subjected to 3C-qPCR. Data are shown in relative cross-linking frequencies compared with that of GAPDH as an internal control. Mean ±SD (n = 3).
Interestingly, loop interactions were already present between ESR1-2 and the majority of promoters (except #2 and #15) in unstimulated MDECs (Fig. 7B, left), and by 24 h after the E2 treatment, these existing loops disappeared. This DNA de-looping process was abrogated in MDECs pretreated with ICI182780. Again, low frequencies of loop formation were observed in the 16p11.2 region in MCF-7 cells, and no DNA movement was observed in these cells treated with E2 (Fig. 7B, right).
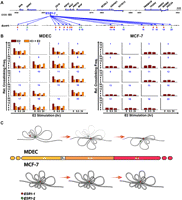
Generation of ESR1-2-mediated DNA loops in the 16p11.2 gene cluster. (A) Diagram of a genomic region interrogated by 3C-qPCR. Twenty reactions (number in blue) of 3C-qPCR were conducted in BamHI-digested DNA samples. Areas (red) selected for 3C-qPCR were frequently localized at the 5′-ends of genes. Inter- and intragenic regions (green) were also selected as negative controls for the assay. (B) Diminution of DNA loop formation by E2 treatment. After E2 stimulation, 3C-qPCR was conducted by using BamHI-treated DNA samples of MDECs (left) and MCF-7 cells (right). Data are shown in relative cross-linking frequencies compared with that of GAPDH. Mean ±SD (n = 3). (C) Proposed DNA loop model of the 16p11.2 gene cluster in normal and cancer cells. In normal breast epithelial cells, estrogen stimulation induces the formation of new DNA loops at ESR1-1 and diminishes the existing DNA loops at ESR1-2. In contrast, this free DNA movement is not present in MCF-7 breast cancer cells treated with estrogen (see detailed description in the text). (Red dot) ESR1-1 binding site; (green dot) ESR1-2 binding site.
These 3C-qPCR data, therefore, suggest that ESR1-regulated transcriptional repression operates through DNA looping in a three-dimensional sphere within the nucleus. This regulatory mechanism is flexible in normal cells, allowing for free chromosome movement in response to estrogen signaling. Looping plasticity is significantly reduced in breast cancer cells, and the establishment of rigid loop structures may coordinately impose long-term repression of multiple loci in a large chromosomal region.
Discussion
Frigola et al. (2006) were first to elegantly describe LRES in colorectal cancer and systematically explore mechanisms leading to this coordinate epigenetic repression in large chromosomal regions. A favored model for LRES is the “silencing and seeding” theory (Bock et al. 2006; Feltus et al. 2006). In a contiguous region, CpG island-related genes are actively transcribed in normal cells, and this active transcription can be a key determinant in protecting CpG islands from de novo DNA methylation (Bock et al. 2006; Feltus et al. 2006; Clark 2007). Loss of transcription activity and thus disruption of the protective mechanism may allow DNA methylation to gradually spread from pre-existing methylated sites to neighboring CpG island shores (Irizarry et al. 2009). These peripheral regions (up to 2 kb) located close to CpG island cores can be frequently methylated in cancer and are associated with the silencing of corresponding genes (Irizarry et al. 2009). In a series of events, increased methylation of CpG sites may serve as seeds to further propagate the repressive information to other nearby regions. Furthermore, de novo DNA methylation may act in concert with histone lysine methylation to promote permanent repression in targeted regions (Lorincz et al. 2002, 2004; Clark 2007). In the case of CpG island-less genes also located in the region, histone methylation may occur independently of DNA methylation to repress these loci (Lorincz et al. 2004; Frigola et al. 2006).
In contrast to this previous concept of epigenetic repression, our present finding of estrogen-mediated LRES offers a different perspective. Unlike the common notion that active transcription of a locus may shield the corresponding CpG island from methylation-mediated repression, the contiguous loci identified in 11 chromosomal regions actually have the propensity to undergo transcriptional silencing in normal cells. Specifically, the expression of the 14-gene cluster on 16p11.2 is concurrently down-regulated in normal breast epithelial cells stimulated with E2 or other estrogen-like ligands. The repression is transiently regulated by the binding of ESR1 to target sites in the nucleus (Supplemental Fig. S8). After the E2 stimulation, ESR1 gradually returns to the cytoplasm (Supplemental Fig. S8A), and the transcription of this gene cluster is restored to the basal level. However, this transient control is deregulated in normal epithelial cells chronically exposed to estrogen or estrogen-like chemicals and in ESR1-dependent breast cancer cells (Stenoien et al. 2001; Hsu et al. 2009). Persistent retention of nuclear ESR1 (Supplemental Fig. S8B) is frequently observed in these neoplastic cells, leading to long-term repression of the 16p11.2 gene locus. Based on the finding presented in Figure 5A, we further suggest that this repression is initiated by the recruitment of the H3K27me3 repressive chromatin mark to target loci. The programmed repression then sets the stage for progressive accumulation of DNA methylation in corresponding CpG islands during tumorigenesis.
Whereas the aforementioned “seeding and spreading” theory may be used to explain progressive accumulation of DNA methylation in individual loci, this two-dimensional linear model is inadequate to support our observation of estrogen-mediated LRES in large chromosomal regions. Our 3C-qPCR data supports an alternative mechanism in the context of three-dimensional chromosome movement. Within the 16p11.2 cluster, existing DNA loops form a sunflower-shaped configuration that coordinately brings these promoters to a common region within ESR1-2 for basal transcription (see the proposed model in Fig. 7C). Upon signaling activation, ESR1 binding to ESR1-2 may “dissolve” this configuration, subsequently releasing active transcription complexes (e.g., p160 coactivator complex and mediators) (Perrissi and Rosenfeld 2005) from the loop structure. A second wave of DNA loops forms another sunflower-shaped structure, shifting the focal interaction with ESR1-1. This newly formed center serves as a docking site for recruiting repressor complexes (e.g., NCoR and SMART) (Perrissi and Rosenfeld 2005) for gene silencing. These long-range interactions are transient, depending upon the timing of signaling activation in epithelial cells. However, chronic exposure of normal cells to estrogen or constant growth stimulation of neoplastic cells results in loss of looping dynamics. In the case of the 16p11.2 cluster, persistent signaling stimulation may drive target genes into epigenetic silencing. The permanent loop structure may represent a previously undescribed epigenetic phenomenon used to establish permanent silencing of genes in breast cancer cells. Within this 14-gene cluster, DNA hypermethylation was only found in 12 of 18 CpG islands examined in breast cancer cells. One plausible explanation is that loop structure may serve as a dominant feature for heterochromatinization of the entire gene cluster, while de novo DNA methylation is a stochastic process, operating at the local level to reinforce epigenetic repression of individual loci.
Our present findings challenge the prevailing concept of “one repressor unit for one silenced gene.” In a nuclear microenvironment, ligand-mediated actions may operate in different transcription units. In our case, upon signaling activation, at least two cis-regulatory elements collaboratively modulate intrachromosomal interactions, resulting in deactivation of a 14-gene unit. Multiple genes located on different chromosomes can also be regulated as an interchromosomal unit (Göndör and Ohlsson 2009; Hu et al. 2009). Further identification of their intra- and interchromosomal copartners may provide insight into complex transcriptional regulation within a cell in response to estrogen stimulation. Integrative epigenomics, as demonstrated in this study, therefore represents a powerful research tool to investigate this type of epigenetic phenomenon in cancer cells.
Methods
Tissue samples and cell culture
Breast samples were collected by the tissue procurement service in accordance with the protocols approved by the Institutional Review Board of The Ohio State University. For isolation of breast progenitors, normal tissue sections were obtained from individuals undergoing reduction mammoplasties mainly due to macromastia. These tissues were dissociated mechanically and enzymatically, and single cells were isolated and grown into mammospheres (2000–10,000 cells per mammosphere) in ultra-low attachment plates (Corning) in serum-free mammary epithelial growth medium (Cambrex) as described (Dontu et al. 2003). These mammospheres could be repopulated in the suspension culture up to six cell passages (Cheng et al. 2008). Mammospheres were then exposed to different estrogen-like chemicals with optimal concentration or DMSO (control) in phenol red-free medium for 3 wk (medium changed twice weekly). After the exposure, mammosphere-containing progenitors were placed on a collagen substratum (BD Biosciences) in phenol red-free medium for 2∼3 wk, with removal of chemicals. Under this culture condition, progenitors were differentiated into breast epithelial cells as described previously (Cheng et al. 2008; Hsu et al. 2009). The MCF-7 breast cancer cell line was obtained from the American Type Culture Collection for in vitro studies.
Chromatin immunoprecipitation (ChIP)
Immunoprecipitated DNA from each of treated MDECs or MCF-7 cells was prepared according to the ChIP protocol published by Lee et al. (2006). Briefly, treated cells were fixed with 1% formaldehyde at room temperature for 10 min. The resultant DNA–protein complexes were sheared with a Bioruptor (Diagenode) to an average of 450 bp as verified on a 1.5% agarose gel, followed by immunoprecipitation using the Dynabeads Protein G (100.04D; Invitrogen) coated with antibodies specific for the POLR2A subunit of RNA polymerase II (Santa Cruz Biotechnology, Inc., sc-899C), ESR1 (Santa Cruz Biotechnology, Inc.), and trimethylation of histone H3 lysine 27 (H3K27me3; Diagenode). Pull-down DNA was subjected to sequencing or quantitative PCR.
ChIP coupled with massively parallel sequencing (ChIP-seq) and data analysis
MCF-7 cells were stimulated with E2 or DMSO for 4 h DMSO- or E2-pre-exposed MDECs and treated MCF-7 cells were subjected to ChIP assay by using anti-RNA polymerase II antibody. Immunoprecipitated DNA was then applied to massively parallel sequencing. The whole-genome sequence analysis was performed by the BC Cancer Agency Genome Sciences Center (BCGSC). Briefly, ChIP DNA (>5 ng) per sample sonicated to the range of from 200 to 600 bp (quantified by the NanoDrop 3300 Fluorospectrometer following the PicoGreen protocol) was submitted to BCGSC for Illumina 1G analysis (two lanes per sample). The library preparation and Illumina Pipeline analysis for ChIP-seq samples were routine and well described in a recent paper from the BCGSC (Robertson et al. 2007). After extracting raw data from the Sequencer, the number of reads sequenced within nonoverlapping kilobase-sized bins was modeled using a normal approximation to a Poisson distribution with mu equal to the mean number of reads per bin (excluding unsampled bins). Modeling was performed on a per-chromosome basis. Enrichment scores were calculated for each bin as 10p(x ≤ X), giving a score with a range from zero (no probability of binding) to 10 (100% probability of binding).
Methylated DNA immunoprecipitation microarray (MeDIP-chip) and data analysis
Methylated DNA from MCF7 cells was prepared according to the protocol published by Weber et al. (2005). Briefly, high-quality genomic DNA was sheared to 300 bp (fragment size ranging from 200 to 600 bp) using Bioruptor. Fragmented DNA was heat denatured to produce single-stranded DNA to enhance the immunoselection step. Antibodies against 5-methyl cytidine (MAb-335MEC-500; Monoclonal Methyl DNA IP-grade; 1 μg/μL; Diagenode) were complexed to the magnetic beads (pan-mouse IgG Dynal Beads, Invitrogen). The immunoprecipitated methylated DNA fragments were processed by the NimbleGen Methylation Microarray Service (NimbleGen Systems, Inc.). Methylation analysis was performed on the NimbleGen Two-Array HG18 Promoter Set. This two-chip array designs relies on RefSeq to span the promoter regions of all well-characterized genes currently available for the human genome with total coverage of 54 Mb with 385 K probes per array. Every sample was hybridized twice, with MeDIP sample labeled with cyanine-5 dye and sonicated genomic DNA labeled with cyanine-3 dye in one of the microarray analyses and the second being a dye-swap analysis. The microarray-scanned images from the HG18 Promoter Set were captured as signal intensity, which were converted to scaled log2-ratio data. The intensity ratio of the MeDIP sample to total DNA was plotted genomic position to identify regions of enrichment. P-value files were generated from the scaled log2-ratio data to test for positive enrichment of DNA methylation for each probe against all probes on the array. Peak files identifying regions of DNA methylation were generated from the P-value files and the enriched peaks were then mapped to the transcription start site of each gene.
Bisphenol A (BPA) exposure in a rat model and microarray analysis
Animal care and use were conducted according to established guidelines approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham. All animals were housed in a temperature controlled facility with a 12 h light/dark cycle. Female Sprague Dawley CD rats (Charles River) were bred and observed for the presence of sperm. Once sperm positive, noted as gestational day 0, females were separated, housed in polypropylene cages with glass water bottles (both polycarbonate/BPA free), and fed the phytoestrogen-free AIN-93G pelleted diet. At birth, offspring were sexed and litters were culled to 10 offspring per lactating dam. Beginning on postnatal day two and continuing through postnatal day 20, the lactating dam of each litter was intragastrically gavaged with sesame oil for controls or 250 μg of Bisphenol A/kg BW/day. The offspring were weaned at 21 d. At day 50, the fourth abdominal mammary glands were dissected from females in the estrous phase of the estrous cycle at the same time each day using ketamine/xylazine anesthesia for live collection. The samples were flash frozen in liquid nitrogen, stored at −80°C, and subsequently shipped under dry ice.
Total RNA (200 ng) from the mammary gland of each 50-d-old rat with prepubertal exposure of BPA or sesame oil was hybridized onto the rat Agilent platform of 60-mer oligo-microarrays (4 × 44 K). The data from each microarray was adjusted for background noise using the normexp procedure, and normalized by fitting the lowess curve to the of log-ratios of Cy5 to Cy3 channel and the average log-intensities of the two as implemented in the limma package (Supplemental Table S4).
RT-qPCR and ChIP-qPCR
Total RNA (2 μg) was reversely transcribed to cDNA with oligo-dT (SuperScript III; Invitrogen). Quantitative RT-PCR was performed by using SYBR Green dye chemistry (Applied Biosystems) on a 7500 real-time PCR System apparatus (Applied Biosystems). Gene expression was calculated by absolute quantitation of standard curves of cloned PCR products. The curves were amplified from universal human reference RNA (Stratagene) with primers for each gene. Results of gene expression are presented as the relative expression level of the given gene to ACTB reference control for each sample. Immunoprecipitated DNA by anti-H3K27me3 or anti-ESR1 antibody was subjected to quantitative PCR analysis. PCR primers targeting H3K27me3 or two ESR1 binding sites were used to amplify DNA samples using the SYBR Green-based detection method. Quantitative values measured by a standard curve (50 to 0.08 ng, fivefold dilution, R2 > 0.99) of input DNA amplified with the same primer set. Results are presented as the mean of triplicates with standard derivation. Details of primer sequence for quantifications are provided in Supplemental Table S5.
Inhibitor treatments
MCF-7 cells were treated with 1 μM 5-aza-2′-deoxycytidine (DAC) for 5 d in MEM medium containing 10% FBS and 6 ng/μL insulin. During the final 72 h of the DAC treatment, cells were hormone deprived by culturing in phenol red-free MEM supplemented with 4% charcoal-dextran-treated FBS. During the final 24 h of hormone deprivation, cells were treated with 1 μM trichostatin A (TSA) and/or an ESR1 antagonist, ICI182780 (1 μM), or DMSO followed by treatment with DMSO (control) or 70 nM E2 for 6 h. Total RNA was collected for RT-qPCR analysis. For 3C-qPCR assay, MDECs without E2 pre-exposure were pretreated with ICI182780 (1 μM) for 24 h prior to E2 stimulation.
Bisulfite sequencing
DNA (0.5 μg per sample), isolated from MDECs or MCF-7 cells, was treated with sodium bisulfite using the EZ DNA Methylation kit (ZYMO Research). Primers flanking the cores of 18 CpG islands in the chr16p11.2 region were designed for PCR amplification of bisulfite-treated DNAs. Details of primer sequence for quantifications are provided in Supplemental Table S5. PCR products were cloned into the TOPO TA cloning kit (Invitrogen). Ten randomly picked clones per CpG island were sequenced using the ABI 3730 DNA analyzer (Applied Biosystems) and then analyzed using the BiQ Analyzer.
Chromosome conformation capture (3C)-qPCR
Chromosome conformation capture assay combined with quantitative PCR analysis (3C-qPCR) was performed as previously described (Hagege et al. 2007). Briefly, E2-pre-exposed MDECs were fixed with 1% formaldehyde. Chromatin was digested using BbvCI (for ESR1-1) and BamHI (for ESR1-2), and then ligated by T4 DNA ligase in diluted condition. Ligated DNA was then de-cross-linked (overnight at 65°C) and purified by classical phenol extraction procedures. Real-time PCR was performed on a 7500 Real-Time PCR System apparatus (Applied Biosystems) using the TaqMan technology (QuantiTect Probe PCR Master Mix, Qiagen). We used a 5′FAM-3′BHQ1 oligonucleotidic probe (IDT). Details of primer sequence for quantifications are provided in Supplemental Table S5. To rule out the possibility of false-negative looping occurrence caused by unsuccessful 3C assay, we pooled three human bacterial artificial clones (BAC), mapping the interested regions as the positive control to the 3C assay. BACs were also used to examine the primer efficiency. Ct values obtained for each chimerical ligation fragment were processed using parameters of a standard curve (slope and intercept) from BAC to obtain quantification values that were normalized to a GAPDH loading control. Results are presented as the mean of triplicates with standard derivation.
Statistical analysis
All data were presented as the mean ±SD of n independent measurements. Statistical comparisons between two groups were made by Student's t-test using SigmaPlot 11. For samples with equal variance, the paired Student's t-test was used. For samples with unequal variance, the Mann-Whitney rank sum test was used.
Acknowledgments
This work was supported by the Gene and Environment Initiative (U01ES015986 to T.H.-M.H and U01ES016003 to C.A.L.), the Integrative Cancer Biology Program (U54CA113001 to T.H.-M.H. and K.P.N.), R01CA069065 (T.H.-M.H.), and R01 ES017594 (T.H.-M.H.) at the U.S. National Institutes of Health and by The Ohio State University Comprehensive Cancer Center (Q.W. and T.H.-M.H.).
Footnotes
-
↵6 Corresponding author.
E-mail tim.huang{at}osumc.edu.
-
[Supplemental material is available online at http://www.genome.org. The sequencing and microarray data from this study have been submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under accession no. GSE21068.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.101923.109.
- Received October 14, 2009.
- Accepted March 17, 2010.
- Copyright © 2010 by Cold Spring Harbor Laboratory Press








