
Radiation pharmacogenomics: A genome-wide association approach to identify radiation response biomarkers using human lymphoblastoid cell lines
- Nifang Niu1,3,
- Yuxin Qin1,3,
- Brooke L. Fridley2,
- Junmei Hou1,
- Krishna R. Kalari1,2,
- Minjia Zhu1,4,
- Tse-Yu Wu1,
- Gregory D. Jenkins2,
- Anthony Batzler2 and
- Liewei Wang1,5
- 1 Department of Molecular Pharmacology and Experimental Therapeutics, Mayo Clinic, Rochester, Minnesota 55905, USA;
- 2 Department of Health Sciences Research, Mayo Clinic, Rochester, Minnesota 55905, USA
-
↵3 These authors contributed equally to this work.
Abstract
Radiation therapy is used to treat half of all cancer patients. Response to radiation therapy varies widely among patients. Therefore, we performed a genome-wide association study (GWAS) to identify biomarkers to help predict radiation response using 277 ethnically defined human lymphoblastoid cell lines (LCLs). Basal gene expression levels and 1.3 million genome-wide single nucleotide polymorphism (SNP) markers from both Affymetrix and Illumina platforms were assayed for all 277 human LCLs. MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] assays for radiation cytotoxicity were also performed to obtain area under the curve (AUC) as a radiation response phenotype for use in the association studies. Functional validation of candidate genes, selected from an integrated analysis that used SNP, expression, and AUC data, was performed with multiple cancer cell lines using specific siRNA knockdown, followed by MTS and colony-forming assays. A total of 27 loci, each containing at least two SNPs within 50 kb with P-values less than 10−4 were associated with radiation AUC. A total of 270 expression probe sets were associated with radiation AUC with P < 10−3. The integrated analysis identified 50 SNPs in 14 of the 27 loci that were associated with both AUC and the expression of 39 genes, which were also associated with radiation AUC (P < 10−3). Functional validation using siRNA knockdown in multiple tumor cell lines showed that C13orf34, MAD2L1, PLK4, TPD52, and DEPDC1B each significantly altered radiation sensitivity in at least two cancer cell lines. Studies performed with LCLs can help to identify novel biomarkers that might contribute to variation in response to radiation therapy and enhance our understanding of mechanisms underlying that variation.
Radiation is widely used in the treatment of cancer. Half of all cancer patients are treated with radiation, either alone or in combination with other forms of therapy (Burnet et al. 2006). However, response to radiation therapy varies widely, ranging from complete eradication of the tumor to severe short- and long-term treatment-dependent adverse reactions in normal tissue (Bentzen 2006; Gerber and Chan 2008). Several clinical factors are known to influence radiation response, including radiation dose, volume, and fraction, but it is also known that genetic inheritance can play an important role in variation in radiation response (Barnett et al. 2009). Many genotyping studies have been performed in an attempt to identify biomarkers for the prediction of radiation response by studying candidate genes known to be involved in radiation effect (Chistiakov et al. 2008; Andreassen and Alsner 2009; Popanda et al. 2009; Pugh et al. 2009). In addition, expression quantitative trait locus (eQTL) studies performed using post-radiation gene expression profiles have identified single nucleotide polymorphisms (SNPs) associated with radiation response through their influence on gene expression (Correa and Cheung 2004; Smirnov et al. 2009). The results of all of these studies suggest that genetic variation plays an important role in individual variation in radiation response.
In this study, we have used approaches previously applied in pharmacogenomic studies involving genome-wide basal gene expression profiles plus genome-wide SNPs for 277 human lymphoblastoid cell lines (LCLs) to identify SNPs/genes that might contribute to variation in radiation response and also to understand the biology underlying the association by performing functional studies of the candidates that we identified. Specifically, we obtained basal gene expression data for all 277 cell lines using Affymetrix U133 plus 2.0 Gene Chips, as well as genome-wide SNP data using Illumina HumanHap 550K and 510S BeadChips together with publicly available Affymetrix SNP Array 6.0 SNP data, resulting in a total of over 1.3 million SNPs per cell line for analysis. We next performed radiation cytotoxicity assays with the same LCLs to obtain the radiation area under the curve (AUC) as an in vitro radiation response phenotype. We then performed a genome-wide association study using the 1.3 million SNPs, basal gene expression array data, and AUC as a radiation response phenotype. We identified 27 loci, defined as at least two SNPs within 50 kb having P-values < 10−4, which were associated with radiation AUC. We also identified genes with expression levels that were associated with radiation AUC with P < 10−3. By then performing an integrated analysis of SNPs, gene expression, and radiation AUC, we selected a total of 23 candidate genes to perform siRNA knockdown with multiple tumor cell lines, followed by MTS and colony-forming functional validation assays to identify genes that influenced radiation exposure sensitivity.
In summary, the series of experiments described subsequently represents the application of genome-wide expression and SNP data from a cell line-based model system to identify genes associated with radiation sensitivity. Those genes, selected based on the association of radiation AUC with SNPs and/or expression, were then validated functionally. Since our major purpose was to identify genes that might provide novel insights into the biology of variation in response to radiation treatment to insure the largest set of genes for validation, the initial selection of genes to be followed up was based on a liberal significance threshold and not the standard genome-wide significance level of 10−7. The functional validation involved siRNA gene knockdown performed with cancer cell lines, followed by MTS cytotoxicity and colony-forming assays. The goals were to identify and functionally validate biomarkers for radiation response and to identify novel mechanisms that might contribute to radiation sensitivity.
Results
Radiation cytotoxicity
Radiation cytotoxicity studies were performed to determine the range of variation in radiation AUC among the individual cell lines studied. Figure 1A shows representative radiation cytotoxicity data for a set of cell lines. AUC values differed significantly among the three racial groups studied, with cells from Han Chinese–American (HCA) subjects appearing to be more sensitive to radiation than were those of the cells from Caucasian–American (CA) subjects (P = 0.007; Fig. 1B). Gender did not have a significant effect on AUC (P = 0.125; Fig. 1C). The distribution of AUC values was skewed, with a median AUC value of 3.17. However, the distribution of values for the natural-log (ln)-transformed AUC was nearly unimodal and symmetric (i.e., “normal” in shape) with a mean value of 1.27 and a range (±2 SD of the mean) of 0.28 to 2.26 (Fig. 1D). We also performed a biological replication study for the cytotoxicity data. Specifically, cytotoxicity assays were repeated 1 yr after the initial studies for 19 randomly selected cell lines. The results obtained at the two different times were significantly correlated (Pearson correlation r = 0.51, P = 0.026).
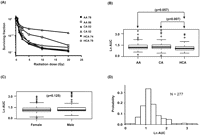
Radiation cytotoxicity. (A) Representative radiation cytotoxicity dose-response curves. Two cell lines from each of the three ethnic groups studied were selected to illustrate a range of radiation cytotoxicity “dose response” curves. Squares indicate African-Americans (AA), triangles Caucasian-Americans (CA), and circles indicate Han Chinese–Americans (HCA). The x-axis indicates radiation dose, and the y-axis indicates the surviving fraction after radiation exposure. (B) Relationship of ethnic group to radiation AUC. (C) Gender effect on radiation AUC. (D) Frequency distribution histogram of natural-log (ln) AUC values for 277 cell lines.
Correlation between expression and AUC
We next performed correlation analysis for the association of expression array and radiation AUC data to identify genes with expression levels that might be associated with radiation AUC (Fig. 2A). The association analysis identified 50 expression probe sets that were associated with radiation AUC with P-values < 10−4 (Q-values < 0.096), and 270 individual probe sets with P-values < 10−3 (Q-values < 0.182). These 270 expression probe sets represented 211 annotated genes, but only one of the probe sets remained significant after Bonferroni correction for multiple testing (P < 0.0002). The top 20 probe sets are listed in Table 1, and the entire list of 270 expression probe sets is found in Supplemental Table 1.
The top 20 expression probe sets associated with radiation cytotoxicity (AUC values)
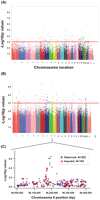
Genome-wide associations with radiation AUC. (A) Association of basal expression with radiation AUC for 277 cell lines. The y-axis represents the –log10(P-value) for the association of individual expression array probe sets. Expression probe sets are plotted on the x-axis based on the chromosomal locations of their genes. If genes had more than one probe set, the one with the lowest P-value was plotted. A P-value of 10−3 is highlighted with a red line. (B) Genome-wide SNP association with radiation AUC for 277 cell lines. The y-axis represents −log10(P-value) for the association of each SNP with radiation AUC. SNPs are plotted on the x-axis based on their chromosomal locations. A P-value of 10−4 is highlighted with a red line. (C) This panel shows the most significant locus on chromosome 8 that was associated with radiation AUC. Blue diamonds indicate SNPs observed by genotyping, while red triangles indicate imputed SNPs. The y-axis represents –log10(P-value) for the association of each SNP with radiation AUC, and the x-axis represents the location of the SNP on chromosome 8.
Genome-wide SNP association with radiation AUC
We also performed an analysis of the association of genome-wide SNPs with radiation AUC (Fig. 2B). A total of 561,298 SNPs on the Illumina 550K SNP array and 493,750 SNPs on the Illumina 510S SNP array had been genotyped using DNA from each of these 277 cell lines. We also had access to publically available Affymetrix 6.0 SNP array data. We performed quality control (QC) for all of these SNPs prior to performing the statistical analysis. Specifically, for data obtained with the Illumina 550K array, we removed 12,261 SNPs that had call rates <95%, 32,550 SNPs with minor allele frequencies (MAFs) <5%, and 4676 SNPs that deviated from Hardy-Weinberg equilibrium (HWE), using a stringent threshold of P < 0.001. Therefore, 511,811 Illumina 550K SNPs were used in the genome-wide SNP analysis. A similar approach was used for the QC analysis of SNPs on the Illumina 510S platform. After removing 10,353 SNPs with call rates <95%, 147,027 SNPs with a MAF <5%, and 3805 SNPs that deviated from HWE (P < 0.001), a total of 332,565 Illumina 510S SNPs remained for analysis. For the publicly available Affymetrix 6.0 SNPs, we first removed SNPs that had already been genotyped using the Illumina platforms, resulting in 643,600 unique SNPs on the Affymetrix 6.0 SNP array for which we performed the QC analysis. After removing 26,140 SNPs with call rates <95%, 107,275 SNPs with a MAF <5%, and 5763 that deviated from HWE (P < 0.001), 504,422 remained. Therefore, after combining data from the two platforms, 1,348,798 SNPs were available for use in the analysis (Fig. 2B).
The P-value for the most significant SNP, Affymetrix marker SNP_A-8538282 (rs7000734), was 3.82 × 10−7 (r = 0.309, MAF = 0.081). The top 16 SNPs, all with P-values < 10−5 are listed in Table 2, and the 151 SNPs that were associated with AUC with P-values < 10−4 are listed in Supplemental Table 2. These 151 SNPs were located within or close to 99 unique genes on the basis of the annotation of genome build 36.3. Among the 151 top SNPs, three were within coding regions, 45 within introns, and 36 and 68 were within 5′- or 3′-untranslated regions (UTRs) or flanking regions, respectively.
The top 16 SNPs that were associated with radiation AUC with P-values < 10−5
For our integrated analyses of SNPs with both expression array and cytotoxicity data, we began with 1335 SNPs that had P-values < 10−3 for association with radiation AUC. As stated previously, we used these less stringent criteria to identify more possible candidates that, even though they did not reach genome-wide significance, might have functional impact on radiation sensitivity. When we determined the locations of these 1335 SNPs, we were able to identify 27 regions/loci that contained at least two SNPs with P-values < 10−4 within 50 kb in each of these regions (a total of 175 SNPs). For purposes of this analysis, we defined these regions as a locus or a “SNP peak region.” All 27 of these loci, and the SNPs within each locus, are listed in Supplemental Table 3. We focused on these loci for the integrated analyses. The most significant locus, locus 8C, mapped to chromosome 8 (Fig. 2C; Supplemental Table 3). This region had six SNPs within 50 kb with P-values < 10−4, including the SNP with the lowest P-value, 3.82 × 10−7. The gene closest to this region encoded PLEKHF2. All six of the SNPs were in tight linkage disequilibrium (LD), with r2 values that ranged from 0.7 to 1.0 in Caucasian-Americans (CAs), with slightly lower values in the African-American (AA) subjects (Supplemental Fig. 1). The allele frequencies for these SNPs were also higher in CA and AA subjects (MAF ≥ 0.065) than in Han Chinese–American (HCA) subjects (MAF ≤ 0.021). We also imputed SNPs not on the genotyping platforms, using the HapMap populations as the reference, that were located 200 kb up or downstream from SNP_A-8538282, the most significant SNP on chromosome 8 (see Fig. 2C). That area contained 222 “observed SNPs” that were on the combined Illumina and Affymetrix platforms, plus 104 SNP markers that we imputed (Fig. 2C). None of the imputed SNPs was more significant than the genotyped SNP_A-8538282.
Integrated SNP, basal expression, and radiation AUC analyses
The effect of genetic variation on radiation-induced cytotoxicity might result, in part, from the regulation of gene expression. Previous studies performed with lymphoblastoid cell lines have shown that post-radiation gene expression is highly influenced by DNA sequence variation (Correa and Cheung 2004; Smirnov et al. 2009). However, few studies have focused on basal gene expression levels and their possible relationship to radiation response, i.e., on information that might be used to predict response. Therefore, we also performed an “integrated analysis” that included data for SNPs, basal expression, and radiation AUC. Specifically, we identified 175 SNPs with P-values < 10−3 that mapped to the 27 loci that we had identified (Supplemental Table 3), and then used data for the 54,000 basal expression array probe sets on the Affymetrix U133 Plus 2.0 platform to identify SNPs within those loci that might be associated with basal gene expression, in either a cis or trans relationship.
Specifically, we observed 2432 SNP expression associations for the 175 SNPs with P-values < 10−4. We then correlated those 2432 expression probe sets with radiation AUC and identified probe sets with P-values < 10−3 for association with radiation AUC. We selected a less-stringent P-value cutoff to capture as much information as possible, realizing that many of the associations would be false-positives. This integrated analysis, moving from loci to SNP markers to expression, identified 50 unique SNPs located in 14 of the 27 loci that were associated with data for 47 probe sets, which represented 39 unique annotated genes, i.e., basal expression of these genes was associated with radiation AUC with P < 10−3 (Supplemental Table 4). None of the SNPs were in cis-regulatory regions, defined as 5 Mb on either side of the gene identified. These 50 unique SNPs mapped to eight different chromosomes, with at least two SNPs on each of those chromosomes. The four most significant loci or “SNP peak regions” mapped to chromosomes 1, 4, 5, and 8, respectively, and contained the LRRN2, IL19, KCNK1, LDB2, NCRNA00290 (also known as LOC728081), DEPDC1B, PPIGP1 (also known as LOC100131033), and PLEKHF2 genes (Supplemental Table 4). The SNPs near PLEKHF2 within the locus on chromosome 8 (Fig. 2C) were particularly striking since they were associated with variation in the expression of six annotated genes, and variation in the expression of those genes was, in turn, significantly associated with radiation AUC. The chromosome 1 locus contained the largest number of SNPs, 19, that were associated with radiation AUC, with P-values that ranged from 10−3 to 10−4. Those 19 SNPs were associated with the expression of 12 annotated genes that were also associated with radiation AUC with P-values that ranged from 10−3 to 10−4. Six annotated genes were associated with the locus on chromosome 8 that contained the most significant six linked SNPs (P-values < 10−4), and those six genes were associated with radiation AUC, with P-values < 10−3. The chromosomes 4 and 5 loci included seven and six SNPs, respectively, that were associated with radiation AUC with P-values that ranged from 10−3 to 10−4, and those SNPs were associated with the expression of 11 and four unique annotated genes, respectively, with P-values that ranged from 10−4 to 10−7. Expression levels for those 15 genes were also associated with radiation AUC, with P-values < 10−3.
Functional validation of candidate genes in tumor cell lines
Our initial association experiments were performed with human LCLs. Since nongenetic factors might confound the results of these association studies, and since gene regulation is tissue specific (Dimas et al. 2009), we next turned to human tumor cell lines, two pancreatic cancer cell lines, MIA-PaCa2 (TP53 mutant), and HupT3 (TP53 mutant), and one cervical cancer cell line, HeLa, which has TP53 wild-type (WT) sequence, but for which the function of TP53 is disrupted by the expression of human papillomaviruse (HPV) E6 (DeFilippis et al. 2003), to functionally validate association results obtained with LCLs. These functional experiments involved siRNA knockdown, followed by MTS cytotoxicity assays and, subsequently, colony-forming assays. We chose these three cancer cell lines because of their relative sensitivity to radiation after testing with MTS assays.
Based on our analysis of the 54,000 basal expression probe sets, 1.3 million SNPs and radiation AUC data, as well as an evaluation of their biological function, we selected 23 candidate genes identified as described in the preceding paragraphs for siRNA screening performed with multiple tumor cell lines. We choose these genes based on their potential biological significance, as well as their expression levels in LCL, which has to be greater than 50 after GCRMA normalization. These genes were chosen based on the following criteria: (1) genes with at least one expression array probe set that had a P-value < 5 × 10−4 or at least two probe sets with P-value < 10−3 for association with radiation AUC, which identified 96 genes expressed in the LCLs. We selected 18 “less studied” genes in terms of their relationship to radiation response from 96 genes for functional studies, since most of them were known to relate to pathways that are important for radiation response, such as DNA repair, cell cycle, cell survival, and apoptosis pathways; (2) genes containing SNPs found within a locus associated with radiation AUC (P < 10−4), which identified 10 genes out of the 27 SNP loci (29 genes) expressed in the LCLs. We selected four genes from the four most significant SNP loci; (3) genes for which expression was associated with both AUC and SNPs (P < 10−3 for AUC, and P < 10−4 for SNPs, i.e., the integrated analysis), which identified 30 genes expressed in the LCLs. We selected seven genes for further functional studies, of which three genes were associated with the three most significant SNP loci, and the other four genes were associated with either at least one SNP with a P-value < 10−5 or at least three SNPs with P-values < 10−4. This selection process resulted in 23 genes after removing redundant genes among different selection categories. This overall selection strategy is depicted graphically in Figure 3.
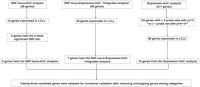
Schematic diagram of the strategy used to select genes for functional validation. Genome-wide association studies for radiation AUC were performed with 1.3 million SNPs or 54,000 expression probe sets. A SNP locus–Expression–AUC integrated analysis was performed using loci that contained at least two SNPs associated with radiation AUC with P-values <10−4, 54,000 expression probe sets and radiation AUC associations were used to identify SNPs associated with radiation AUC through their influence on gene expression (SNP-expression P-value < 10−4, expression-AUC P-value < 10−3). We excluded genes that had expression levels less than 50 and have previously been known to be involved in pathways related to radiation sensitivity. Finally, 23 candidate genes were selected for functional validation studies performed with multiple cancer cell lines.
For functional validation, we used either two unvalidated siRNAs or one validated siRNA based on QIAGEN data to knockdown each of the 23 candidate genes. If two siRNAs were available, we defined “significance” as a gene with a significant change in apparent AUC for both siRNAs in comparison with a control siRNA. In order to screen candidates efficiently, MTS assays were used with all three tumor cell lines for all 23 of the genes selected for study, although we understood the limitations of short-term cytotoxicity assays for the prediction of radiation survival and that we might miss some associations using this assay (Carmichael et al. 1987; Anoopkumar-Dukie et al. 2005). Knockdown of seven genes had a significant effect on radiation sensitivity in one cell line, three genes were positive for two cell lines, and knockdown of two genes significantly altered radiation sensitivity in all three cell lines (Table 3; Fig. 4). We then selected five genes for further study for which knockdown with specific siRNAs significantly altered radiation sensitivity in at least two cancer cell lines, specifically C13orf34, MAD2L1, PLK4, TPD52, and TTF1 (Table 3; Fig. 4). We also included DEPDC1B, even though this gene only showed an effect of knockdown on radiation sensitivity in HeLa cells, since two SNPs located 2.5 and 8.4 kb downstream of this gene (rs4326096 and rs2409791) were significantly correlated, in the cell-line model system, with the expression of C13orf34 (P = 9.97 × 10−5 and 4.86 × 10−5, respectively) (Supplemental Table 4), a gene that displayed a functional effect on radiation-induced cytotoxicity in all three cancer cell lines (Table 3; Fig. 4). These two SNPs were highly linked (r2 > 0.9).
Candidate genes selected for siRNA screening, with MTS assay results as well as colony-forming assay results when appropriate
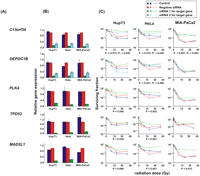
siRNA screening of candidate genes by MTS assay in multiple cancer cell lines. Data are shown for five of the top 23 candidate genes that were studied functionally in HupT3, MIA-PaCa2, and HeLa cancer cell lines by MTS assay after siRNA knockdown performed with two “unvalidated” or one “validated” siRNA when available. (Blue) Data for nontransfected cells; (red) negative control siRNA; (green, light blue) data for specific siRNAs. “Significance” was defined as a gene with a significant change in apparent AUC in comparison with control siRNA and was indicated by the P-value. If two siRNAs were available, “significance” was defined as a gene with significant changes for both siRNAs, and P1 and P2 represented the P-values for siRNA 1 and siRNA 2, respectively. At least three independent experiments were performed in triplicate. Error bar, SEM of at least three independent experiments. (A) Candidate gene symbols. (B) qRT-PCR. The y-axis indicates relative gene expression after siRNA knockdown when compared with “all star negative” siRNA. (C) MTS assays. The x-axis indicates the radiation dose, and the y-axis indicates the surviving fraction after exposure to radiation.
As the next step in the analysis, and to further confirm results obtained with the MTS assay, we also performed colony-forming assays for these same six genes in the MIA-PaCa2, HupT3, and HeLa cells used to perform the MTS assays, plus a lung-cancer cell line, A549, since radiation is commonly used to treat lung cancer. As shown graphically in Figure 5, knockdown of C13orf34, DEPDC1B, and TPD52 desensitized all four cell lines to radiation treatment. MAD2L1 knockdown had a significant impact on radiation effect in HeLa and HupT3, but not in the A549 cell line. However, knockdown of MAD2L1 resulted in lack of colony formation in MIA-PaCa2 cells in the absence of radiation treatment, indicating that this gene might be essential for cell proliferation. Knockdown of PLK4 only desensitized radiation response in HupT3 cells, consistent with the MTS assay results, and TTF1 knockdown did not alter radiation sensitivity in any of the four cell lines (data not shown). These results indicated that this surrogate MTS assay can provide valid leads for identifying genes that might play a role in radiation sensitivity.
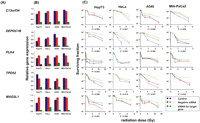
siRNA screening of candidate genes by colony-forming assay performed with multiple cancer cell lines. Data for five of the 23 top candidate genes were selected for colony-forming assays using HupT3, MIA-PaCa2, HeLa, and A549 cancer cell lines. qRT-PCR was also performed to determine knockdown efficiency. At least three independent experiments were performed in triplicate. Error bar, SEM of at least three independent experiments. Significance was defined by P-values. (A) Candidate gene symbols. (B) qRT-PCR to assess expression levels for each candidate gene after knockdown in each cell line. (C) Colony-forming assays performed with HupT3, MIA-PaCa2, HeLa, and A549 cancer cell lines. The x-axis indicates the radiation dose, and the y-axis indicates the surviving fraction after radiation exposure.
Taken together, through the initial screening studies in LCLs and further functional validation in multiple tumor cell lines, we found that the expression of five genes, C13orf34, MAD2L1, PLK4, TPD52, DEPDC1B, were involved in radiation-induced response. We computed the percentage of variation in the AUC of LCLs that was explained by the variation of these five genes' expression (with and without inclusion of these two SNPs, rs4326096 and rs2409791, close to DEPDC1B), which were 11.4% and 16.1%.
Discussion
Radiation therapy plays an important role in the treatment of cancer. However, response to radiation therapy varies widely. The therapeutic efficacy of radiation is determined mainly by total dose, but the use of high doses has been limited because of the severity of side effects in normal tissue. It is estimated that nearly 80% of interindividual variation in normal tissue response to radiation might be due to genetic factors (Turesson et al. 1996). Radiation therapy also has a relatively narrow therapeutic index (Turesson 1990; Bentzen et al. 2008). Therefore, understanding the biology underlying the variation in response to radiation might help us to maximize radiation efficacy in the tumor, while minimizing side effects in normal tissues. Several radio-genetic studies have already shown that genetic polymorphisms in genes within known radiation response pathways are significantly associated with radiosensitivity. Those pathways include endogenous oxidative stress defense, inflammatory response, cytokine activity related to fibrosis, DNA damage signaling, cell cycle control, and DNA repair (Chistiakov et al. 2008; Andreassen and Alsner 2009; Popanda et al. 2009; Pugh et al. 2009). These previous studies raised the possibility that genetic variation might contribute to individual variation in radiation response. In the present study, we performed a genome-wide association analysis using 277 LCLs for which we had 1.3 million SNPs, basal gene expression data, and an ionizing radiation cytotoxicity AUC phenotype to identify genes that might be responsible for radiation sensitivity. LCLs have been used successfully in many pharmacogenomic studies to identify and understand genetic variation associated with variation in drug response phenotypes (Huang et al. 2008; Li et al. 2008, 2009; Bleibel et al. 2009). In addition, in vitro lymphocyte radiosensitivity has been found to be significantly correlated with clinical response to radiation therapy, although biological differences obviously exist between lymphocytes and LCLs (West et al. 2001; Dikomey et al. 2003).
Based on the analysis of SNP, basal gene expression and radiation cytotoxicity (AUC) data, our study yielded a total of 240 candidate genes, including 211 identified as a result of either expression versus AUC associations (P < 10−3) or an integrated analysis that included SNP markers, expression, and radiation AUC data. We also identified 29 genes based on the association of SNPs with AUC (P < 10−4). Although only one probe set was statistically significant after Bonferroni correction, and none of the SNPs remained significant after Bonferroni correction, which might be due to our limited sample size used for our GWA analyses, when we performed Ingenuity Pathway analysis for these 240 genes, the top three networks all involved “cell death” and centered around NFKB, PI3K/Akt and MAPK/ERK as “network hubs” (Supplemental Fig. 2). Similarly, the NFKB pathway has been also found to be involved in the response to a higher dose of radiation (10 Gy) in TP53-null or mutant lymphoblast cell lines (Lu et al. 2010). Many candidate genes identified during our study have been reported to have altered levels of expression in response to radiation exposure in the NCI-60 cell lines or in lymphoblastoid cell lines, especially TP53-dependent genes such as CDK1, PHLDA3, and PTPRC (Amundson et al. 1999, 2003, 2005, 2008; Jen and Cheung 2005). In addition, genes such as MEF2B, NRF1, PHPT1, ZMAT3, CHEK1, and GALR3 that are up- or down-regulated by ionizing radiation exposure, were also found to be associated with radiation AUC from our study (Amundson et al. 2004; Jen and Cheung 2005; Dressman et al. 2007; Paul and Amundson 2008; Rzeszowska-Wolny et al. 2009; Westbury et al. 2009). These results suggested that our association study performed with 277 human lymphoblastoid cell lines was capable of generating biologically relevant candidates for follow-up study.
The LCL system used in our initial screening studies, like all model systems, has limitations. EBV transformation might cause chromosomal instability and cellular changes in the LCLs (Sie et al. 2009), and variation of the response of these cells might be influenced by nongenetic factors such as cell growth rate or baseline ATP levels (Choy et al. 2008). In addition, the regulation of gene expression is tissue specific (Dimas et al. 2009; Kwan et al. 2009). Therefore, to validate the initial association results obtained using LCLs, we selected 23 top candidate genes (see Table 3; Fig. 3) to perform functional validation studies using siRNA knockdown performed with MIA-PaCa2, HupT3, Hela, and A549 cancer cell lines (Figs. 4, 5). We removed genes that were not highly expressed in the LCLs, as well as genes that are involved in pathways such as DNA repair, cell cycle, cell survival, and apoptosis, which are already known to influence radiation sensitivity. We understood that these selection criteria were arbitrary, but they provided a way to narrow down the candidates for functional follow up. Five genes were shown to alter the radiation response with MTS and colony-forming assays (Figs. 4, 5), and all five genes contributed ∼11% of variation in AUC observed. These results suggested that, like many complex traits, response to radiation also involves multiple genes in different pathways, and, indeed, many of the genes at the top rank in our association study have been shown to play important roles in pathways related to radiation response. However, this is not unique. Even with clinically established pharmacogenomic examples such as CYP2C19 *2, only 12% of the variation in platelet aggregation can be explained by this variant (Shuldiner et al. 2009). Although the five final genes contributed 11% of the total variation, they also provided us with new insights into novel mechanisms in radiation response. For example, among the genes for which we performed colony-forming assays (Fig. 5), two altered radiation sensitivity in all four cancer cell lines tested. The first, C13orf34, mapped to chromosome 13q22, and has been implicated as a putative breast cancer susceptibility locus (Rozenblum et al. 2002). This portion of the genome is also a common site for somatic deletions in many malignant tumors (Rozenblum et al. 2002). As a cell cycle protein, C13orf34 enhances the initial activation of Polo-like kinase 1 (Plk1) in an Aurora A-dependent manner at the G2/M transition, and facilitates G2/M transition (Macurek et al. 2008, 2009; Seki et al. 2008a,b; Pomerening 2009). However, no previous study had linked C13orf34 to the DNA damage response pathway or to radiation sensitivity. The fact that we found that C13orf34 expression was correlated with radiation cytotoxicity and that knockdown of C13orf34 desensitized cancer cells to radiation exposure suggests a potential role for C13orf34 in the DNA damage response pathway, in addition to its role as a cell cycle regulator. This hypothesis will need to be tested further in future experiments.
The second gene that influenced radiation response in all four cell lines was TPD52, also known as “Tumor protein D52.” TPD52 is a coiled-coil motif bearing protein that plays a role in cell proliferation, apoptosis, vesicle trafficking, tumorigenesis, and metastasis (Lewis et al. 2007; Ummanni et al. 2008). TPD52 is highly overexpressed in multiple cancers as a result of gene amplification (Balleine et al. 2000; Rubin et al. 2004; Byrne et al. 2005; Zhu et al. 2007; Petrova et al. 2008). In breast cancer patients, TPD52 overexpression was correlated with lymphocyte radiosensitivity (Sims et al. 2007), an observation consistent with our association study in LCLs and our functional validation experiments performed with tumor cell lines.
In summary, although variation in radiation response might result from multiple gene effects and, possibly, gene-environment interactions, our results provide important insight into novel genes and mechanisms that may contribute to variation in response to radiation therapy.
Methods
Cell lines
EBV-transformed LCLs from 93 African-American (AA), 89 Caucasian-American (CA), and 95 Han Chinese–American (HCA) unrelated healthy subjects (sample sets HD100AA, HD100CAU, HD100CHI) were purchased from the Coriell Cell Repository. These samples had been collected and anonymized by NIGMS, and all subjects had provided written consent for their experimental use. This study was reviewed and approved by the Mayo Clinic Institutional Review Board. Human pancreatic cancer MIA-PaCa2 and HupT3 cell lines were gifts from Daniel D. Billadeau (Mayo Clinic, Rochester, MN). Human cervical cancer HeLa and non-small-cell lung cancer A549 cell lines (ATCC no. CCL-185) were obtained from the ATCC.
LCLs were cultured in RPMI 1640 medium (Mediatech) supplemented with 15% heat-inactivated Fetal Bovine Serum (FBS) (Mediatech). According to the protocol from the Coriell website (http://ccr.coriell.org/sections/support/global/lymphoblastoid.aspx?pgid=213), LCLs were maintained at a density of 2–8 × 105 cells/mL, and were split or refed with fresh medium every 3 d, depending on the growth status of each cell line. HeLa and MIA-PaCa2 cell lines were cultured in DMEM medium containing 10% FBS. HupT3 and A549 cell lines were grown in RPMI 1640 medium with 10% FBS.
Cell proliferation assay
Cell proliferation assays were performed in triplicate at each radiation dose. Specifically, 100 μL of cells (5 × 105 cells/mL) were plated into 96-well plates (Corning) (Li et al. 2008) and were treated with ionizing radiation at 0, 0.156, 0.3125, 0.625, 1.25, 2.5, 5, 10, and 20 Gy using 137Cesium gamma-rays (J.L. Shepherd and Associates Mark I Model 25 Irradiator). After incubation for 3 d, 20 μL of CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay solution (Promega Corporation) was added to each well. Plates were read in a Safire2 plate reader (Tecan AG). Nineteen LCLs were selected randomly, and the MTS assay was repeated ∼1 yr after the initial assay. Cytotoxicity for human tumor cell lines was determined in a similar fashion, except the cells were incubated overnight before radiation treatment at 0, 0.25, 0.5, 1, 2.5, 5, 10, and 20 Gy, followed by MTS and colony-forming assays.
Expression array assays
Total RNA was extracted from each of the cell lines using Qiagen RNeasy Mini kits (QIAGEN, Inc.). RNA quality was tested using an Agilent 2100 Bioanalyzer, followed by hybridization to Affymetrix U133 Plus 2.0 Gene-Chips. A total of 54,613 probe sets were used in the analyses. Expression array data were obtained for all of the cell lines, 166 of which had been used in previous studies (Li et al. 2008, 2009) plus 111 new cell lines that were used in the present study.
Genome-wide SNP analysis
DNA from all of the LCLs was genotyped using Illumina HumanHap 550K and 510S BeadChips, which assayed 561,298 and 493,750 SNPs, respectively. Genotyping was performed in the Genotype Shared Resource (GSR) at the Mayo Clinic. We also obtained publicly available Affymetrix SNP Array 6.0 Chip SNP data for the same cell lines, which involved 643,600 SNPs unique to the Affymetrix SNP array. The genotyping data from 166 of 277 LCLs using Illumina HumanHap 550K BeadChips had been used in a previous study (Li et al. 2009). SNPs that deviated from Hardy-Weinberg Equilibrium (HWE), based on the minimum P-value from an exact test for HWE (Wigginton et al. 2005) and the stratified test for HWE (Schaid et al. 2006) (P-values < 0.001); SNPs with call rates <95%; or SNPs with MAFs <5% were removed from the analysis.
Transient transfection and RNA interference
siRNAs for the candidate genes and “all star negative control” siRNA were purchased from QIAGEN. The human HeLa cervical cancer cell line and the human pancreatic cancer cell lines, MIA-PaCa2 and HupT3, were used for the siRNA knockdown experiments. Reverse transfection was performed in 96-well plates. Specifically, ∼3000–4000 cells were mixed with 0.1 μL of lipofectamine RNAiMAX reagent (Invitrogen) and 10 nM siRNA for these experiments.
Colony-forming assays
“All star negative control” and specific siRNAs were transfected into MIA-PaCa2, HupT3, HeLa, and A549 cell lines. Twenty-four hours later, ∼400–1000 cells were plated in triplicate in 6-well plates. After exposure to radiation, cells were incubated for up to 7 d. Colonies in each well were fixed with methanol and stained with crystal violet. All colonies were counted visually or by the use of Quantity One (Bio-Rad).
Real-time quantitative reverse transcription-PCR (qRT-PCR)
Total RNA was isolated from cultured cells transfected with control or specific siRNAs with the Qiagen RNeasy kit (QIAGEN, Inc.), followed by qRT-PCR performed with the one-step, Brilliant SYBR Green qRT-PCR master mix kit (Stratagene). Specifically, primers purchased from QIAGEN were used to perform qRT-PCR using the Stratagene Mx3005P Real-Time PCR detection system (Stratagene). All experiments were performed in triplicate with beta-actin as an internal control. Reverse transcribed Universal Human reference RNA (Stratagene) was used to generate a standard curve. Control reactions lacked RNA template.
Statistical methods
The radiation cytotoxicity phenotype, AUC, was calculated based on a logistic model. Three different logistic functions (a four parameter logistic model, a three parameter logistic model with a fixed asymptote at 0%, and a three parameter logistic model with a fixed asymptote at 100%) were used to fit the data with the R package “drc” (http://cran.r-project.org/web/packages/drc/drc.pdf). The best model fit (i.e., lowest mean square error) from the three logistic models was used to determine the cytotoxicity AUC phenotype. The AUC phenotype was determined by numerically computing the area under the estimated dose-response curve, from dose 0 to 20 Gy. AUC values were then natural-log transformed. Expression array data were normalized on a log2 scale using GCRMA (Wu et al. 2004). The normalized expression data and log transformed AUC values were then regressed on gender. Since the LCLs were from multiple races/ethnic groups, before completing the genetic association analysis, population stratification was assessed using the method developed by Price et al. (2006). This approach uses an eigen analysis for detecting and adjusting for population stratification, in which the eigen analysis was performed within each of the three racial groups. Using the top five eigenvectors within each race, the individual genotypes were adjusted using the model Gij = αj + γkj + ɛij, with Gij representing the genotype for the ith cell line in racial group j (j = 1, 2, 3), αj the race effect for race j, and γkj the kth eigenvector (k = 1, 2, 3, 4, 5) effect for race j. Similarly, for genetic analysis of SNPs with AUC or expression, the log transformed AUC values and normalized log2 expression values were also adjusted for race using the five eigenvectors, in addition to gender.
Association analyses of expression-AUC, expression-SNP, and SNP-AUC were then completed using Pearson correlations and the adjusted AUC, SNP, and expression data. False Discovery Q-values (Storey and Tibshirani 2003) were also computed for each test. Pairwise LD was estimated using r2 (r-squared) statistics and was displayed graphically using the Haploview software (Barrett et al. 2005). Genes and SNPs were annotated using NCBI Build 36.3. The pathway analysis, for the top genes, was performed using Ingenuity Pathway Analysis (IPA; Ingenuity Systems).
To integrate the genotype, expression, and drug cytotoxicity data, we first identified loci containing at least two SNPs associated with AUC with P-values < 10−4 within 50 kb of each other and identified all SNPs located within the specific loci with P-values < 0.001. Next, we determined which expression probe sets were associated with these SNPs (P-values < 10−4). Finally, to determine whether the expression probe sets associated with these SNPs were also associated with radiation AUC values, we identified which expression probe sets were associated with radiation AUC (P-values < 0.001). A similar approach has been used successfully to detect novel candidate genes for functional follow-up (Li et al. 2009).
For the most significant locus on chromosome 8, SNPs were imputed for a region 200 kb in length on either side of the most significant SNP. Imputation was performed using MACH 1.0 (Li and Abecasis 2006), with HapMap data (http://hapmap.ncbi.nlm.nih.gov/) as the reference panel. Specifically, SNPs for AA were imputed using both CEU and YRI data, SNPs for CA were imputed based on CEU data, and SNP markers for HCA were imputed based on the CHB and JPT data. In addition, the percent of variation in the drug response phenotype explained by a set of genes was estimated using r2 from a multivariable linear regression model. Lastly, significance of the AUC values between negative control siRNA and gene-specific siRNA was determined by a two-tailed unpaired t-test.
To determine association between SNPs and expression using the data sets generated from our 300 human lymphoblastoid cell lines, visit http://pgxcms.mayo.edu/AR.
Acknowledgments
This work was supported in part by National Institutes of Health (NIH) grants K22 CA130828, R01 CA138461, and U01 GM61388 (The Pharmacogenetics Research Network), an ASPET-Astellas Award, and a PhRMA Foundation “Center of Excellence in Clinical Pharmacology” Award.
Footnotes
-
↵5 Corresponding author.
E-mail wang.liewei{at}mayo.edu; fax (507) 284-4455.
-
[Supplemental material is available online at http://www.genome.org. The microarray data and SNP data from this study have been submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under SuperSeries accession no. GSE24277.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.107672.110.
- Received March 12, 2010.
- Accepted July 15, 2010.
- Copyright © 2010 by Cold Spring Harbor Laboratory Press








