
Region-specific detection of neuroblastoma loss of heterozygosity at multiple loci simultaneously using a SNP-based tag-array platform
- John M. Maris1,
- George Hii1,
- Craig A. Gelfand2,6,
- Shobha Varde2,7,
- Peter S. White1,
- Eric Rappaport1,
- Saul Surrey3, and
- Paolo Fortina4,5,8
- 1 Division of Oncology, The Children's Hospital of Philadelphia, and Department of Pediatrics, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania 19104, USA
- 2 Orchid Biosciences, Inc., Princeton, New Jersey 08540, USA
- 3 Cardeza Foundation for Hematologic Research Department of Medicine, Jefferson Medical College, Thomas Jefferson University, Philadelphia, Pennsylvania 19107, USA
- 4 Center for Translational Medicine, Department of Medicine, Jefferson Medical College, Thomas Jefferson University, Philadelphia, Pennsylvania 19107, USA
- 5 Dipartimento di Medicina Sperimentale e Patologia, Universita' degli Studi “La Sapienza,” 00198 Roma, Italy
Abstract
Many cancers are characterized by chromosomal aberrations that may be predictive of disease outcome. Human neuroblastomas are characterized by somatically acquired copy number changes, including loss of heterozygosity (LOH) at multiple chromosomal loci, and these aberrations are strongly associated with clinical phenotype including patient outcome. We developed a method to assess region-specific LOH by genotyping multiple SNPs simultaneously in DNA from tumor tissues. We identified informative SNPs at an average 293-kb density across nine regions of recurrent LOH in human neuroblastomas. We also identified SNPs in two copy number neutral regions, as well as two regions of copy number gain. SNPs were PCR-amplified in 12-plex reactions and used in solution-phase single-nucleotide extension incorporating tagged dideoxynucleotides. Each extension primer had 5′ complementarity to one of 2000 oligonucleotides on a commercially available tag-array platform allowing for solid-phase sorting and identification of individual SNPs. This approach allowed for simultaneous detection of multiple regions of LOH in six human neuroblastoma-derived cell lines, and, more importantly, 14 human neuroblastoma primary tumors. Concordance with conventional genotyping was nearly absolute. Detection of LOH in this assay may not require comparison to matched normal DNAs because of the redundancy of informative SNPs in each region. The customized tag-array system for LOH detection described here is rapid, results in parallel assessment of multiple genomic alterations, and may speed identification of and/or assaying prognostically relevant DNA copy number alterations in many human cancers.
Human malignancies are characterized by accumulation of somatically acquired alterations in the tumor cell genome (Vogelstein and Kinzler 2004). There is a large literature documenting association of genomic alterations with tumor phenotype, and regional deletions are often reported to be a marker for disease aggressiveness, particularly in human solid tumors such as neuroblastoma (Maris and Matthay 1999; Vandesompele et al. 2001; Mora et al. 2002; Brodeur 2003). As there are many more regions of loss of heterozygosity (LOH) than isolated tumor-suppressor genes, it is becoming increasingly evident that the biological consequence of hemizygous deletions may often be the result of haploinsufficiency and/or targeting multiple genes within a region of LOH. Thus, detection of LOH will likely remain a cornerstone of predicting tumor aggressiveness for many human cancers.
Neuroblastoma provides the paradigm for the clinical utility of tumor-specific molecular genetic data (Brodeur et al. 1984; Seeger et al. 1985; Bowman et al. 1997; Matthay et al. 1998; Maris and Matthay 1999; Schmidt et al. 2000; Brodeur 2003). This solid tumor is the most common malignant disease of infancy and the third most common cause of pediatric cancer mortality (Brodeur and Maris 2002). It is a markedly heterogeneous disease for which age at diagnosis and presence of disease metastasis are strongly associated with prognosis. In addition, hemizygous deletions in human neuroblastoma may lend further prognostic information (Maris and Matthay 1999; Maris et al. 2000; Brodeur 2003; Maris and Shusterman 2003; Spitz et al. 2003). Such deletions have been well characterized at 1p36 (Caron et al. 1996a; Maris et al. 2000), 11q14–23 (Guo et al. 1999, 2000; Luttikhuis et al. 2001; Plantaz et al. 2001; Spitz et al. 2003), 3p14–25 (Ejeskar et al. 1998; Breen et al. 2000; Spitz et al. 2003), 4p15-p16 (Caron et al. 1996b; Perri et al. 2002), 5q (Meltzer et al. 1996), 9p21 (Marshall et al. 1997), 14q32 (Thompson 2000), 16p12–p13 (Maris et al. 2002), 18q21 (Reale et al. 1996), and 19q13 (Mora et al. 2001).
Given the complexity of somatically acquired genomic alterations in human neuroblastoma, and the proven clinical utility of at least a subset, we designed a customizable platform to survey multiple genomic regions for LOH in parallel. Other factors to be considered were the relatively small size of many diagnostic biopsies, intratumoral heterogeneity, and incomplete compliance with submission of a matched blood sample for constitutional DNA in many cases. We therefore sought to design a high-throughput assay to detect LOH in primary neuroblastoma biopsy samples that met each of the following requirements: (1) ability to reliably detect LOH at six or more genomic regions simultaneously; (2) low input amount of tumor DNA; (3) obviation of the requirement for matched constitutional DNA; (4) flexibility to modify regions surveyed; (5) scalability; and (6) relative insensitivity to “contaminating” normal DNA present in the stromal component of most solid tumor biopsies. We now report on a SNP-based tag-array strategy that meets each of these requirements, and provide proof of principle in human neuroblastoma-derived cell lines and archival primary tumor biopsy specimens.
Results
Neuroblastoma SNP tag-array strategy and design
The tag-array-based genotyping protocol is diagrammed in Figure 1. First, genomic DNA encompassing the SNP region (A/C in Fig. 1A) under interrogation is amplified. PCR products then are used as templates for primer-directed, solution-phase, single-base extension using two different fluorescently tagged ddNTPs (Fig. 1B). The extended product is then captured on a GenFlex Tag Array chip via hybridization of complementary barcode sequences (Fig. 1C), which enables the simultaneous interrogation of up to 2000 extension products and 50 controls. The array (Fig. 1D) is scanned for dye fluorescence, and software deconvolutes the data generating genotype calls.
SNP performance and selection of SNPs for LOH detection
Expected copy changes in patients with neuroblastoma are listed in Table 1. We first identified 467 SNPs known to map within our regions of interest (Table 1, column 10; Supplemental Table 1). We then genotyped 16 control DNA samples obtained from two CEPH families to test for SNP performance and ensure proper inheritance of polymorphic alleles. Performance was assessed by determining adequate fluorescence intensity (log relative fluorescence values <2.0 were scored as “failed”) and appropriate “clustering” of homozygous and heterozygous SNPs according to the relative intensity of labeling dyes. Finally, proper inheritance was ensured through three generations in both CEPH families with SNPs showing non-Mendelian inheritance patterns eliminated. Homozygosity and heterozygosity thresholds for some SNPs were redefined according to the clustering patterns formed by the 16 CEPH individuals. A total of 121 SNPs (26%) were excluded from further analysis based on these criteria. No region was preferentially affected by this process, and the median density of markers within regions being assayed for LOH was 293 kb (range 286 kb for the 10.8-Mb region surveyed at 1p36 to 1499 kb for the 51.9-Mb regions surveyed at 3p) (Table 1).
Neuroblastoma tag-array design
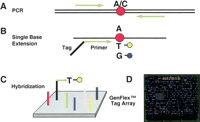
GenFlex Tag Array genotyping. The system consists of 2050 tag probe locations, designed to have approximately equal hybridization characteristics. (A) DNA encompassing each SNP is amplified with a set of primers and then (B) interrogated in a single-base extension reaction in solution with a primer attached to a tag complementary to a unique tag probe on the GenFlex Tag Array chip (C). Different alleles of a given SNP can be discriminated by use of sequence-specific dideoxynucleotides labeled with either fluorescein or biotin. The biotinylated positions are then labeled indirectly by reaction with streptavidin-conjugated phycoerythrin or SAPE after hybridization of the tag-SBE products to the tag probes. (D) Laser-induced emissions from the chips are then scanned at both 530 and 570 nm.
Tag-array detection of LOH in neuroblastoma cell lines
We applied our tag-array platform to a panel of six neuroblastoma cell lines, two of which had a matched constitutional DNA sample available for comparison. LOH was detected in at least one locus in each cell line (range 1–5 loci) (Table 2). Representative results are shown in Figure 2 for neuroblastoma cell line SK-N-AS. For each of the six cell lines studied, there was 100% concordance between the tag-array and conventional methods of microsatellite-based genotyping and FISH.
Tag-array results for neuroblastoma cell lines and primary tumors
Sensitivity of tag-array system to contaminating normal DNA
We next assessed the required degree of tissue purity in order to make a definitive LOH call. Spiking experiments were done using blood and tumor DNA. Approximately 40–50 SNPs were typed in four chromosomal regions in both DNAs (Fig. 3, top panel, 100% T and B columns). Previous typing showed only two SNPs in tumor DNA were heterozygous compared to 43 in blood for the regions interrogated. These data were corroborated using the GenFlex Tag Array system. The number of heterozygous SNPs detected in mixtures containing varying amounts of the two DNAs is shown in Figure 3 (bottom bar graph) with the number of heterozygous SNPs indicated in each region in the associated table (top panel). Our results show that the total number of heterozygous SNPs increases above that seen in tumor DNA (only two) when tumor DNA is contaminated with 15% normal (blood) DNA. This number increases linearly as the percentage of contaminating normal (blood) is increased to 45%. At 45% contamination, the number of detectable heterozygous SNPs was 34 compared to 43 for 100% blood-derived DNA. These results suggest that a definitive call of LOH in primary tumor tissue can be made in the presence of contaminating normal tissue provided that the degree of contamination is kept to a minimum (ideally <15%). However, even at higher degrees of contamination (45% normal), LOH was able to be inferred from our results. Results from higher contaminations necessitate comparison of results with those from DNA from the patient's blood to infer LOH in the tumor. For example, if 50 SNPs were detected as being heterozygous in blood DNA and only 20 were detected using the “tumor” sample containing >15% normal cell contamination, then LOH is inferred in tumor DNA.
Tag-array detection of LOH in neuroblastoma primary tumors
Lastly, we assayed for LOH in archival primary neuroblastoma using the tag-array platform (Table 2; Fig. 4). LOH was detected at a minimum of one locus in 11/14 of the primary tumor samples. As expected, 11q23 LOH was the most commonly observed aberration (detected in nine samples), with concomitant loss of 11p material in 6/9 samples. LOH at 1p36 was detected in four INSS Stage 4 cases and both cases with MYCN amplification. There was 100% concordance between array results and those, when available, generated by independent analyses (e.g., microsatellite, FISH).
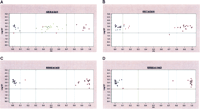
Cluster analysis of GenFlex Tag Array hybridization results. SNP calls are shown comparing (A) constitutional DNA to (B) primary tumor DNA from the same neuroblastoma sample and SK-N-AS cell line (C,D) DNA interrogating 1p36 and 11q23 regions both showing LOH. Blue circles and purple squares represent homozygous SNPs, while green triangles represent heterozygous SNPs. The X-axis represents the fraction of X allele signal [value = (X signal/(X + Y signals)], such that points near 1 represent XX homozygotes, those near 0 represent YY homozygotes, and points toward the middle XY heterozygotes. The software first uses a generic set of thresholds (the dashed vertical lines) to differentiate regions along the X-axis into each genotype cluster, leaving regions for potentially ambiguous data to remain as a “no call” (red data points). The Y-axis represents the log of the sum of both allele signals, leading to a confidence threshold or signal-to-noise cutoff (horizontal solid line), with points below this level representing “no call” data as well. The graphs show all of the SNP assays that were designed and attempted, thus although failed assay designs are still included in this data view, it is clear that several meaningful data points are gathered.
Discussion
We demonstrate development of a tag-array assay to detect LOH. Briefly, up to 2000 allele-specific single-base extension reactions are analyzed on a generic high-density oligonucleotide probe array (Fan et al. 2000). SNPs mapping within target regions are PCR-amplified, and the amplimer then serves as a template for single-base extension using dye-tagged dideoxynucleotide terminators. SNE is performed with an oligonucleotide primer stopping one nucleotide 5′ of the SNP under interrogation, with the primer having 5′ and 3′ complementarity to the array tag and the SNP amplimer, respectively. After hybridization of pooled SNE products to the tag array, fluorescence signals define allelic representation for each SNP.
Allelic alterations can be surveyed using this approach at 2–6 kb density (depending on the ultimate number of human SNPs in a region-specific manner). This resolution is an order of magnitude greater than the 6.5 Mb and 23 kb density of informative SNPs on the 10K and 100K HuSNP chips (Affymetrix), respectively. These commercial SNP chips are not compatible with defining smaller regions undergoing LOH, only interrogate SNPs that are present on the chip, and allow for no flexibility in expanding SNP density. Our approach is completely flexible in terms of SNP choice and density for interrogation, and can be designed to obviate either the need for matched constitutional DNA, or alternatively, to survey both tumor-derived and constitutional DNA on the same chip. This method allows additional SNPs to be evaluated based on newly reported regions undergoing LOH, or as new prognostically relevant loci become known. Other platforms for typing are available (e.g., Illumina, ParAllele); however, comparative studies have not been done to assess advantages/disadvantages using the same large sample sets to evaluate sensitivity, specificity, and cost efficiency.
Although this report shows development of an assay using a maximum of 500 SNPs and a single tumor DNA sample, our assay could be designed to simultaneously interrogate four samples in parallel by using four different barcode sequences for each SNP. In addition, future versions of the chip could allow for quantification of allele hybridization intensity and thus quantification of DNA copy number loss, gain, or amplification. It remains to be determined if this SNP-based approach is superior to other methods commonly used to survey the cancer genome such as array-based comparative genomic hybridization. In addition, because LOH can occur without change in DNA copy number, generally owing to mitotic recombination, polymorphism-based techniques offer more potential than CGH in detecting such events.
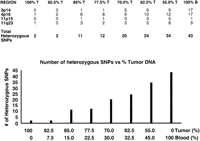
Quantitative allele frequency estimation based on analyses of mixed templates. (Upper panel) The number of heterozygous SNPs detected in mixtures containing varying amounts of the constitutional and tumor DNAs is shown with the number of heterozygous SNPs indicated in each of the four regions. (Bottom panel, bar-graph) The total number of heterozygous SNPs increases above that seen in tumor DNA (only two) when tumor DNA is contaminated with 15% normal (blood) DNA. The number increases linearly as the percentage of contaminating normal (blood) is increased to 45%.
In conclusion, this platform offers many advantages over traditional approaches and provides a clinical diagnostic assay for patients with neuroblastoma. The approach is flexible, uses a multiplex PCR strategy, and results in parallel, cost-effective sensitive screening, and is readily adaptable to monitoring LOH in a wide variety of human diseases.
Methods
Samples
Six human neuroblastoma-derived cell lines were selected from the Children's Hospital of Philadelphia (CHOP) neuroblastoma cell line repository, and two had matched lymphoblastoid cell lines established from the same patient available for comparison (Table 2). In addition, 14 primary neuroblastoma tissue samples obtained at the time of original diagnosis were randomly selected, and matched constitutional DNA was available for each of these samples. Clinical and biological covariates available for each primary tumor sample generally included age at diagnosis, disease stage by the International Neuroblastoma Staging System (INSS) (Brodeur et al. 1993), Shimada histopathologic classification (Shimada et al. 1984), MYCN amplification status (Mathew et al. 2001), and DNA index (Look et al. 1991). All specimens showed >90% tumor content by flow cytometric evaluation of disassociated tissue and analysis of pilot frozen sections stained by hematoxylin and eosin. Tumor tissues were homogenized, and genomic DNA was extracted by anion-exchange chromatography (Qiagen). Sixteen normal DNA samples from two CEPH families were used for SNP validation and quality control. The CHOP Institutional Review Board approved this research.
Neuroblastoma SNP tag-array strategy and design
The GenFlex Tag Array chip consists of 2000 oligonucleotide barcode sequences. We identified 13 regions of interest for this assay (Table 1), including nine reported in the literature as showing frequent copy number loss, two showing copy number gain, and two “control regions” that remain copy-number neutral in the vast majority of studied cases (Plantaz et al. 1997, 2001; Van Roy et al. 1997a,b; Vandesompele et al. 1998; Bown 2001). Because breakpoints defining the regions of copy number alterations are heterogeneous, we conservatively identified the most consistently deleted/gained region from the literature and concentrated our SNPs within these regions. For example, because the majority of deletions involving Chromosome 1 in neuroblastoma show LOH for all markers in 1p36, we defined an 8.2-Mb region within 1p36.33–1p36.22, defined by the microsatellite markers D1S243 and D1S1401, that was deleted in >99% of reviewed cases (White et al. 2005).
SNP selection and primer design
After selecting regions to be interrogated, we next sought to identify between 20 and 50 informative (heterozygous) SNPs/region (20 for control regions, 50 for regions of LOH) in order to have redundancy, and thus increase the likelihood and statistical power of correctly identifying a copy number aberration in tumor tissue. We used the SNP Consortium Web interface (http://snp.cshl.org/) and genome assembly build 30 to first identify 2870 SNPs with the A/G or C/T polymorphisms within these 13 regions. We then further prioritized the SNPs to be used in the assay by assuring that the heterozygosity score was known and was reported to be >30%. We then manually parsed the list to ensure relatively uniform coverage across the region being interrogated. Finally, the assay was run on two separate CEPH families (eight family members each) to assess SNP performance and ensure proper inheritance through three generations (see below). The final primer sequences were designed using Auto Primer (Beckman Coulter, Inc.) and are shown in Supplemental Table 1.
Multiplex PCR
PCR reactions were performed in an MJ Engine PTC-200 Thermalcycler (MJ Research) and multiplexed so that 12 SNP sequences were amplified simultaneously in the same well (pools used for amplification available upon request). SNPs were grouped together to ensure that each SNP being multiplexed had the same variant base sequence to be interrogated. PCR was done in 5 μL of total volume containing 50 nM each primer, 75 μM each dNTP, 50 mM KCl, 10 mM Tris-HCl (pH 8.3), 5 mM MgCl2, 0.5 U of TaqGold, and 2 ng of genomic DNA. PCR amplification conditions were 95°C for 5 min followed by 95°C for 30 sec, 50°C for 55 sec, and 72°C for 30 sec, repeating for four times, then 95°C for 30 sec; a ramping protocol was followed starting at 50°C for 55 sec, adding 0.2°C per cycle and 72°C for 30 sec. This was repeated 24 times and followed by 95°C for 30 sec, 55°C for 55 sec, and 72°C for 30 sec, repeated four times, followed by a final 72°C for 7 min.
Post-PCR processing and single-base primer extension
PCR products were treated with exonuclease I and Shrimp Alkaline Phosphatase (USB) to digest unextended PCR primers and eliminate unincorporated nucleotides, respectively, at 37°C for 30 min, followed by enzyme inactivation at 100°C for 10 min. Then 12-plex, two-color single-base extension reactions were performed using two labeled nucleotide terminators (Fluorescein or Biotin; Perkin Elmer) and two unlabeled terminating nucleotides, but complements to the SNP alleles being interrogated. The extension reaction was 96°C for 3 min, 94°C for 20 sec, 40°C for 11 sec, repeated 45 times using Thermosequenase I (Amersham Biosciences).
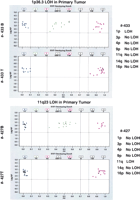
Primary tumor data interrogating SNPs in the region of 1p and 11q23. Tumors 433 and 427 were interrogated for the seven commonly altered regions in neuroblastoma (NB) and compared to results from the corresponding constitutional DNAs. Blue circles and purple squares indicate homozygous SNPs, while green triangles indicate heterozygous SNPs. All heterozygous SNPs in the tumor sample, when compared to the normal sample, have been converted to hemizygous SNPs in both regions, thus showing LOH. Typing results (LOH/No LOH) for the seven different regions are listed on the far right.
Hybridization and detection
The 12-plexed SNE reactions were pooled and concentrated into a single small volume for hybridization. Prechilled (–20°C) 1.2 mL of absolute ethanol was mixed with 19.85 μL of 8 M LiCl and 33 μL of glycogen (100 mg/mL) and added to the pool of 12-plex reactions, vortexed, and centrifuged (16,000g) for 15 min. Supernatant was then carefully removed and the pellet was dried at 42°C for 10 min, and resuspended in 100 μL of hybridization buffer (Orchid Biosciences). The sample was then heated to 100°C for 10 min, snap-cooled on ice for 2 min, and then hybridized to the GenFlex Tag Array chip in an Affymetrix Hybridization Oven 640 at 42°C for 16–18 h with rotation set at 50 rpm. The chip then was processed on the Affymetrix fluidics station using protocol “GenFlex,” with the following wash and stain buffers: non-Stringent Wash Buffer A (6× [1 M] SSPE, 0.01% Tween 20), Stringent Wash Buffer B (3× [0.5 M] SSPE, 0.01% Tween 20), and Stain Buffer (6× SSPE, 1× Denhardt's solution, 0.01% Tween 20), 5 μg/mL streptavidin, and 5 μg/mL SAPE (streptavidin-conjugated phycoerythrin from Molecular Probes).
Data analysis
The analyzed fluorescent intensity values were imported into SNPCode (Orchid Bioscience) for signal deconvolution and genotyping call using a proprietary algorithm. Plots are generated with log of relative fluorescence intensity (Log RF) and p values (PV) on the Y- and X-axes, respectively. By default, any value <2.0 on the Y-axis is scored as “Fail,” whereas on the X-axis, values between 0.1 to 0.3 and 0.7 to 0.8 also are scored as “Fail.” By default again, on the X-axis, any values between 0.0 and 0.1 are scored as “XX,” those between 0.3 and 0.7 as “YX,” and those between 0.8 and 1.0 as “YY.” However, as more samples are collected and run through the software, clustering patterns for each individual SNP emerge. If an SNP happens to fall in the Fail zone but with Log RF >2.0, and the sample shows that it is within the cluster, then one can manually change the calling of the SNP from Fail to the appropriate genotype call.
Data were extracted using a customized analysis program, which uses simple processes for balancing and normalizing the signal from both fluorescence channels of the Affymetrix GeneChip scanner (Fan et al. 2000). Once balanced, the P value (percent of allele X for each genotype) is calculated as X/(X + Y), with X and Y representing the corrected fluorescence intensities of each SNP allele determined for any one genotype. Genotypes are extracted from the data, which graphically is in the form of three clusters. The left and right clusters represent Y homozygous and X homozygous genotypes, respectively, and the central cluster represents heterozygotes (Figures 2 and 4).
Acknowledgments
This work was supported in part by NIH Grants R33-CA83220 (P.F., J.M.M., and S.S.), R01-HL69256 (S.S.), R01-CA87847 and R01-CA78545 (J.M.M.), and U01-CA98543 (Children's Oncology Group).
Footnotes
-
[Supplemental material is available online at www.genome.org.]
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3865305.
-
↵6 Present address: Preanalytical Systems, Becton, Dickinson and Company, Franklin Lakes, New Jersey 07417, USA
-
↵7 Present address: Johnson & Johnson, Milltown, New Jersey 08850, USA.
-
↵8 Corresponding author. E-mail paolo.fortina{at}jefferson.edu; fax (215) 955-6905.
-
- Accepted May 17, 2005.
- Received February 22, 2005.
- Cold Spring Harbor Laboratory Press








