
Impact of genomics on research in the rat
Abstract
The need to translate genes to function has positioned the rat as an invaluable animal model for genomic research. The significant increase in genomic resources in recent years has had an immediate functional application in the rat. Many of the resources for translational research are already in place and are ready to be combined with the years of physiological knowledge accumulated in numerous rat models, which is the subject of this perspective. Based on the successes to date and the research projects under way to further enhance the infrastructure of the rat, we also project where research in the rat will be in the near future. The impact of the rat genome project has just started, but it is an exciting time with tremendous progress.
Importance of the rat in biomedical research
The dominant power of the laboratory rat is the biological characterization of the >500 strains (http://rgd.mcw.edu/strains/), most of which were developed as models for complex, common diseases. However, while the rat is primarily known as a “physiological” model, there has been a steady increase in the use of the rat for genomic and genetic studies over last 14 years. Given the need to annotate the human genome with function, linking the rat into this process via its own genome project is a logical and necessary requirement for accelerating improvements in health care, as virtually every drug is tested in the rat before humans. Since 1966, there have been more than 1 million publications using the rat, with nearly 37,000 published annually for the last eight years. While the vast majority of rat papers remain mechanistic in nature, there are increasing numbers of genetic studies. From 1966, when PubMed started its coverage of the literature, there were 9657 genetic papers that include rat. Since 1991, when the first quantitative trait locus (QTL) was mapped in the rat (Hilbert et al. 1991; Jacob et al. 1991), there have been 26,064 papers published with rat genetics as a component. A CRISP search of the National Institute's funded grants (http://crisp.cit.nih.gov/) identified 41 currently funded grants on topics such as alcohol, hypertension, cancer, and autoimmune disease. There have been 406 grants funded relating to rat genetics since 1991. Given that the publication and funding rates in the rat remain strong in mechanistic studies, concurrent deployment of the genetic infrastructure to place these critical biological parameters on the rat genome will enable us to translate our understanding and treatment of disease from rat to human, through comparative genomics.
The rat genome project
In 1987, Robinson reported the current status of genetic linkage in the rat, the first major report (Robinson 1987). At that time, there were 10 identified linkage groups and four named chromosomes, constructed with 39 phenotypes (coat color, eye color, growth, tumors, teeth, etc.) and 33 electrophoretic and coat color markers. A major contributor to the genetic mapping revolution in the human genome project began with Weber and May's 1989 publication for the use of simple sequence length polymorphisms (SSLPs), also known as “CA-repeats”and “microsatellites” (Weber and May 1989). This new class of genetic markers was also deployed in the rat. Since then, the rat genome project has yielded a tremendous wealth of genomic resources including genetic maps; radiation hybrid (RH) cell lines and the associated RH maps (>5000 genetic markers and 19,500 genes and ESTs mapped); cDNA libraries generating >683,500 ESTs (with more being generated) clustered into >40,000 UniGenes; >10,033 genetic markers; and a published draft (∼6.8×) sequence of the genome based on the inbred BN/NHsdMcwi (Brown Norway) strain (Gibbs et al. 2004). The novel sequencing strategy combined whole-genome shotgun (WGS) with bacterial artificial chromosome (BAC) sequencing and covered 90% of the rat genome. Moreover, Celera recently released another 1.5× of draft sequence from the Sprague-Dawley rat (Kaiser 2005). The sequenced rat genome is estimated to be 2.75 Gb, distributed across 21 of the 22 chromosomes (the Y-chromosome is not yet complete), and is predicted to encode ∼20,973 genes, with 28,516 transcripts and 205,623 exons (Gibbs et al. 2004). The exact number of genes and transcripts will take several more years to resolve, but the bulk of the data is available for investigators to use now. Because of the success of the rat sequencing project and the value of the rat for functional genomics, the Mammalian Gene Collection (Gerhard et al. 2004) (full-length cDNA project) decided to sequence 6000 full-length genes from the same BN strain that was sequenced, with >4500 nonredundant genes completed. Most of these resources are publicly available through NCBI, the Rat Genome Database (RGD), RatMap, UCSC, Ensembl, and other genome databases (Table 1).
List of major rat resources
The data from the rat genome sequence provide researchers with a precise knowledge of the rat gene content, essential for the advance of biomedical research. It also improves physical and genetic map resolution, since chromosomal position no longer depends on recombination rates and statistical analysis. However, it must be noted that other lines of evidence may be required, that is, genetic linkage analysis or other forms of mapping, to ensure that local regions of the genome under investigation have been assembled correctly. The genomic tool box is now nearly complete and, as outlined throughout this review, is having an important impact in ongoing research using the rat.
Strain characterization
Many rat strains have been selectively bred for multifactorial disease (polygenic with environmental influence) and then bred to isogeneity. Currently 1015 rat strains are found in the Rat Genome Database, >50% of which are inbred strains for complex traits (538 strains). The diseases studied in these strains range from arthritis to cancer to hypertension to multiple sclerosis to seizures, including 168 different diseases and 393 phenotypes, as defined by RGD's strain disease and phenotype ontologies. In some cases, there are multiple inbred strains for a single multifactorial disease. For example, five different rat strains (BUF, DA, F344, LEW, and PVG) have an increased risk of multiple sclerosis (MS). Crosses between two of these disease strains (DA and LEW) and resistant control strains have resulted in the identification of 18 QTLs involved in experimental allergic encephalomyelitis, an animal model of MS (Dahlman et al. 1999a,b; Roth et al. 1999; Bergsteinsdottir et al. 2000). Overlapping QTL confidence intervals, for the same trait in multiple strains (e.g., Eae2 and Eae11), may then allow for identification of shared haplotypes between the disease strains, which can facilitate positional cloning of the disease allele.
As inbred strains are developed, multiple genes conferring disease may be concurrently fixed, resulting in multiple disease models within a single inbred strain, although some of these traits may remain unidentified. Because of this, there is a need to better characterize strains, both at the phenotypic and at the genomic levels, that is, generate a rat phenome resource. Major efforts are focusing on generating a rat phenome; Mashimo et al. (2005), from the National Bio Resource Project for the Rat (NBRP), have characterized 109 traits in 54 inbred rats, while PhysGen (http://pga.mcw.edu) has characterized 11 different strains (nine inbred and two outbred) for >280 different traits, and have generated and characterized two chromosome substitution panels (44 strains derived from the sequenced BN and the FHH and SS hypertensive strains) (Jacob and Kwitek 2002). An important aspect of these types of studies is that all experiments are performed using the same methodology.
To complement the detailed phenotypes generated by these efforts, alleles of 48 common inbred strains have been determined for 4328 SSLPs spanning the rat genome, as part of the U.S. Rat Genome Project (Steen et al. 1999). Furthermore, the NBRP has determined 357 SSLP genotypes in 98 strains, including the 54 strains from their Rat Phenome Project (Mashimo et al. 2005). These data allow the construction of haplotypes across all major rat strains using publicly available tools such as the ACP Haplotyper (http://rgd.mcw.edu/ACPHAPLOTYPER/) in order to identify common haplotypes within models with similar diseases. From these data, one can determine the “evolutionary” relatedness of the various inbred strains of rats (Thomas et al. 2003). These allele data are now being greatly supplemented by the addition of >45,000 single nucleotide polymorphisms (SNPs) identified across multiple rat strains (http://www.ncbi.nlm.nih.gov/SNP/snp_summary.cgi; Zimdahl et al. 2004; Guryev et al. 2005). These numbers will grow rapidly with two major SNP discovery projects underway in Europe (the Functional Genomics Group in the Netherlands and the Max-Delbruck-Center for Molecular Medicine in Germany) and a recent NHGRI White Paper to identify SNPs in an additional eight strains. Integration of the detailed phenotype information with haplotypes will provide an incredibly powerful tool for complex disease gene discovery.
Genetic mapping and positional cloning
QTL mapping
Quantitative trait locus (QTL) mapping is a proven useful resource to assign the biology of the rat onto the genomic sequence by identifying chromosomal regions that contain genes affecting complex phenotypes. While a QTL is a rather large genetic locus, the genes within this interval are responsible for a component of the trait variation, enabling the genome to be annotated with physiology. Importantly, most rat models reflect a clinical phenotype, and several comparative mapping studies have determined that common phenotypes often map to conserved genomic regions between rat and human (outlined in detail below). The ultimate goal of QTL mapping is to identify the genes, by positional cloning, that underlie complex phenotypes and diseases and to gain a better understanding of their physiology and pathophysiology.
To date, there have been 536 QTL papers published with >1000 QTLs reported for different physiological and pathophysiological traits. These papers include investigations of the genetic basis of blood pressure (Rapp 2000), diabetes (Jacob et al. 1992; Galli et al. 1996; Pravenec et al. 1996), cardiovascular disease (Stoll et al. 2001; Moreno et al. 2003), stroke (Rubattu et al. 1996), ethanol preference (Murphy et al. 2002), behavioral conditioning and anxiety (Fernandez-Teruel et al. 2002; Flint 2003), fat accumulation (Tanomura et al. 2002), arthritis (Olofsson et al. 2003a), copper metabolism (de Wolf et al. 2002), pituitary tumor growth (Wendell and Gorski 1997), aerobic capacity (Ways et al. 2002), and chemical carcinogenesis (De Miglio et al. 2002). Most of the QTLs have been mapped in the last three years, mainly because of advances in technologies that allow for high-throughput genotyping and an accelerated development of genetically modified strains. Table 2 shows a classification by phenotype category of the QTLs found in rat to date.
QTLs mapped in rat
QTL mapping is often followed by confirmation of the loci by the development of congenic lines, in order to evaluate the QTL in the absence of other mapped QTLs, and as a step in positional cloning (Flint et al. 2005). To date, 118 QTLs mapped in rat have been confirmed by congenic lines, many of which have narrowed the critical genomic interval to a handful of candidate genes. More than 50% of these congenics (59 strains) were developed for studying blood pressure control, followed by congenics for non-insulin-dependent diabetes mellitus (29 strains). From the 118 congenic lines developed following QTL mapping, 61 congenic lines have been published only since 2002. Following this trend, it is expected that we will see acceleration in the rate of gene discovery in the rat, reflecting the availability of the rat sequence, the accessibility to high-throughput sequencing for the search of sequence variants, as well as microarray technologies for gene identification, pathway analysis, and mapping of cis and trans regulatory elements. These resources will greatly facilitate the identification of genes underlying the hundreds of QTLs mapped for complex diseases and phenotypes.
Genes positionally cloned
Traditional positional cloning efforts in the rat have been coming to fruition in identifying disease genes—many over the past two years. Numerous genes have now been identified in the rat by positional cloning, concurrent with the great increase in rat genomic resources. These include genes for cancer (Flcn, Tsc2) (Yeung et al. 1994; Okimoto et al. 2004), type 1 diabetes (Gimap5, Cblb) (MacMurray et al. 2002; Yokoi et al. 2002), type 2 diabetes (Cd36) (Aitman et al. 1999), neurological disorders (Cct4, Reln, Unc5h3) (Lee et al. 2003; Yokoi et al. 2003; Kuramoto et al. 2004), arthritis (Ncf1) (Olofsson et al. 2003b), renal disease (Pkhd1, Rab38) (Ward et al. 2002), bleeding disorders (Rab38, Vkorc1) (Oiso et al. 2004; Rost et al. 2004), retinal degeneration (Mertk) (Gal et al. 2000), and hypotrichosis (Dsg4, Foxn1) (Segre et al. 1995; Jahoda et al. 2004). Many of these genes were cloned from spontaneous mutants with Mendelian inheritance of disease, for example, the Pkdh1 mutation in the PCK rat causes autosomal recessive polycystic kidney disease (ARPKD). However, the number of identified genes involved in complex traits is on the rise.
One of the most challenging tasks in genomics is the prediction of gene function, and the study of the interactions between genes in the genome—what is called functional genomics. DNA microarray studies provide the potential to greatly enhance our knowledge of the genes and pathways involved in the physiological responses to physiological stressors, drugs, and environmental stimuli, and in pathogenesis of diseases. Microarray studies in rat have been used in conjunction with other genetic strategies, like QTL analysis, congenic mapping, or transgenic techniques to accelerate the search for genes underlying various phenotypes (Aitman et al. 1999; Monti et al. 2001; Liang et al. 2003; Vitt et al. 2004). The cloning of the Cd36 gene is one of the first examples of cloning a complex trait gene in the rat using a combined approach of introgressing a QTL to generate congenic strains and profiling their expression patterns compared to the parental strain. A more recent study looked at gene expression in a panel of BXH/HXB recombinant inbred (RI) rat strains (Hubner et al. 2005) to identify eQTLs (expression QTLs) in the rat genome. eQTLs that overlap with previously identified QTLs for metabolic syndrome have provided nearly 76 candidate genes to be evaluated.
One application of using animal models for the purpose of gene identification of disease is the translation of that gene to the human clinical setting, although some are skeptical that disease genes translate well between humans and animals. For instance, mutations in the leptin/leptin receptor gene cause dramatic obesity in rodents but do not contribute to the common form of human obesity but, rather, to rare Mendelian forms (Montague et al. 1997; Clement et al. 1998; Strobel et al. 1998). However, the entire field of obesity research was opened up with the cloning of the Lep and Lepr genes. This scenario is not unlike the cloning of rare monogenic forms of hypertension and hypotension in humans (Lifton et al. 2001). Although the specific genes may not significantly contribute to essential hypertension, they nevertheless generate understanding of the pathways related to sodium handling in the kidney, which certainly play a role in the mechanisms leading to the more common form of the disease. The strength that the rat provides is a platform on which to study these mechanisms and pathways in much more detail.
Transgenetics and translating
Transgenic rats
For more than 15 years, genetics studies have followed two tracks: positional cloning from genetic mapping to gene identification, and transgenesis (random insertion of gene, knockouts, knock-ins, and conditional knockouts) to unravel gene function. Traditional rat transgenesis by pronuclear injection has been established since 1990 (Hammer et al. 1990; Mullins et al. 1990). However, because the rat lacks viable ES cell lines, knockout and knock-in technology is unavailable, somewhat limiting the adoption of the rat for gene-manipulation studies. Nonetheless, there have been >200 transgenic rats generated.
As in the mouse, the major purpose of generating rats via transgenesis was to study a particular gene of interest. It is now relatively straightforward to alter the expression of specific rat genes as well as to use rats as surrogate hosts for expression of genes from other species. Many transgenic rat strains have been “humanized,” by using a gene from human, providing a bridge between genetic linkage studies in humans, and functional association of a (mutant) gene with particular pathological features. For example, humanized rats were used to dissect complex diseases such as heart hypertrophy (Tian et al. 2004), end-organ damage (Hocher et al. 1996), and hypertension (for review, see Pinto-Sietsma and Paul 1997; Liefeldt et al. 1999; Bohlender et al. 2000). These examples provide a proof of principle that human disease modeling in rats is valuable, and one can expect that the etiology of other diseases will be similarly illuminated through sequence knowledge and transgenesis. Furthermore, the availability of transgenic rats expressing human genes makes it possible to follow up disease progression in longitudinal in vivo studies, to monitor the effects of long-term treatments, cell implantation, or antisense approaches on the course of disease. Finally, the need to validate a gene cloned by position via transgenic rescue will further increase the use of transgenic technologies in two ways, functional cloning and target validation.
Functional cloning
The availability of large insert clones enables their use for transgenesis in rats, with the advantage of screening multiple genes with their cis-regulatory elements intact. The rat is fortunate to have several different types of large insert clones from a variety of strains, including one PAC and two BAC libraries from BN (two substrains), and BAC libraries from SS, FHH, and F344 strains. Functional cloning was initially coined by Eddy Rubin, who used a human YAC to determine that there was a functional element within the human YAC that influenced asthma (Symula et al. 1999). The ability to modify the phenotype of an animal with a large insert clone, in advance of knowing the causal gene, demonstrates that a gene or element within that clone is capable of modifying function. Furthermore, it is becoming increasingly evident that not only are multiple QTLs responsible for complex disease, but also that what was thought to be a single QTL may actually contain multiple linked QTLs playing a role in a mapped phenotype. In this situation, a single gene transgenic or a knockout would be insufficient to affect a phenotype. If this is the case, then functional cloning may provide an alternative strategy to help identify the causal genes.
Target validation
Once a gene has been positionally cloned or implicated to be causal, there is a need to prove the causality, a phase of gene discovery termed target validation. The gold standard for proving that a gene is causal is to knock in the particular allele or mutation responsible for the trait, or replace the defect with a “normal” allele to demonstrate that this specific substitution changes the phenotype. While there can be no doubt that conducting this type of experiment can provide conclusive proof, it is an onerous and expensive process that is not likely feasible for all 30,000+ genes. The release of the rat genome sequence has facilitated an alternative approach to target validation—transgenic rescue— whereby the phenotype is normalized via a transgene, particularly when the trait shows a recessive mode of inheritance (Pravenec et al. 2001; Jacob and Kwitek 2002). Recently, cloning of fertile adult rats has been achieved by nuclear transfer (Zhou et al. 2003), which opens the door for targeted gene manipulations techniques such as knockout technology. However, it will be some time before this can be done routinely as the efficiency is too low to be used as a general methodology.
Comparative mapping
The primary motivation for the rat genome project was to leverage the deep biological history to annotate the human sequence with common complex diseases (Jacob and Kwitek 2002), with the goal of understanding the pathogenesis of human disease. Consequently, most rat research is ultimately translational, aimed at improving human health through the understanding of key genetic and physiological factors in common disease pathways. Using the homologous regions between genomes (Brudno et al. 2004; Gibbs et al. 2004; Wilder et al. 2004) to map disease-causing genes or regions from one organism to another has begun to bear fruit in humans, a result of the investments into the rat genome project.
The availability of the rat genomic sequence along with the genomic sequence of many other species introduces the possibility of comparative genome analysis at a nucleotide level rather than the ordering of large blocks of genes used previously. The utility of comparative analysis is based on the hypothesis that functionally important sequences will be conserved across species. In 2000, Stoll et al. (2000) reported that QTLs in the rat could be used to predict the locations at which human QTLs were likely to exist. Since then, numerous other studies have demonstrated that there are evolutionarily conserved regions in human, mouse, and rat that are linked to the same phenotype in all three species (Stoll et al. 2000; Sugiyama et al. 2001; Jacob and Kwitek 2002; Korstanje and DiPetrillo 2004). Over the next five years, we can expect a large increase in the number of studies using cross-species comparisons to find causes of common complex diseases (Glazier et al. 2002; Korstanje and Paigen 2002). As an example, Peter Harris's group at the Mayo Clinic had sought the gene responsible for autosomal recessive polycystic kidney disease (ARPKD) in humans. Comparative genomics showed ARPKD mapped to the conserved region of the PKC rat and human, and subsequent studies showed the same gene to be the cause of PKD in both species (Ward et al. 2002). Another example of a successful translation of a gene between rat and human includes CD36 (Aitman et al. 1999; Febbraio et al. 2002) in non-insulin-dependent diabetes mellitus. Approximately 100 papers report that a particular disease trait maps to the same conserved region in rat and human, illustrating the near-term benefits of the rat genome project. However, one must keep in mind that QTLs are large and numerous for multifactorial traits, such as behavior and metabolic syndrome. Therefore, overlapping QTLs may sometimes be merely a chance event. To address this issue, the rat can be extensively evaluated for disease subphenotypes to better match QTLs by intermediate phenotype. Furthermore, with the promise of a rat SNP map, finer-resolution mapping may reduce the size of a QTL. Finally, comparative mapping data from additional species such as dog, cow, or other models might be used to confirm conserved QTL. Integration of the genome sequence with existing mapping data and the biological data attached to those maps, plus the creation and annotation of a comprehensive catalog of gene products, will increase the use of such comparative studies and the impact the rat has on translational research.
Gene therapy
The rat has already served as a useful model for gene transfer experiments of optic nerve disease (for review, see Martin and Quigley 2004), Parkinson's disease (Klein et al. 2002; Lo Bianco et al. 2002; Yamada et al. 2005; Zheng et al. 2005), treatment of cerebral ischemia (Tsai et al. 2002), gene transfer into rat heart (Most et al. 2004; Schroder et al. 2004; O'Donnell and Lewandowski 2005; Ross et al. 2005; Schmidt et al. 2005), erectile dysfunction in diabetic rats (Bennett et al. 2005), and testing naked DNA gene transfer and therapy (Herweijer and Wolff 2003). Moreover, the application of RNA interference to efficiently and specifically knock down expression of mammalian gene products has opened a novel avenue for experimental and therapeutic applications, designed to reduce the levels of an undesirable protein (Shi 2003). A second RNA technology, mRNA reprogramming by trans-splicing, offers the ability to repair mRNAs, and thus proteins (Mansfield et al. 2000). In the future, we expect the rat to play a more active role in improving the relatively poor results in human clinical trials to date.
The “new” genetic models
Various approaches to generating novel animal models for complex disease provide an additional way to map phenotypes to the genome, and facilitate the process of gene identification, by narrowing the region where linkage to a phenotype resides and by fixing the effect of a gene in a homogeneous genetic background. In the sections below, we describe several means to generate new or “designer” rat models to follow up genetic linkage or QTL studies.
Congenic strains
In order to validate the functional importance of a genomic region, initially identified by genetic linkage analysis, congenic techniques were originally developed to study the MHC in the mouse by Nobel Prize winner G. Snell (Snell 1948) at the Jackson Laboratory. This strategy remains a common way to study genes nearly 60 years later. Since congenic strains differ only in a short chromosomal segment from their background strain, it is possible to investigate the phenotypic effect of the locus, isolated from other effects caused by other loci on the original genetic background (Fig. 1). The development of congenic strains can be accelerated by genotyping the whole genome and selecting the breeders that, besides containing the target region of the donor strain, have a greater proportion of alleles from the recipient strain throughout the genome. This process of whole-genome marker-assisted selection, also called “speed congenics,” can reduce the breeding time by half (Visscher 1999). With this method, generation of a congenic strain is reduced from 4–5 yr to 2–3 yr. In complex diseases, with multiple QTLs determining a trait, generation of double or triple congenic strains is sometimes necessary in order to confirm a causative locus. These multiple congenics are typically constructed one at a time and then assembled onto multicongenics.
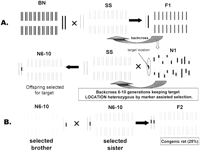
Schematic representation of the generation of a congenic strain from two genetically different rat strains. (A) Parental strains Brown Norway (BN) and Dahl salt-sensitive (SS) are intercrossed for the generation of a heterozygous F1 population. The F1 is then crossed with the parental background of interest (in this example, the SS) to generate an N2 population. The N2 rats are then backcrossed six to 10 generations using marker-assisted selection of the offspring, in order to substitute a selected genomic region from the BN rat. (B) A male and female rat, selected by genotyping for this specific target region containing the phenotype of interest, are then mated. Twenty-five percent of the offspring from this cross will be homozygous for this region. These rats are then inbred to produce a stable inbred congenic strain. Reprinted with permission from Blackwell Publishing © 2004, from Cowley Jr. et al. (2004).
Consomic strains
Consomic strains are rat strains in which a whole chromosome from one strain is transferred to the genomic background of another strain, by a methodology similar to that of congenic generation. The Medical College of Wisconsin has assembled two complete panels of consomic strains, using the BN strain that was sequenced as the donor strain. In these consomic strains, a chromosome from the BN rat was substituted, one at a time, into the genetic background of the SS/JrHsdMcwi (Dahl salt-sensitive; SS) or FHH/EurMcwi (Fawn Hooded Hypertensive; FHH) rats. The SS rat is a model for salt-sensitive hypertension (Rapp 1982), insulin resistance (Kotchen et al. 1991), hyperlipidemia (Reaven et al. 1991), endothelial dysfunction (Luscher et al. 1987), cardiac hypertrophy (Ganguli et al. 1979), and glomerulosclerosis (Roman and Kaldunski 1991). The FHH rat is a model for systolic hypertension, renal disease, pulmonary hypertension, a bleeding disorder, alcoholism, and depression (Provoost 1994). The two consomic panels capture nearly 50% of the genetic variation in the rat (Steen et al. 1999), and provide a foundation of genetic resources for the study of disorders of heart, kidney, lung, and vasculature. Given the 50% genetic variability, it is reasonable to assume that a similar level of biological variability can be expected, making the consomic strains a powerful tool for mapping additional complex traits.
One major advantage of using consomic strains is to rapidly generate congenic lines. Generation of congenic rats from consomic rat strains takes at most three generations of breeding, following an intercross with the consomic and parental strain (Fig. 2). There are additional applications for consomic rat strains; for example, they can be used to assess the role of a genomic region in different backgrounds, assessing whether background effects modify gene function. Consomic rat strains can also be used to develop polygenic models to study gene–gene interactions. The contribution of genes on each chromosome to the observed traits cannot only be assessed both by phenotyping but also by expression profiling. Comparisons between the consomic and parental strains provide valuable insights into the genomic pathways (clustered gene expression patterns) that differ between strains and how these differences might be connected to a particular pathogenic phenotype of the animal.
Recombinant inbred strains
Recombinant inbred (RI) strains provide an additional tool for mapping phenotypes to the genome. This strategy is based on the generation of a panel of inbred strains derived from an F2 population (Pravenec et al. 1996). The RI strategy generates lines that contain more than one QTL, permitting the analysis of gene interaction and detection of weak loci. However, the presence of a unique genome background in each strain prevents their use in rapid generation of congenic animals.
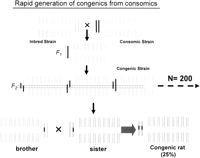
Generation of congenic rats from consomic strains. The parental strain is crossed with the consomic strain, to generate an F1 population with identical genetic background and a heterozygous target chromosome. These F1 rats are intercrossed to generate an F2 population of rats whose target chromosome will be congenic, because of recombination events. Two similar F2 rats are selected (by genotype) and mated to fix the region of interest. Reprinted with permission from Blackwell Publishing © 2004, from Cowley Jr. et al. (2004).
One of the largest rodent recombinant inbred panels is the reciprocal HXB/BXH recombinant inbred strains, derived form the Spontaneously Hypertensive (SHR) and the BN rat strains (Pravenec et al. 1989, 1999). These strains are a great resource for genetic analysis of cardiovascular and metabolic phenotypes. The use of this RI strain panel has facilitated the mapping of several traits, including blood pressure (Pravenec et al. 1995), reproductive traits (Zidek et al. 1999), metabolic traits (Pravenec et al. 2002), behavior (Conti et al. 2004), and susceptibility to cancer (Bila and Kren 1996). Other RIs have also been developed, such as the LExF (Shisa et al. 1997) and the SWXJ (Svenson et al. 1995) panels, although phenotypic characterization of this panel not been as extensive as that for the HXB/BXH.
ENU mutagenesis
For many years, the genetics community relied on spontaneous mutations as a source of rat models, which is a major limiting factor in the use of rat for structure–function studies, particularly for disease gene validation. An alternative approach includes inducing mutation through the use of chemical mutagens such as N-ethyl-N-nitrosourea (ENU). While ENU mutagenesis has been used for many years, the last 10 years has seen an increase in its use to generate gene mutations (Guenet 2004). The typical ENU strategy is to treat males with ENU, inducing mutations in the spermatogonial stem cells (predominantly loss-of-function mutations), breed them to untreated females, and screen the offspring for phenotypic effects. As the ENU approach has advanced, phenotyping is now emerging as the rate-limiting step in such large-scale studies. These studies produce large numbers of animals having mutations with phenotypic effects. However, the vast majority of these mutants are never identified because sufficient phenotypic screening is not applied. There has been an increasing emphasis on large-scale ENU screens for systematic and comprehensive gene function analyses of the mouse genome (Hrabe de Angelis et al. 2000; Nadeau 2000; Nolan et al. 2000; Brown and Balling 2001). The mutagenesis programs are phenotype-driven, using appropriate screens on mutagenized animals to identify novel mutant phenotypes, particularly those that model human disease, followed by mapping and isolation of the underlying causal genes. However, large-scale phenotypic screens are very expensive and consume a tremendous amount of animal per diem charges. As such, there has not been a similar attempt at this type of screen using rats.
In 2003, two groups published on the use of ENU in combination with a gene screen to generate rat gene knockouts. The major difference in this strategy is that the genes of interest are screened to determine which animals have a mutation in a gene of interest (Zan et al. 2003; Smits et al. 2004). Only the animals that have a mutation are kept. In this way, per diem costs are minimized, although the gene screening also involves a cost. This strategy has been used to knock out several genes in the rat (Zan et al. 2003; Homberg et al. 2005). The strategy has one other major advantage; the ENU mutagenesis can be done in the strain of choice (following the determination of the appropriate ENU dosing), preventing problems associated with the genome background effects that are relatively common when knockouts are generated on a limited number of ES cell lines. We expect this methodology to gain in popularity until such time as there are ES cells for rats, and remain an alternative approach when genome backgrounds affect a phenotype.
Heterogeneous stocks
Heterogeneous stocks (HS) are derived from crossing eight inbred strains followed by continuous outbreeding for several generations (Hansen and Spuhler 1984). While this strategy was developed before the human and rat genome projects, the resources of the rat genome project make this collection of rat strains extremely powerful and another alternative for investigators using rats in their research programs.
The chromosomes of the HS progeny represent a random mosaic of the founding animals with an average distance between recombination events close to a single centimorgan. This high degree of recombination enables the fine-mapping of QTLs into sub-centimorgan intervals and the identification of multiple QTLs within what was previously identified as a single QTL (e.g., Ariyarajah et al. 2004; Stylianou et al. 2004). The HS rat colony was derived by the NIH in 1984 for alcohol studies (Pandey et al. 2002). Fine-mapping of QTL to sub-centimorgan intervals has been successful for traits of anxiety (Mott et al. 2000), ethanol-induced locomotor activity (Demarest et al. 2001), and conditioned fear (Mott et al. 2000) in HS mice. Studies are currently underway to detect QTLs for multiple traits including behavior and diabetic traits using HS rats.
Embryonic stem (ES) cells in rats
The development of ES cells is the final tool needed for the rat genomic and genetic toolbox. To some investigators this remains the holy grail of genetics and functional genomics. While having ES cells in the rat would facilitate some specific research questions, it is not an essential requirement, as evidenced by the continued use of the rat in the absence of this technology. Nonetheless, having the technology would remove the temptation to switch model systems based on the technology rather than the biology. At this point it is difficult to know when there will be ES cell lines for the rat. Over the last decade, numerous groups have attempted to develop ES cells, but thus far to no avail. However, given the current level of interest, it is likely that there will be ES cells developed over the next decade. Whether they are true ES cells or some other extrapolation from advances in stem cell research remains to be seen. Another potential solution to generating knockouts may come from conditionally expressed siRNA probes or similar inhibitors. In the interim, the rat will continue to be effectively deployed using available technologies and alternative study designs to the need for ES cells.
Genome databases
The rapid increase in rat genetic and genomic data facilitated the need for a centralized database to efficiently and effectively collect, manage, and distribute a rat-centric view of this data to researchers around the world (Table 1). The Rat Genome Database (http://rgd.mcw.edu, RGD) works in collaboration with the Mouse Genome Database (MGI), NCBI, UCSC, EBI, RGSC, Baylor College of Medicine, SWISS-PROT, BIND, RatMap, and the PhysGen PGA (de la Cruz et al. 2005). RGD coordinates all nomenclature for genes, strains and QTLs in collaboration with Ratmap (http://ratmap.gen.gu.se), a European database aimed to manage rat related data, and the International Rat Genome and Nomenclature Committee (http://rgnc.gen.gu.se). RGD curates and integrates rat genetic and genomic data and provides its free access to support research using the rat as a genetic model for the study of human disease. It was created to serve as a repository of rat genetic and genomic data, as well as mapping, strain, and physiological information. It also facilitates investigators' research efforts by providing tools to search, mine, and analyze these data sets. Curated data in RGD include rat genes, QTLs, microsatellite markers and expressed sequences (ESTs), rat strains, genetic and radiation hybrid maps, sequence information, and ontologies. Ontologies provide standardized vocabularies for annotating molecular function, biological processes, cellular components, phenotypes, and disease associations. These ontologies allow for searching across genes, QTLs, and strains, and provide a basis for cross-species comparisons. RGD also contains a variety of tools to search and analyze both the data incorporated into RGD and data generated in an investigator's own laboratory. The list below identifies the major tools provided at RGD.
Genome Browser
The Genome Browser (http://rgd.mcw.edu/sequenceresources/gbrowse.shtml) allows rapid visualization of different types of information beneath genome coordinate positions. Information includes genes, genetic markers, ESTs, and mapped QTLs for different phenotypes. Other genome browsers, with numerous tracks and links, are listed in Table 1.
Genome Viewer (GViewer)Tool
GViewer Tool (http://www.rgd.mcw.edu/gviewer/Gviewer.jsp) provides users with a complete genome view of genes and QTLs annotated with function, biological process, cellular component, phenotype, disease, or pathway information. GViewer will search for terms from the Gene Ontology, Mammalian Phenotype Ontology, Disease Ontology, or Pathway Ontology.
Virtual Comparative Map (VCMap)
VCMap (http://rgd.mcw.edu/VCMAP/) is a dynamic sequence-based homology tool that allows researchers of rat, mouse, and human to view mapped genes and sequences and their chromosomal locations in the other two organisms. The comparative maps are based either on radiation hybrid maps or genome sequence-based conservation between species, and allow sequence-related information derived from one species to be mapped to the conserved region in another. The maps are clickable, allowing the view of information on markers, chromosomal regions, and mapped QTLs.
ACP Haplotyper
ACP Haplotyper (http://rgd.mcw.edu/ACPHAPLOTYPER/) allows visualization of chromosomal segments that are conserved between 48 different inbred strains and provides a measurement of relatedness between these inbred strains. This tool creates a visual haplotype that can be used to identify conserved and nonconserved chromosomal regions between any of the 48 rat strains characterized as part of the Allele Characterization Project. For the selected chromosome, the tool compares the allele size data between selected strains, for microsatellite markers on genetic, RH, or sequence maps.
Genome Scanner
Genome Scanner (http://rgd.mcw.edu/GENOMESCANNER/) combines the strain allele data with the position of the genetic marker on the genetic and radiation hybrid maps. By selecting the strain of interests, Genome Scanner will select polymorphic markers between the selected rat strains, which can be further used to perform genetic linkage studies or to increase map density in a specific chromosomal region.
Gene Annotation Tool
The purpose of the Gene Annotation Tool (http://rgd.mcw.edu/gatool/) is to gather information about genes by parsing several databases available online (Entrez, SWISS-PROT, KEGG, and RGD) to provide the user with a comprehensive file of gene descriptions and annotations.
In addition to these tools, there is a wealth of genomic information and bioinformatics tools for the rat, freely available on the Web (Table 1).
Conclusions
Throughout this paper, we have discussed the deployment of genetics and genomics in the rat. We have demonstrated the increase in use of these resources within the various sections. However, we believe the real numbers are much larger, as there is always a lag in science between adoption of new strategies and the publication of the results. We believe there will be expansive growth in the use of the rat over the next five years for the following reasons. First, the ability to clone genes by position is poised to explode, based on the availability of the genomic sequence and the large number of genetic mapping studies and related congenic strains already completed. The identity of these genes will drive interest in finding other genes for other diseases, and the wealth of rat models will continue to attract other investigators. Second, comparative genomics continues to show that the QTLs in rats match the evolutionarily conserved regions where the QTLs map in human. This means that the genes found in the rat have increased likelihood to contribute to the disease process in humans. With the rat's genomic sequencing being ∼90% identical to that of the human at the gene level, we can expect the rat to continue to add value to the annotation of function onto the human genome. There will, of course, be instances where the results in the rat are unique to the rat. In these instances, the results are likely to be important to the evolutionary biologists. Third, where the disease gene is shown to play a role in both rat and human, we can predict that mechanism-based studies and the development of new therapeutics will quickly follow suit. Fourth, with respect to the mechanistic-based studies, the genetic models, notably the congenics, consomics, and ENU mutagenesis strains, offer the first real controls for mechanistic-based studies of common, complex diseases. The reason these models are advantageous is quite simple—the genome background of the disease strain and control strain are the same; the only issue for these models is how different are they, phenotypically, from the control. For example, in the case of a consomic, the maximum difference between the two strains studied would be the size of the largest chromosome, the minimum the size of the smallest chromosome. Therefore, the genetic relatedness of the consomic models to their respective control strain would range from ∼90% to ∼98% identical. For congenics and ENU mutagenesis on an inbred genome background, the identity will be even higher between the “case” and control strains. The ability to provided genome background control strains, rather then simply comparing two or more different strains or the use of outbred strains, will be a revolution. Fifth, once found, genes involved in complex traits will then be studied in a variety of model systems, for example, (1) transgenic rescue using genes (Pravenec et al. 2003) and large insert clones (Michalkiewicz et al. 2004); (2) mutagenesis using a combination of ENU and a gene-specific screening assay now provides the ability to create rat gene knockouts (KOs) for use in functional studies (Zan et al. 2003; Smits et al. 2004). H.J. Jacob and A.E. Kwitek and colleagues at the Medical College of Wisconsin (MCW) have been funded to generate KO rats to complement the consomic panels; M.N. Gould was funded to make an additional 20 KO rats; Ingenium Pharmaceuticals (http://www.ingenium-ag.com) has an ENU program for rats. (3) An alternative strategy involves knocking the gene out in cell culture and then cloning the rat (Zhou et al. 2003). While not yet efficient enough for mainstream research it offers promise. (4) Continued efforts are under way to develop ES cells for rats within both the academic and industry settings. (5) The availability of RNAi probes for the rat offers another means to alter phenotype using transgenics. Sixth, the sequenced strain (BN) has been extensively studied, physiologically, and compared to 10 commonly used strains of rats; furthermore, it has been used as the donor strain to produce 44 chromosome substitution strains (consomics). These consomics capture an estimated 50% of known genetic variance in the rat and offer the ability to genetically map new traits and to generate congenics at an unparalleled pace (http://pga.mcw.edu). Seventh, there is a single nucleotide polymorphism (SNP) project underway in the European Union, and another similar project in the United States. This SNP project will generate 100,000s of SNPS and facilitate development of haplotype maps. For the U.S. project, a White Paper has been accepted by the NHGRI to generate SNPs from eight strains of rats. The strains were selected to capture the maximal degree of known genetic variation (Thomas et al. 2003) and significant numbers of QTLs. Table 3 shows how many QTLs were identified in crosses involving the most commonly used strains (and their derivatives). Clearly, the five most commonly used strains account for the majority of the QTLs. Finally, in the last five years, the rat community has grown from a handful of investigators with a biannual meeting for ∼50 people to being between one-third and one-half the size of the mouse community in terms of data generation and grants, and holding a meeting annually with ∼150 participants. The science and interest have grown to such a level that Cold Spring Harbor Laboratory hosts the rat meeting every other year.
QTLs per strain for strains included in SNP identification
In conclusion, the sequencing of the rat has resulted in an explosion of research using the rat. Given that the rat is a model known and studied for its biology, it is likely that the biology of the rat will now greatly facilitate the knowledge potential of the human genome project. Indeed, the rapid response of the community of investigators to the rat genomic tools and the important results generated to date using these resources are impressive. Perhaps it is time to consider a sequencing project of up to 10 more strains at much lower density coverage and the construction of a high-density SNP haplotype chip. It would markedly accelerate the discovery of genes in these critical rat models, thereby accelerating our understanding of disease in humans.
Footnotes
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3744005.
-
↵1 Corresponding author. E-mail AKwitek{at}MCW.edu; fax (414) 456-6516.
- Cold Spring Harbor Laboratory Press








