
Many Paths to Many Clones: A Comparative Look at High-Throughput Cloning Methods
Abstract
The creation of genome-scale clone resources is a difficult and costly process, making it essential to maximize the efficiency of each step of clone creation. In this review, we compare the available commercial and open-source recombinational cloning methods with regard to their use in creating comprehensive open reading frame (ORF) clone collections with an emphasis on the properties requisite to use in a high-throughput setting. The most efficient strategy to the creation of ORF clone resources is to build a master clone collection that serves as a quality validated source for producing collections of expression clones. We examine the methods for recombinational cloning available for both the creation of master clones and their conversion into expression clones. Alternative approaches to creating clones involving mixing of cloning methods, including gap-repair cloning, are also explored.
Functional genomics and proteomics offer the promise of examining the roles of all genes and proteins in an organism in a controlled format. These studies depend on the availability of cloned copies of the genes in a format conducive to protein expression. Historically, this need has been fulfilled by pooled cDNA clone libraries. Despite numerous improvements in library construction methods, however, it has not been possible to construct cDNA libraries that have comprehensive representation of the expressed genes for any organism, particularly higher eukaryotes. A secondary issue is that it is frequently necessary to fish individual clones of interest out of the library in order to permit their use in experiments. Accordingly, researchers around the world have recently exploited the availability of completed genome and cDNA sequences to create comprehensive, arrayed collections of individual cloned genes for a number of organisms.
The earliest comprehensive open reading frame (ORF) clone collections were constructed with gap-repair cloning, a method of cloning that uses homologous recombination in the yeast Saccharomyces cerevisiae (Martzen et al. 1999; Uetz et al. 2000; Ito et al. 2001; Zhu et al. 2001). Gap repair is a simple and inexpensive method of cloning, in which the desired ORF is amplified by use of ORF-specific primers with flanking sequences that are homologous to the ends of a linearized yeast-cloning vector. Yeast transformed by a mixture of the PCR product and the linearized vector will recombine the homologous ends to incorporate the ORF into the vector in vivo. Aside from PCR amplification of the ORF DNA, no in vitro manipulations are required for the cloning of the ORF DNA. Methods for construction of clones via other homologous recombination systems, such as the Escherichia coli-based cloning methods that utilize λ Red/recET-mediated recombination, have been described (Datsenko and Wanner 2000; Zhang et al. 2000; Poteete 2001; Court et al. 2003), but have not been utilized for cloning on a large scale.
There are several practical limitations to gap-repair cloning. The long sequence tails required for homologous recombination (∼50 bp) require long PCR primers that are both expensive and prone to synthesis errors. In addition, there is a high false (empty) clone rate (Ito et al. 2001; Zhu et al. 2001). Because of this, it is essential for clones constructed in this manner to be sequence validated. Most importantly, once constructed, the clones are effectively locked into the configuration of the original vector. Moving the ORFs to a different vector would require starting again at the PCR step, with its inherent incorporation errors as well as the relatively high failure rate of recombination. Thus, it would be necessary to sequence validate all newly created expression clones in each subsequent expression collection.
Recently, site-specific recombination-based cloning has emerged as an alternative and more general method of constructing large ORF clone collections (Walhout et al. 2000; Reboul et al. 2001, 2003; Brizuela et al. 2002; Yamada et al. 2003). Although use of these methods can sometimes result in greater expense when assembling collections of clones (due in part to the high cost of the recombinase used in some cloning systems), the site-specific recombination cloning systems are ultimately less costly for transferring ORFs to many different expression vectors. Recombinational cloning systems utilize an approach in which archival master clones (as E. coli plasmids) are created that, once sequence validated, can be used directly to create a broad variety of cognate expression clones. Like restriction enzyme-based sub-cloning, the site-specific recombination reactions are effectively “cut and paste” (albeit in a concerted reaction), and the transferred DNA is perfectly conserved. As such, sequence validation of the resulting expression constructs created in this way is unnecessary.
Highly efficient site-specific recombination-based systems are available from commercial suppliers, such as the Gateway cloning system from Invitrogen and the Creator cloning system from Clontech, as well as the Univector cloning system developed by the lab of Stephen Elledge (Baylor College of Medicine; Liu et al. 1998, 2000), which make the use of recombinational cloning in a high-throughput setting practical.
Why ORF Clones?
Historically, expression clone collections have relied on cDNA libraries cloned as a pool into specific vectors. To generate such a library, mRNA is collected from an appropriate cell source, converted to DNA molecules with reverse transcriptase, and then introduced en masse into a protein-expression vector. Expression libraries have found considerable success in the study of prokaryotes and simple eukaryotes, but several issues have limited their application for genetic studies in more complex species, most notably mammalian cells. The presence of the 5′ and 3′ untranslated regions (UTRs) does not allow the efficient attachment of polypeptide tags to either end of the proteins (as is often required in high-throughput applications). In addition, the translational reading frame of the tag with respect to the coding sequence is not known, and the UTR sequences themselves may contain in-frame stop codons. Thus, it is better if the UTRs are omitted during the cloning process, so that fusion sequences can directly abut the gene-coding sequences.
Creating clones that contain only coding sequences, often referred to as ORF clones, requires the PCR amplification of the coding sequence with ORF-specific primers, which must be generated with information from an annotated sequence resource such as RefSeq (Maglott et al. 2000; Pruitt et al. 2000). Typically, four types of starting material may be used as template to generate clones as follows: (1) genomic DNA; (2) first-strand cDNA; (3) cDNA library; or (4) pre-existing sequence validated full-length cDNA clones (such as those available from the Mammalian Gene Collection [MGC; Strausberg et al. 1999, 2002] or RIKEN [Okazaki et al. 2002]). Genomic DNA is the easiest of the templates, because all genes are equally represented, but it is only applicable to organisms with little or no splicing, limiting it to prokaryotes and simple eukaryotes. First-strand cDNA and cDNA libraries are useful starting material for mammalian collections, particularly if the genes of interest are not already available as clones, but they suffer from the limitation that the representation of genes in the library will reflect the representation of the mRNAs in the cell of origin. Some housekeeping genes may be therefore vastly over-represented in the library, and large genes or rare transcripts may not be represented at all. It follows that even carefully curated clone resources that eliminate redundant and defective clones, such as the Mammalian Gene Collection and the Riken collection, are populated to a large extent by clones derived from more abundant transcripts. Nevertheless, the challenges inherent in creating ORF clones are clearly outweighed by the advantages of their use, such as the opportunity to execute highly parallel experiments, in which all or a subset of genes in a genome can be queried in a controlled setting under identical conditions. Moreover, because the identity of all clones is known in advance, information regarding all clones (even those with no response) can be recorded.
Necessary Characteristics of High-Throughput Cloning Systems
There are a few broad parameters on which cloning systems can be effectively judged, that is, fidelity of the cloning process, ease of use and reliability of the cloning system, validation of the cloned products, and flexibility of use of the cloned products. Another important consideration is that the cloning system itself should confer no undesirable properties on the clones. Finally, when comparing recombinational cloning systems, it is important to consider the costs and efficiencies of creating a master clone collection (capturing ORF sequences from PCR products) separately from those of creating expression clone collections (transferring ORF sequences from master clones to expression vectors).
Fidelity
From the standpoint of the end-users, the defining requirement for a high-throughput cloning system is a high fidelity of transfer of cloned DNA from master clones to expression plasmids. Moreover, if creating a collection of expression clones requires thousands of transfers, the transfer efficiency of cloned DNA must approach or equal 100%. The transfer mechanism should be conservative (cut and paste), thereby avoiding mutations that could be introduced during a replication step during transfer of the cloned ORF, and should result in the ORF situated reliably in the correct orientation and in the correct translational reading frame. With a conservative transfer mechanism, once a master clone is produced and sequence validated, there is no need to repeat the sequencing on any expression clones produced from them.
For a production facility that creates master clones, an efficient capture reaction is also desirable, although the requirements at this stage are not as critical, because the creation of master clones will happen only once, and the resulting clones can be validated by sequencing. The creation of ORF master clones is an inherently mutagenic process, due to the high error rate of the DNA polymerase systems used for PCR, the reverse transcriptase used in making the cDNA template for PCR, as well as the chemical synthesis of oligonucleotides used in PCR amplification. As such, all master clones should be clonally selected and sequence validated. (Clone pools can also be kept to preserve multiple splice variants [Reboul et al. 2001, 2003]; however, it might ultimately be desirable to clone purify and sequence a representative clone for each splice variant.) Master clones serve as the permanent quality-controlled archive clone for each ORF.
Ease of Use
The ideal cloning system should be simple to use and involve a minimal number of steps. Pilot studies and clone transfers of small groups of ORFs should be achievable by a junior-level scientist using a multichannel pipette. In creating very large ORF clone collections that include many thousands of genes, however, the use of automation to assist the cloning process is inescapable. Removing human handling of samples from the cloning process helps to create a more systematic process, results in a process with fewer errors, which integrates better with a clone-tracking database.
When a cloning process is adapted to automated methods, there are several important parameters to consider. First, and most importantly, the reaction should be efficient. If the efficiency does not approach 100%, the error rate will become intolerable for large projects. For example, if the efficiency of creating expression clones was only 95%, the resulting collection will be missing 5% of the target genes, requiring sequence validation of each expression collection to determine which 5% are missing. Second, for any process that must be applied in a high-throughput setting, the reaction should be simple, streamlined, and easy to execute. For instance, pipeting steps should be minimized and easily adapted to robotic platforms. Third, the chemistry must be robust. Given the many reactions that will be processed simultaneously, there is usually a wide range of concentrations of certain reagents (e.g., PCR products) and no capacity for individualized adjustment. Thus, the cloning reaction must be intrinsically tolerant to such variability. Finally, it is important that one set of cloning conditions applies to all ORFs, and that the system has little or no bias with respect to ORF size.
Reliability
If the cloning system is available as a kit, the components of the kit must be stable and reliable. It is especially desirable if the reagents can be purchased in large quality-controlled lots, so that once optimal conditions are established, they do not have to be readjusted in the middle of a production run.
Validation
Sequence validation of master clones is arguably the most expensive stage of the cloning process. The goal of sequencing is to determine which clones are acceptable for use in protein-expression experiments. Even small changes in the amino acid sequence of a protein can have profound effects on its biochemical activity and function, highlighting the importance of carefully evaluating cDNA clones intended for protein expression. Therefore, it is an essential part of the validation process to (1) document the accurate sequences of all clones, (2) indicate where those sequences vary from reference sequences, (3) evaluate and annotate the biological consequences of any variations with respect to the predicted protein sequence, and (4) apply fitness requirements to the clones to either accept or reject them from the project. Ideally, only a single clone for each attempted gene would need sequencing. However, given the unavoidably mutagenic nature of the methods used in the construction of cDNA clones (i.e., errors in oligonucleotide synthesis, reverse transcription, and PCR amplification), several isolates for each gene must often be collected and evaluated to find a good clone. In this light, it is particularly useful if the cloning vector works well in DNA sequencing reactions (see discussion of the Gateway cloning system below).
Flexibility
A powerful advantage of recombinational cloning systems is their ability to support the transfer of genes into virtually any type of expression vector, supporting the broadest possible range of experimentation. For some cloning systems, a varied collection of expression vectors have already accumulated. Nevertheless, the vectors available may not be suitable for novel experiments, so it is especially desirable for users to be able to easily create their own expression vectors and adapt them for use in the cloning system.
Minimizing Undesirable Properties of Cloning Systems
In many cases, particularly in high-throughput experiments, it is desirable to add polypeptide tags to one or both ends of the expressed proteins that can be used as purification tags (His6, GST, CBP, etc.), marker proteins (LacZ, GFP, luciferase, etc.) or as epitopes (HA, Myc, Flag, etc.). The use of fusion tags is, however, somewhat complicated by the contribution of the recombination sequences to the tags, and how stop codons are handled. It is thus important that the design of the master clones is informed by the user's needs with respect to the tags used in any expression clones that will be created. For example, if carboxy-terminal tags are desired, then the natural stop codon must be omitted, or there will not be read-through into the tag. However, the removal of stop codons for some cloning systems will prohibit the production of native protein, because all expressed proteins will have amino acids corresponding to the recombination site fused to the end of the protein. As the inclusion or exclusion of the stop codon results in different versions of the 3′ PCR primer used in amplifying the ORF, this decision must be made at the very beginning of the project.
Cost
The importance of cost should not be underestimated. Although a reaction cost of a few dollars per sample is rarely a problem for a handful of reactions, when many thousands of samples are involved, truly daunting costs can accrue. This is especially true for the reaction costs involved in transferring ORFs from one vector to another, which will be incurred each time a new expression collection is created.
Available Site-Specific Recombinational Cloning Systems
Each of the three major cloning systems described in detail in this review makes use of site-specific DNA recombination. For the sake of convenience, the discussion of these cloning systems will be broken down into capture of PCR-amplified ORF sequences into master clones (which is error prone), and transfer of the DNA from master clones to expression vectors (which, in all systems, is accurate and efficient). The discussion will feature the Gateway (Invitrogen) and Creator (Clontech) systems that utilize conservative transfer in which the ORF DNA is swapped from the master clone backbone to the expression vector backbone, and also the Univector system (developed in the laboratory of Stephen Elledge), in which a cointegrate expression plasmid is created by the recombination-mediated fusion of the master clone with the expression vector.
Gateway Cloning Systems
The Gateway recombinational cloning system available from Invitrogen utilizes a modified version of the site-specific recombination system of bacteriophage λ (Hartley et al. 2000; Walhout et al. 2000). The Gateway system utilizes a minimal set of the components of the λ system for in vitro transfer of DNA, the λ Integrase protein (Int), the λ Excisionase protein (Xis), the E. coli protein IHF, and the att recombination sequences embedded in the DNA to be recombined. In the Gateway cloning system, the orientation of cloned DNA is maintained through vector transfers by the use of two nearly identical, but noncompatible versions of the λ att recombination site (Fig. 1). Hence, attB1 can recombine with the corresponding attP1 (upstream site on donor vector), but not attP2 (downstream site on donor vector).
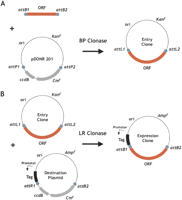
Overview of the Gateway site-specific recombination cloning system. (A) Cloning of ORF attB-PCR products by Gateway BP Clonase-mediated recombination. The blue circles represent λ att recombination sites. (B) Transfer of ORF coding sequences from the Entry vector to create an expression clone by Gateway LR Clonase-mediated recombination.
To select for the desired recombinant product and against the parental plasmids and undesired recombination intermediates, the Gateway system uses an E. coli death gene, ccdB, in combination with differential drug-resistance markers on the master (Entry) and Destination plasmids. The ccdB gene, taken from the E. coli F plasmid segregation control system, allows for negative selection in E. coli by virtue of its ability to inhibit E. coli DNA gyrase (Bernard and Couturier 1992). When the products of Gateway recombination reactions are used to transform E. coli, cells transformed by a Gateway Donor or Destination plasmid or by the cointegrate intermediate of the Gateway recombination reaction are thus unable to grow. Only the desired recombinant product, which lacks the ccdB gene and has the appropriate drug selection marker (e.g., ampicillin resistance for the expression plasmid product), can give rise to transformants.
Creating Gateway Master Clones Using BP Recombination
Although it is possible to create Gateway master (Entry) clones by subcloning with restriction enzymes, this approach can be cumbersome to implement for large-scale cloning projects, as even rare-cutting restriction enzyme recognition sites can be found in coding sequences. The primary means of creating Gateway master clones is BP recombination, a reaction in which the ORF with flanking attB sites (usually generated by PCR) is recombined into a vector with the corresponding attP sites. BP recombination is accomplished by a simple in vitro recombination reaction that requires the λ Int and the IHF protein (a mixture marketed as BP Clonase by Invitrogen), and is usually complete within hours (see Fig. 1A). For large-scale projects requiring high efficiency, it can be advantageous to allow an overnight incubation. Gateway BP recombinational cloning is both efficient and relatively insensitive to target DNA concentration, making it amenable to automation. However, this method of capture does exhibit some size bias, with reduced efficiency for fragments larger than 3 kb (D. Hill, pers. comm.; A. Rolfs, pers. comm.; G. Marsischky and J. LaBaer, unpubl.).
An alternative method for capturing ORFs into the master vector for Gateway is to use a Gateway TOPO Vector (Invitrogen), a modified version of the Entry vector in which the topoisomerase enzyme has been covalently attached to the two free ends of the linearized vector. By adding a short sequence tail to the 5′ end of one of the PCR primers, this method favors directional insertion of the ORF at a reported efficiency of 90%. This efficiency is somewhat less than the BP reaction and the method is considerably more expensive, but it can be useful for genes that are otherwise difficult to capture. Finally, it should also be noted that if a gene is already available in a Gateway-modified expression vector (i.e., flanked by attB sites), it is possible to transfer it backward into an Entry vector by recombining it with a vector containing the corresponding attP sites and BP Clonase. This can be a useful option, as it can be more straightforward to create and sequence validate an expression clone than an Entry clone (see Discussion, below). It also permits the creation of clones that are ready for use in experiments, but can be used to create master clones.
Creating Gateway Expression Clones Using LR Recombination
The transfer of ORF sequences cloned into Gateway expression clones is accomplished by LR recombination in a simple in vitro reaction that requires the phage λ Int and Xis proteins together with the IHF protein (LR Clonase), an expression plasmid modified with attR sites, and an Entry plasmid ORF clone in which the ORF to be transferred is flanked by attL sites (see Fig. 1B). As with BP recombination, the LR recombination reaction is complete within hours. The LR recombination reaction is streamlined, easily adapted to robotic manipulations, and exceedingly reliable with the efficiency of transfer of cloned DNA to expression vectors approaching 100%.
Once a library of master clones is established, it is straightforward to convert them to any expression vector that has been modified for use in the Gateway system. The Gateway system allows for the expression of ORF-encoded proteins with tags at either the N or the C terminus, the latter requiring omission of the natural stop codon during the original cloning of the master clones. Because the recombination sites directly flank the cloned ORF coding sequence, expression of proteins with C-terminal fusions is possible without splicing; thus, C-terminal fusions are available under all circumstances. An important consequence of this arrangement is that there is an addition of short 8 amino acid linkers (encoded by the 25 bp attB recombination sites) to any protein expressed from a Gateway expression clone with N- or C-terminal tags. These linkers are positioned between the ORF-encoded protein and the N- and C-terminal tags.
There exist a variety of tested expression vectors available from Invitrogen and from the research community (Loftus et al. 2001; Karimi et al. 2002; Curtis and Grossniklaus 2003; Helliwell and Waterhouse 2003; Parr and Ball 2003; Renesto and Raoult 2003; Van Mullem et al. 2003) that are adapted for use with the Gateway cloning system. They allow expression of proteins in a wide range of organisms (including bacteria, yeast cells, insect cells, and mammalian cells), using both plasmid and viral expression vectors (adenovirus, retrovirus), with a variety of available promoters. If a suitable expression vector is not available, essentially any expression vector can be modified by inserting a Gateway recombination/selection cassette by blunt-end ligation at the ORF position in the vector. This cassette (available from Invitrogen) contains the attR recombination sites flanking a DNA fragment carrying the ccdB gene (a negative selection gene) and the chloramphenicol selection marker. Note that because of the ccdB gene, these manipulations must be executed in an appropriately modified bacterial strain.
Sequencing Gateway Master Clones
The ability to sequence validate master clones is an essential feature of any high-throughput cloning system. Historically, the original Gateway cloning vectors (e.g., pDONR 201) were low copy and difficult to sequence, in part due to sequences within the attL sites. These problems have now been corrected with the release of new vectors (e.g., pDONR 221) and sequencing protocols utilizing blocking primers for the problematic attL sequences (Esposito et al. 2003). Project planners should ensure that they are using these improved reagents and protocols.
Clontech Cloning Systems
The Clontech cloning approach uses two different enzyme systems for the capture of PCR products to create master clones and for the transfer of genes from master clones to expression clones. For the capture reaction, Clontech uses a proprietary enzyme, In-Fusion, which mediates DNA cloning by the use of short stretches of sequence homology. The Clontech Creator cloning system, which is used to transfer the cloned ORFs from master clones to expression vectors, is based on the Cre-loxP-based site-specific recombination system of bacteriophage P1 (Sternberg et al. 1981). Both Clontech systems are well-suited to automated methods.
Creating Master Clones With In-Fusion
Like the Gateway system, Clontech master (Donor) clones can be assembled using restriction enzymes; however, for large-scale cloning projects, the In-Fusion system is the straightforward choice. The In-Fusion system uses a proprietary enzyme that has intrinsic strand displacement and exonuclease activities, and when the ends of two linear DNA fragments share the same sequence (any homologous sequence suffices), promotes their pairing. Once transformed into bacteria, the resected and paired DNA fragments are readily converted into circular plasmids. In this case, by ensuring that the ends of each PCR product contains 15 bp of homology to the corresponding ends of the vector, the PCR products are captured readily into the vector. One advantage of the In-Fusion reaction is that it is agnostic with respect to the sequences used for recombination. Thus, it can be used as a general method for inserting DNA fragments into any vector. In-Fusion recombinational cloning entails a brief in vitro incubation in which the ORF-specific PCR product and pDNR-Dual are mixed with the In-Fusion enzyme. This results in the amplified DNA cloned within the loxP sites of pDNR-Dual (see Fig. 2A). A simple blue-white screen identifies E. coli transformants of pDNR-Dual with cloned inserts. Because the cloned DNA disrupts the vector lacZ gene, clones with inserts are easily identifiable as white colonies on plates containing IPTG and XGAL. Advantageously, the In-Fusion cloning reaction exhibits only minimal size bias.
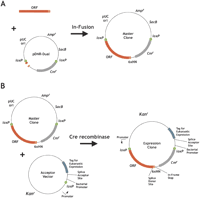
Overview of Clontech recombinational cloning systems. (A) Cloning of ORF PCR products by In-Fusion-mediated recombination. The yellow and orange rectangles represent the 15-bp sequence homology required for In-Fusion cloning at the ends of PCR product and the linearized vector. (B) Transfer of ORF coding sequences from the master clone to create an expression clone by Creator cloning using the Cre site-specific recombinase. The green circles represent loxP sites.
Creating Expression Clones With Creator Cloning
Clontech Creator recombinational cloning utilizes the Cre-loxP site-specific recombination for the transfer of ORFs from the master vector to the expression (Acceptor) vectors (Fig. 2B). The plasmid segment bearing the ORF coding sequence is flanked on the upstream end by a loxP site; on the downstream end, there is a splice donor site to permit the expression of ORFs cloned without native stop codons with C-terminal fusion domains. For expression contexts where splicing is not possible, such as expression of the cloned ORFs in E. coli, the splice donor site is followed by a sequence encoding a C-terminal 6xHN tag (where H = His, and N = Asn) and an in-frame stop codon to prevent read-through. After the ORF, there is a promoterless chloramphenicol resistance selection marker followed by a second loxP site. The successful transfer of the sequences between the loxP sites to an expression vector places the chloramphenicol gene in proximity to a bacterial promoter, conferring resistance to that antibiotic. Correct orientation of the cloned DNA through vector transfers is enforced primarily by the polarity of the loxP site itself, but also by the position of the expression vector-encoded E. coli promoter that drives expression of the chloramphenicol marker.
As with Gateway and In-Fusion cloning, the Creator cloning reaction is a simple one-step in vitro reaction that requires only the Donor clone, an Acceptor vector, and the Cre recombinase. Counter-selection against parental plasmid is accomplished by selecting for recombinants on growth medium containing sucrose (by means of the Bacillus subtilis SacB gene that confers sucrose toxicity on the master clone plasmid); selection for recombinant clones with chloramphenicol resistance and for the drug selection marker from the Acceptor plasmid (usually kanamycin resistance). A collection of vectors that express proteins in bacteria, yeast, and mammalian cells, with a variety of tags are available from Clontech. Moreover, any existing expression vector can be converted to a Creator-compatible vector by inserting a blunt-ended fragment (available from Clontech) at the desired position of the ORF.
Like Gateway cloning, once a library of master Creator clones is established, it is simple to convert them to any Creator-based expression vector. The Creator system allows for the expression of ORF-encoded proteins with tags at either the N or the C terminus, although in contrast to Gateway cloning, most C-terminal fusions are added by RNA splicing. Thus, if ORFs are cloned without stop codons, tags can be efficiently added by incorporating them into the expression vector with a splice acceptor site (but only in vivo in eukaryotes). If no tag is included in the vector, or if the protein is expressed in a setting without splicing, translation will continue into a prearranged 6xHN tag, and then stop. As with Gateway cloning, a recombination site-encoded linker will be present in any N-terminal fusion protein expressed using Creator expression clones. This linker is 11 amino acids, encoded by the 34-bp loxP site, and is positioned between the ORF-encoded protein and the N-terminal fusion tag.
Univector Cloning System
Like the Clontech Creator system, the Univector cloning system, which was developed in the laboratory of Stephen Elledge at the Baylor College of Medicine, uses the Cre-loxP site-specific recombination system for transfer of cloned ORF DNA to expression vectors (Liu et al. 1998, 2000). The Univector cloning system is also available from Invitrogen as the Echo cloning system. The Univector cloning system uses in vitro Cre-loxP-mediated recombination to create a single cointegrate ORF expression plasmid from a master clone (a pUNI plasmid) and an expression vector (a pHOST plasmid; Fig. 3). The approach of plasmid fusion by site-specific recombination to create expression vectors that is found in the Univector system is unique among the high-throughput cloning systems available.
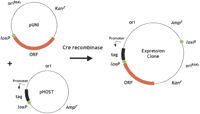
Overview of the Univector site-specific recombination cloning system. The fusion of the ORF master clone (pUNI) with an expression vector (pHOST) to create a cointegrate expression clone using the Cre site-specific recombinase. The green circles represent loxP sites.
Creating Univector Master Clones
To create master clones in a pUNI plasmid (e.g., pUNI50), ORF-specific PCR products are cloned using rare-cutting restriction enzymes (e.g., SfiI-A-SfiI-B) via the pUNI plasmid polylinker, or by using TOPO cloning (Echo cloning system, Invitrogen).
Univector master clones have a conditional origin of replication from the E. coli conjugal plasmid, R6Kγ, which is only able to replicate in an E. coli host expressing the pir gene (which encodes the R6Kγ replication protein, π), and must be created and maintained using a replication-permissive pir+ E. coli strain.
Creating Univector Expression Clones
As with Creator cloning, creating an expression clone with the Univector cloning system is accomplished by Cre-loxP site-specific recombination. Like the other systems described here, the Univector cloning reaction is a simple one-step in vitro reaction that requires only the master clone (pUNI clone), an expression vector (pHOST vector), and the Cre recombinase. The Univector cloning system utilizes two loxP sites which are found separately on the master clone (a single loxP site flanks the start of the ORF coding sequence), and on the expression vector (a single loxP recombination site follows the expression promoter), to mediate the formation of the cointegrate expression plasmid. Correct orientation of the cloned DNA in the cointegrate expression plasmid is enforced during recombination by the polarity of the two loxP sites. Selection for the cointegrate expression plasmid is accomplished by simply transforming an E. coli strain that does not express the pir gene (i.e., where plasmid replication is driven by the expression vector origin of replication, which does not depend on the pir gene), and selecting for transformants on medium containing kanamycin to ensure the presence of ORF coding sequences from the master clone.
Once a library of Univector master clones is established, it is simple to transfer them to any of the Univector-based expression vectors that are available for E. coli, yeast, plants, insect, and mammalian cells (Liu et al. 2000, 2003; Berthold et al. 2003). In principle, the Univector cloning reaction should be as amenable to automated methods as either the Gateway or Creator cloning systems. Also, because the Univector system uses the Cre-loxP recombination, the low cost of Cre recombinase is an important consideration when multiple expression clone collections are to be created. One serious drawback to the Univector system is that it allows only for the expression of ORF-encoded proteins with tags at the N terminus (an 11 amino acid loxP-encoded linker will be present in any N-terminal fusion protein expressed from a Univector clone). Although homologous recombination in recBC mutant E. coli has been used to add C-terminal tags to Univector expression clones (Liu et al. 1998), this approach requires additional sequence validation of the resulting expression clones, and so is less suitable as a high-throughput method for creating expression clones from sequence-validated master clones.
It would be preferable if the Univector system could be modified to allow C-terminal fusions to ORF-encoded proteins in the same manner as the Creator system. This could be accomplished simply by embedding a splice-donor site immediately following the cloned ORF sequence in the pUNI-derived master clone. As a second option (which parallels the Creator system), if no tag for C-terminal fusions is included in the expression vector, or if the protein is expressed in a context that does not permit splicing, translation of an ORF cloned without a stop codon would continue though a short sequence (which could encode a fusion tag) before reaching a predetermined stop.
Experience of Cloning Facilities With Recombinational Cloning Systems
There are several established projects that are using the Invitrogen Gateway, Clontech In-Fusion, and Univector systems to create ORF clone collections (LaBaer and Marsischky 2004; Rual et al. 2004). Of these efforts, the Caenorhabditis elegans ORFeome project of the laboratory of Marc Vidal is certainly the most complete and has garnered the most notoriety (Reboul et al. 2001, 2003). This project targeted the cloning of ∼19,500 ORFs identified in the C. elegans genome, an effort that has resulted in the production of ∼12,000 successful ORF clone pools. The majority of the ORF cloning failures in this cloning effort were a result of difficulties with ORF prediction from the genomic sequence, but a significant number were due to a somewhat decreased efficiency of cloning of ORFs larger than 3 kb using Gateway BP recombination (D. Hill, pers. comm.). Whereas evidence of cloning size bias might be considered to be alarming (as it is likely that many interesting proteins are encoded by larger ORFs), it should be pointed out that the cloning of large ORFs is less efficient for any extant cloning method, including standard restriction enzyme ligation-based cloning. Instead, this issue reflects the problems of implementing state of the art technology on a high-throughput basis. In any case, reduced cloning efficiency by BP recombination is not expected to interfere with the successful completion of the ORFeome project, as most (73%) of the C. elegans ORFs are <1.5 kb (Reboul et al. 2003), and cloning of larger ORFs is possible by changes in procedure, such as the use of specialized BP reaction conditions.
Like the Vidal lab, the Institute of Proteomics at Harvard Medical School (HIP) is also creating ORF clone collections using the Gateway cloning system. HIP is also using the In-Fusion system to build Creator system Master clones (the experience of HIP with these cloning systems will be described in the next section). In addition to the projects in the Vidal lab and at HIP, and other projects described in this issue of Genome Research, a variety of other academic and commercial groups are creating ORF clone collections using Gateway cloning, although for the most part, they have not yet published the results of their cloning efforts. These groups include GeneCopoeia (http://www.genecopoeia.com/orfexpress.php), which offers 15,000 end-read sequence-validated human Gateway ORF clones, an EMBL effort that has used Gateway cloning to create a smaller collection of cloned human ORFs (1800 ORFs) for use in protein localization studies (Simpson et al. 2000), and the Berkeley Drosophila Genome Project, which has converted a small number of its sequence-validated full-length cDNA clones (Stapleton et al. 2002) to the Gateway cloning system. Finally, Invitrogen itself offers its Ultimate ORF Clone collection, fully sequence-validated human and mouse ORF clones in the Gateway cloning format. The extent of this collection is thus far limited to genes of high interest, such as GPCRs and kinases, and comprises ∼9000 human and mouse ORFs.
In addition to HIP, there are two other groups that are creating master clones in the Clontech master cloning vector pDNR-Dual. First, the MRC Geneservice (Cambridge UK, http://www.hgmp.mrc.ac.uk/geneservice/customservices/cloning_services.shtml) makes Creator system-compatible clones using the In-Fusion cloning system. These clones are created from existing full-length MGC cDNA clones, or from existing cDNA clones provided by the individual requesting the ORF clone. The second group is the Berkeley Drosophila Genome Project (BDGP), which has undertaken the transfer of ORFs from their full-length DNA clones into pDNR-Dual using the In-Fusion system (M. Stapleton, pers. comm.). The In-Fusion system was chosen for this project over the Gateway system primarily because the recombination sites surrounding the cloned ORF in Creator Master clones do not have inverted repeat sequences, and thus, are easier to sequence than Gateway Entry clones.
Finally, there is one well-developed effort with the goal of creating ORF clones in the Univector system. The SSP Consortium (http://signal.salk.edu/SSP) has created a sequence-validated, full-length cDNA clone collection comprising 10,500 genes from the thale cress, Arabidopsis thaliana (Yamada et al. 2003). All of the finished clones have been used to create ORF clones in the Univector system master vector pUNI51 by PCR amplification of the ORFs and cloning by use of standard methods. These ORF clones were also sequence validated.
Experience of HIP With Recombinational Cloning Systems
The Institute of Proteomics at Harvard Medical School (HIP) has used both the Invitrogen Gateway and Clontech In-Fusion/Creator cloning systems extensively. HIP has created over 13,000 master clones in Gateway and 5000 in the Creator cloning system representing genes from human, S. cerevisiae, and Pseudomonas aeruginosa (Brizuela et al. 2002; LaBaer and Marsischky 2004; LaBaer et al. 2004). All of these master clones have been validated by end-read sequencing, and clones representing ∼6500 genes have been fully sequenced. HIP has also processed thousands of Gateway LR transfers and hundreds of Creator transfers to create expression clone collections.
For both cloning systems, failure rates for the transfer of cloned ORF DNA to expression vectors are exceedingly low (HIP, unpubl.). As a check of transfer fidelity, plasmid DNA from independent transformants (rather than pools) of a group of several hundred of Creator expression clone transfers was checked by restriction digestion. Likewise, the fidelity of a large number of Gateway LR clone transfers has also been checked. No failed transfers were detected for either cloning system by this validation method. These results have been corroborated by end-read sequence validation of selected expression clones. Generally speaking, the transfer process for both systems is sufficiently robust that pools of transformants can be used in experiments rather than picking individual transformants. This approach, where it is feasible, greatly streamlines the process of creating expression clones to support experimentation.
The most failure-prone step in building large ORF clone collections is the process of creating master clones. Although it would be best to compare the available cloning systems on the basis of published results of their use in creating master clone collections, this is not possible because there have been no large-scale cloning efforts published to date that utilize the Clontech In-Fusion system. We therefore performed a controlled pilot study that compared the efficiency of assembly of master clones by the two best-featured systems, Gateway and In-Fusion/Creator. For this comparison, a common set of full-length ORF clones representing 288 human genes was selected from the Mammalian Gene Collection (MGC; Strausberg et al. 1999, 2002). All of the ORFs were amplified by PCR from the MGC clones in a manner appropriate for each cloning system (i.e., such that tails were added to the ends of the PCR products to provide appropriate recombination sites for cloning). The PCR products were then used for in vitro recombination reactions with the Gateway and In-Fusion cloning systems, which were themselves used to transform E. coli.
The Gateway and In-Fusion cloning systems performed similarly in this functional comparison. Overall, the efficiency of producing PCR products and obtaining master clone transformants was found to be similar for the two cloning systems (see Fig. 4). Likewise, it was found that the overall fidelity of cloning was high for both the Gateway and In-Fusion methods. For each system, the full-length DNA sequence was determined for two independent transformants from 200 ORF cloning attempts per cloning system, and compared with the sequence of the original MGC clone. There were no false (empty) clones found among the sequenced clones for either method. Importantly, the sequenced clones did not contain an unacceptable number of sequence changes; the frequency of sequence discrepancies was 1/3500 bp for the Gateway clones and 1/4100 bp for the In-Fusion clones. An important limitation of this pilot study is that the maximum ORF size in the study was ∼2.5 kb, and the majority of the ORFs were smaller than 2 kb.
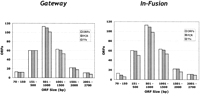
Pilot study: comparison of Gateway BP and In-Fusion cloning methods. Plasmid DNA from 288 MGC clones was PCR-amplified using Platinum Pfx Polymerase (Invitrogen) with end sequences suitable for use in either the Gateway BP or In-Fusion cloning systems. The PCR products were then used in in vitro recombination reactions with the plasmids pDONR 201 (Gateway) or linearized pDNR-Dual (In-Fusion). The products of the recombination reactions were then used to transform E. coli. (White bars) Total MGC ORFs; (dark-gray bars) successful PCR reactions; (light-gray bars) successful transformations.
In cloning efforts at HIP outside of the controlled pilot study, it has been observed that both the Gateway and In-Fusion systems have some bias against the capture of larger ORF DNA fragments (G. Marsischky and J. LaBaer, unpublished results from the S. cerevisiae and human ORF cloning projects). Because failure of cloning by both methods can result in false (empty) clones, it is necessary to compare the results of sequence-validated clones. It is also important that the clones used in such a comparison be created in both systems by identical methods in so far as that is possible. This excludes from analysis many of the yeast clones that were created prior to the introduction of key laboratory automation methods at HIP. We thus compared the sequence validation results from ∼2000 human ORFs (∼4000 isolates) cloned using the In-Fusion cloning system with the results of ∼1600 yeast ORFs (∼6400 isolates) from Phase 2 of the S. cerevisiae project cloned using the Gateway BP cloning system. In each group, the ORFs targeted for cloning range in size from 0.25 kb to ∼4 kb. Despite differences in the templates used for PCR amplification (individual MGC clones, and yeast genomic DNA) the efficiency of PCR amplification was nearly 100% in each group. As with the pilot study described above, ORFs smaller than 2 kb were cloned with ∼90% efficiency in both systems. For ORFs larger than 2 kb, however, the In-Fusion system was somewhat more efficient than Gateway BP cloning (76% vs. 50% for 2-3 kb, and 63% vs. 29% for 3-4 kb). For both systems, the number of false (empty) clones increases with increasing ORF size in addition to a decreased yield in transformants.
Subsequent to these cloning efforts, an optimized In-Fusion reaction developed by Clontech led to an increase in the efficiency of cloning ORF PCR products larger than 3 kb. We have successfully cloned ∼200 large ORFs (3-11 kb) using the In-Fusion system. The efficiency of cloning ORFs 3-5kbwas ∼80% (A. Rolfs, pers. comm.). Thus, although both cloning systems perform similarly for cloning most ORFs, it appears that the In-Fusion system may be advantageous for the cloning of larger ORFs.
DISCUSSION
For many of the key parameters set out earlier, both of the allied recombinational cloning systems, Gateway and In-Fusion/Creator, are functionally equivalent or at least competitive. Most of the traits they share are positive. For instance, both systems are broadly optimized and ready for standardized high-throughput cloning manipulations involving automation. Reagents supplied by the vendors with the cloning systems are stable and reliable. Master clones from both systems can also be easily sequence validated and transferred into a variety of expression-clone formats. On the other hand, both systems share a common disadvantage in that they introduce translated sequence from the recombination site as a linker between the ORF translation product and any expression vector-encoded tags.
There are some significant points of difference between the two cloning systems, however, which reduce to issues regarding size bias, configuration of tags, and cost. The most significant difference is that the Gateway BP recombination system appears to be less efficient than the In-Fusion recombination system for cloning of PCR products of large ORFs (>3 kb). It should be pointed out, however, that cloning of large ORFs is possible using either cloning system, but that DNA sequencing of more transformants will be required to obtain a useful master clone.
With respect to polypeptide tags, the configuration of the Gateway system might be considered to be advantageous, because it allows for a choice of tags on the carboxyl terminus in all circumstances, including use in bacterial expression and in vitro transcription/translation systems. However, a potential disadvantage of the Gateway system is that it adds eight amino acid linkers derived from the attB recombination site to both N- and C-terminal fusion proteins (although this foreign linker sequence has not yet been demonstrated to be a systematic problem). From this perspective, the Creator system might be considered to be a better system. Although it is similar to Gateway in that it gives rise to expression plasmids encoding N-terminal fusion proteins with an 11 amino acid loxP-derived linker (a limitation shared by the Univector system), it produces C-terminal fusions by RNA splicing to expression vector-encoded tags, or by creating an ORF clone without a stop codon to allow read-though to a 6xHN tag. Thus, a potential advantage of the Creator system is its use of splicing as a mechanism to add tags that, in principle, can directly abut the C terminus of the ORF-encoded protein (albeit this option is limited to in vivo expression in eukaryotes). Finally, it should be pointed out that the size of the attB-and loxP-derived linker sequences is similar in all three cloning systems. The consequence of inclusion of these linkers in ORF expression clones is unknown, particularly in the context of large-scale experimentation where they are most likely to be used.
In addition to the technical differences among the various systems, it is also worth considering cost, particularly the cost of transferring genes from master to expression vector. If only a handful of genes are to be moved, this is rarely an issue, but when whole libraries are moved, the costs multiply quickly. This effectively marginalizes smaller (and many larger) laboratories with respect to experiments that would require the creation of new expression clone collections. The cost of these essential reagents will be higher if they are distributed exclusively by one manufacturer than if they can be obtained from multiple suppliers or produced easily in one's own laboratory, such as Cre recombinase. It is best to consider this and all other issues at the start of the project, because once a clone collection is produced and validated in a particular cloning system, all downstream steps are restricted to that system.
The Future of High-Throughput Cloning
Despite the strengths of the recombinational cloning systems available for high-throughput cloning available today, there are great pressures to speed the creation of large ORF clone resources and to leverage their use. It is likely that the research community will develop cheaper, more flexible, and possibly more reliable approaches to creating large ORF clone resources than those presented to us by the commercial vendors. One direction that is likely to be explored is the use of very low-cost homologous recombinational-cloning methods to create master clones for use in the available site-specific recombination systems. This approach could be applied, for instance, to the creation of master clones, complete with recombination sites, for use in the Creator and Univector Cre-loxP-based cloning systems. Alternatively, proprietary cloning methods available from different vendors might be combined to create a process with improved efficiency. For instance, for the creation of Gateway master clones, it might be preferable to use the Clontech In-Fusion method, as it is more efficient for cloning large DNA fragments than Gateway BP recombination.
Finally, another future direction in the creation of ORF clone resources might be a return, with one important modification, to the previous approach of creating expression clones directly. This could be done as before using homologous recombination methods, but with recombination sites properly embedded in the clones for use in one of the site-specific recombinational cloning systems. Thus, sequence-validated expression clones could be ready for immediate use in experiments, and these clones could be used later to create a master ORF clone resource. This approach is already available within the Gateway cloning system, as BP recombination can be used to create master clones from Gateway expression clones.
Footnotes
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2528804.
-
↵1 Corresponding author. E-MAIL gmarsischky{at}hms.harvard.edu; FAX (617) 324-0824.
- Cold Spring Harbor Laboratory Press








