
Identification of Mammalian microRNA Host Genes and Transcription Units
Abstract
To derive a global perspective on the transcription of microRNAs (miRNAs) in mammals, we annotated the genomic position and context of this class of noncoding RNAs (ncRNAs) in the human and mouse genomes. Of the 232 known mammalian miRNAs, we found that 161 overlap with 123 defined transcription units (TUs). We identified miRNAs within introns of 90 protein-coding genes with a broad spectrum of molecular functions, and in both introns and exons of 66 mRNA-like noncoding RNAs (mlncRNAs). In addition, novel families of miRNAs based on host gene identity were identified. The transcription patterns of all miRNA host genes were curated from a variety of sources illustrating spatial, temporal, and physiological regulation of miRNA expression. These findings strongly suggest that miRNAs are transcribed in parallel with their host transcripts, and that the two different transcription classes of miRNAs (`exonic' and `intronic') identified here may require slightly different mechanisms of biogenesis.
MicroRNAs (miRNAs) are an evolutionarily conserved large class of noncoding RNAs (ncRNAs) 18-22 nucleotides long that mediate posttranscriptional silencing of genes. miRNAs were first discovered in Caenorhabditis elegans with the identification of the lin-4 and let-7 miRNA genes, which act as posttranscriptional repressors of target genes by antisense binding to their 3′ untranslated regions (UTRs; for review, see Ambros 2000). Shortly thereafter, hundreds of other miRNAs were found in worms as well as in flies, plants, and vertebrates (for review, see Carrington and Ambros 2003; Bartel 2004). Rapid progress has begun to unravel the genetic roles of miRNAs in development and other biological processes. For example, in C. elegans, let-7 and lin-4 miRNAs function as heterochronic genes, and mutations in either disrupt proper specification of cell fates (Ambros 2000). In Drosophila, a mutation in miR-14 leads to a disruption in normal patterns of cell death and also defects in fat metabolism (Xu et al. 2003). In mammals, ∼230 miRNAs have been identified from a vast array of tissues and cell types (Lagos-Quintana et al. 2001, 2002, 2003; Mourelatos et al. 2002; Dostie et al. 2003; Houbaviy et al. 2003; Lim et al. 2003; Michael et al. 2003; Kim et al. 2004).
Current models for miRNA biogenesis and maturation suggest that compartmentalized stepwise processing of miRNAs takes place first in the nucleus and then in the cytoplasm. The prevailing view is that primary transcripts of miRNAs (pri-miRNAs) are processed in the nucleus by the RNase III enzyme, Drosha, to stem-loop intermediates known as pre-miRNAs (Lee et al. 2003). These pre-miRNAs are then transported to the cytoplasm for cleavage by Dicer and maturation to their active forms (Lee et al. 2003). Although RNAs of 22-, 75-nt as well as longer pri-miRNA transcripts have been detected by Northern blot analyses for a handful of miRNAs, the transcription units (TUs) or gene hosts that give rise to the vast majority of miRNAs have not been examined in great detail.
In the present study, we set out to identify the modes of transcription for mammalian miRNAs by annotating their positions in the human and mouse genomes. We found that more than half of all known mammalian miRNAs are within introns of either protein-coding or noncoding TUs, whereas ∼10% are encoded by exons of long nonprotein-coding transcripts, also known as mRNA-like noncoding RNAs (mlncRNAs; Erdmann et al. 2000). Our annotation illuminates the modes of transcription for miRNAs and has allowed the identification of `intronic' as well as `exonic' transcription classes of miRNAs. We curated the expression patterns of well defined microRNA host genes to illustrate likely expression patterns for microRNAs in wide-ranging biological contexts.
RESULTS AND DISCUSSION
The miRNA Registry contains 232 mammalian miRNAs (http://www.sanger.ac.uk/Software/Rfam/mirna/; Griffiths-Jones 2004). Of these we identified 117 miRNAs located in introns of protein-coding genes or long ncRNA transcripts (Table 1, Supplemental Tables A,B). Approximately 40% (90) of all miRNAs are found within introns of protein-coding genes, whereas ∼10% (27) are located within introns of long ncRNA transcripts (Table 1). Interestingly, 30 miRNAs overlap with exons of ncRNAs (Supplemental Table C). In some cases (14), miRNAs are located in either an exon or an intron (`mixed') depending on alternative splicing of the host transcript (Table 1, Supplemental Table D). Where clusters of miRNAs overlap with a single host transcript, the vast majority of miRNAs are located in the same intron or exon (Supplemental Tables A-D). Additionally, 32 miRNAs overlap with two or more TUs transcribed on opposite DNA strands (Supplemental Tables A-D). This observation indicates that miRNAs are commonly associated with complex transcriptional loci. The remaining 70 miRNAs are of uncertain transcriptional origin and were not analyzed further. To sum up, a total of 161 miRNAs are linked to the transcription of mRNAs or ncRNAs. Based on these observations, we propose that miRNAs be classified as exonic (exon-derived) miRNAs or intronic (intron-derived) miRNAs.
Distribution of Mammalian miRNAs Found in Introns and Exons of Host Transcripts
The numbers given represent total numbers of miRNAs overlapping with transcripts in the human or mouse genome (see Methods). For comparison purposes, miRNAs in introns of transcripts are separated according to the nature of the host gene (protein coding vs. ncRNA). Nine of the `mixed' miRNAs are transcribed in introns or exons of alternatively spliced mlncRNA hosts the rest are associated with protein-coding host genes as well as mlncRNAs of alternatively spliced transcripts. Asterisk (*) denotes the number of mixed miRNAs derived from mlncRNA transcripts only.
A partial listing of protein-coding miRNA host genes is shown in Table 2. These miRNA host genes encode proteins with a broad spectrum of biological roles ranging from embryonic development, to the cell cycle, and physiology (Supplemental Tables A,D). To derive a perspective on the classes of protein-coding genes possibly utilized by miRNAs for their transcription, we surveyed the Gene Ontology (GO) classifications of all miRNA host genes (Gene Ontology Consortium 2001; http://www.geneontology.org/). The two most commonly identified `biological process' classifications for GO annotated miRNA gene hosts are `metabolism' (GO:0008152; 19 of 90; 21%) followed by `cellular physiological process' proteins (GO:00050875; 14 of 90; 16%) and the most common GO `molecular function are `purine nucleotide binding' (GO:0017076; 8 of 90; 9%) and `DNA binding' (GO:0003677; 7 of 90; 8%). Comparison of the most commonly identified GO classifications for miRNA host genes and the entire collection of GO annotated mammalian genes does not reveal disproportionate representation of these classes of genes (data not shown). We also note that several genes involved in human disease are hosts to miRNAs. For instance, nonsense, splicing, frameshift, or deletion mutations in the Chloride Channel Protein 5 (CLCN5) gene cause Dent disease and nephrolithiasis, an X-linked recessive disorder in human patients (OMIM: 300008; Yamamoto et al. 2000). A causative role for the CLCN5-encoded miR-188 has not been explored in this disease.
miRNAs in Introns of Protein-Coding Genes
A significant number of intron-based miRNAs reside within related families of protein-coding genes (Table 3, Supplemental Tables A,D,E). This allowed us to group specific sets of miRNAs into novel families based on host gene identity. In some cases, host gene families aid in the identification of related sets of intronic miRNAs (Table 3). For instance, miR-148b and miR-152 are sequence-related and located in two genes encoding subunits of the coatomer transporter (see Table 3, Supplemental Table E). In other examples, we note sequence-unrelated miRNAs that are found within related families of proteins. For example, miR-105 and miR-224 are located in two genes encoding subunits of the ligand-gated ion channel γ-aminobutyric-acid receptor-α-3 and -ϵ respectively, although the mature miRNAs are unrelated by sequence (Table 3).
Families of miRNAs Found in Introns of Related Protein-Coding Genes
In addition to the miRNAs in protein coding genes, we also noted 66 miRNAs within TUs that lack a significant protein-coding potential (Table 1). More detailed analysis of these miRNA host TUs revealed a number of previously classified long ncRNAs (see Tables 4,5 and Supplemental Tables B-D). These include: BIC (Tam 2001), Deleted in Leukemia 2 (DLEU2; Migliazza et al. 2001), Noncoding RNA in Rhabdomyosarcoma (NCRMS; Chan et al. 2002), Synapse-Specific-7H4 (7H4; Velleca et al. 1994), FLJ34037 (Ota et al. 2004), and numerous FANTOM2 long ncRNAs (Okazaki et al. 2002; Numata et al. 2003). Only the BIC RNA has previously been noted as a carrier for a miRNA (Lagos-Quintana et al. 2002). These types of ncRNA transcripts are sometimes referred to as mRNA-like ncRNAs (mlncRNAs; Erdmann et al. 2000) because they share properties with mRNAs such as splicing (e.g., BIC: Tam 2001), polyadenylation (e.g., Air: Sleutels et al. 2002), long transcription units (e.g., Xist: Hong et al. 1999), and possibly also spatio/temporally restricted expression (e.g., Ntab: French et al. 2001). The molecular/genetic function of most noncoding mRNA-like RNAs is unclear but some, such as Xist and Air, have been studied in greater detail.
miRNAs in Introns of Noncoding Transcription Units
miRNAs Found in Exons of Nonprotein-Coding Transcription Units
As well as the previously defined ncRNA TUs noted above, we identified dozens of novel TU hosts for miRs with little protein coding potential (see Methods, Tables 4,5, and Supplemental Tables B-D). For example, EST and cDNA data in ENSEMBL allowed the identification of an mlncRNA TU host containing miR-100, let-7a-2, and also miR-125b-1 in an intron (Table 4, Fig. 1A). Sequence alignment of the human/mouse host transcript 1 (ENSESTT00000036713/ENSMUSESTT00000030830; data not shown); and transcript 2 (ENSESTT00000036715/2610203C20Rik) does not reveal significant homology (Fig. 1B). Nevertheless, the miR-100, let-7a-2, and miR-125b-1 transcription locus is remarkably similar in human and mouse in both arrangement and structure (Fig. 1A). The possibility exists that some miRNA hosts of this type may be so-called `inside out' genes analogous to some small nucleolar RNA (snoRNA) hosts (Tycowski et al. 1996), in which transcription of the host gene serves purely to yield the intron-encoded RNAs. It is also plausible that some mlncRNA hosts containing miRNAs in their introns serve as functional RNAs.
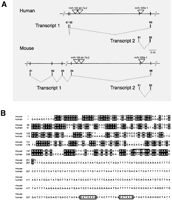
Genomic organization of the mammalian miR-100, let-7a-2, miR-125b-1 locus and sequence alignment of human and mouse miR-125b-1 host transcripts. (A) Identified transcripts are shown with exons represented as vertical lines and labeled above in numerical order. Dotted lines denote splicing of transcripts. Positions of miRNA precursors are denoted with a triangle. Exons are not to scale. Human/Mouse Transcript 1 corresponds to ENSESTT00000036713 and ENSMUSESTT00000030830, respectively. Transcript 2 corresponds to human ENSESTT00000036715 and mouse 2610203C20Rik transcripts. (B) A DNA sequence alignment for Transcript 2 is shown. Identical bases are denoted in black. Consensus polyadenylation sites are boxed in. Note that nucleotides 597-955 of the human Transcript 2 are not shown.
A significant number of miRNAs overlap exons of novel noncoding transcripts (Tables 1,5; Supplemental Tables C,D). For example, the miR-22-predicted hairpin precursor is contained entirely within exon 2 of a noncoding TU, and the splicing pattern is generally conserved in human and mouse, despite the lack of protein-coding potential (Fig. 2A,B). Significant sequence conservation between the mouse and human miR-22 TU is observed within the 5′ end and within the miR-22-predicted hairpin precursor (Fig. 2B). Such high overall conservation in the mammalian miR-22/TU may indicate an important role in biogenesis or localization. Although high sequence conservation within the predicted hairpin precursors of mammalian miRNAs is a common feature (Lim et al. 2003), it is unclear whether the extended sequence similarity observed in miR-22/TU is a more common phenomenon for the exonic class. An alignment of the mouse and human miR-155/BIC does not show such extended conservation beyond the predicted hairpin precursor (data not shown). In addition to miR-22, a further 29 other miRNAs are contained within exons of transcripts (Table 5). In the majority of cases, exonic miRNAs were identified within mlncRNA transcripts, but in two rare cases we noted that the miRNA overlapped with the 3′ UTR of a protein-coding transcript (Supplemental Table D).
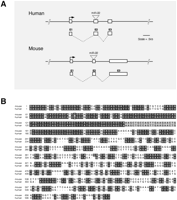
Genomic organization of the mammalian miR-22 locus and sequence alignment of human and mouse miR-22 host transcripts. (A) Exons are shown as white rectangles and labeled above. The human/mouse miR-22 host transcript corresponds to ENSESTT00000065902 and 2210403K04Rik, respectively. (B) DNA sequence alignments of the miR-22 human/mouse host transcripts are shown. Identical bases are black. The mature miR-22 sequence is highlighted in gray. The conserved splice site junction between exons 1 and 2 is also highlighted in gray. Note that nucleotides 688-1658 of the mouse transcript are not shown.
The prevalence of miRNAs within introns or exons of TUs prompted us to ask whether it might be possible to derive reliable miRNA expression patterns from host gene expression data. By analogy to mammalian snoRNAs, intronic miRNAs may be cotranscribed with the host gene by inclusion in introns of their pre-mRNAs and excision from debranched introns by an exonuclease (Tycowski et al. 1993). Similarly to yeast snoRNAs, mammalian miRNAs of exonic transcription origin could be derived from ncRNA transcripts by endonucleolytic cleavage within duplex regions (Chanfreau et al. 1998). If parallel transcription of host genes and miRNAs takes place, similar expression patterns for host transcript and miRNAs would be observed. To test this possibility, we analyzed the expression of five predicted miRNA host transcripts by RT-PCR from several organs in adult mice and compared their expression pattern to that of their corresponding exonic or intronic miRNA (see Methods and Table 6). In four out of five cases, we note an exact correlation between the host gene/transcript expression (Table 6) and the previously reported miRNA expression in mouse adult organs (Sempere et al. 2004). This result suggests that miRNA expression may be reliably derived from host transcript expression information, and strongly suggests that overlapping transcripts in the genome are likely hosts for miRNAs.
Comparison of miRNA Expression and Host Transcript Expression
a miRNA expression is derived from Sempere et al. (2004).
b An ethidium bromide-stained gel picture shows RT-PCR results from mouse adult brain (B), liver (Li), heart (H), lung (Lu), kidney (K), and spleen (S).
The above result impelled us to derive likely miRNA expression pattern information from all host genes identified here. We curated host gene expression data from a number of publicly available sources including primary literature and from public gene expression pattern databases (see Methods). This analysis allowed the identification of physiologically regulated, signal transduction, and tissue/organ specifically transcribed miRNA hosts (see Supplemental Tables A-D for a full description of expression patterns of miRNA hosts). For example, miR-33 is in an intron of SREBP2 (Table 2), an important gene in cholesterol homeostasis that is tightly regulated by sterols at the transcriptional level (Sato et al. 1996). In addition, we identified a cluster of three intronic miRNAs (miR-25, -93, and -106b) that are likely linked to the transcription of DNA Replication Licensing Factor MCM7 (Table 2), a gene that is quiescent in nondividing cells and is upregulated just prior to S phase of the cell cycle (Fujita et al. 1996). Interestingly, human miR-7-3 is contained within intron 2 of Pituitary Gland Specific Factor 1 (Table 2), a gene that is expressed in the pituitary gland (Tanaka et al. 2002). This mature miRNA has two other identical copies in the genome, one of which (miR-7-1) is possibly linked to the expression of a ubiquitously expressed gene, HNRPK (Supplemental Table E). This illustrates that expression pattern information for identical miRNAs may be confounded by distinct but overlapping patterns of transcription. In such cases where multiple paralogous miRNAs exist in the genome, it may prove useful to distinguish the expression of individual miR copies by analyzing the expression of their respective host genes (see Supplemental Table E for a list of similar/paralogous microRNAs contained in different host genes).
The current dogma in the miRNA field is that compartmentalized processing of all miRNAs to mature miRs occurs with Drosha in the nucleus and Dicer in the cytoplasm (for review, see Bartel 2004; He and Hannon 2004). In two seminal studies, it was demonstrated that miRNAs are dependent on Drosha for processing in HeLa cells (Lee et al. 2003) and that enrichment of a particular pri-/pre-miRNA occurs in the nucleus while the mature miR is found in the cytoplasm (Lee et al. 2002). However, we note that the miRNAs tested in these studies are intronic class miRNAs (i.e., miRs-15a, -16-1, -20, -30a; Supplemental Tables A,B), a `mixed' miRNA (miR-21) (Supplemental Table D), and several undefined transcription class miRNAs (data not shown). In lieu of the findings in this study of two types of transcription miRNA classes in mammals, it will be very interesting to revisit these experiments in the context of experimentally verified exonic transcription-class miRNAs. It seems likely that commonalities in processing and maturation by Drosha and Dicer occur for all transcription classes of miRNAs; however, the existence of exonic miRNAs in mammals implies that differences of biogenesis may also take place. In this regard, it is noteworthy that we found miR-206 within an exon of the synapse-associated mlncRNA, 7H4 (Table 4, Supplemental Table C; Velleca et al. 1994; Numata et al. 2003). 7H4 was originally identified by Velleca et al. (1994) as an RNA selectively found in the motor endplate of the rat skeletal neuromuscular junction. Detection of 7H4 at the neuromuscular junction raises the intriguing possibility that the unprocessed form of miR-206 may be transported outside of the nucleus as a pri-mir-206/7H4 RNA. Future studies will be required to determine whether some miRNAs can be actively transported to subcellular compartments outside of the nucleus embedded within their host transcripts.
Conclusion
The findings presented here clearly show that the expression of a large subset of mammalian miRNAs may be transcriptionally linked to the expression of other genes, coding for both proteins and ncRNAs. The identification of two distinct classes of miRNAs, overlapping introns of other transcripts, and encoded in exons, suggest that the maturation of miRNAs may be more complex than previously thought. Mammalian snoRNAs, responsible for modification of ribosomal and spliceosomal RNAs, were shown to be excised from introns of protein-coding (often ribosomal protein) genes (Qu et al. 1995). The suggestion that approximately half of mammalian miRNAs are expressed by a similar mechanism has important and far-reaching implications for our understanding of gene expression, regulation, and the basic definition of what constitutes a gene. Previous studies have demonstrated unexplained levels of conservation in intronic and other apparent noncoding sequence in mammals (Bejerano et al. 2004). There has also been recent discussion about the regulatory possibilities of parallel output arising from exonic and intronic sequences in genes (Mattick 2003). Although we are far from categorical evidence of parallel transcription on a global scale, and the data presented here account for only a small percentage of noncoding conservation, the presence of miRNAs in host genes, together with the reported roles of intronic snoRNAs, provides a tantalizing suggestion of this phenomenon on a larger scale.
METHODS
miR Databases
miRNA hairpin sequences were retrieved from the microRNA Registry release 3.1 (http://www.sanger.ac.uk/Software/Rfam/mirna/; Griffiths-Jones 2004). Some homologs were identified using pairwise similarity of the hairpin sequence and by matching of mature miRNAs. The ability of flanking regions to adopt a hairpin structure was determined using the mfold program (http://www.bioinfo.rpi.edu/applications/mfold/).
Genome Analysis and Curation
miRNAs were mapped to the human (NCBI Build 34) and mouse (NCBI Build 32) genome assemblies using WUBLASTN. To safeguard against concerns about mapping errors in the mouse NCBI Build 32 assembly, we also made use of the mouse NCBI Build 30. ENSEMBL IDs from mouse Build 30 are provided in several cases in which errors in the newer assembly were noted. For ease of manual annotation, the results of these searches were made accessible via the ENSEMBL genome graphical interface (http://www.ensembl.org/). Annotation of mouse and human miRNAs was aided by using a combination of genome annotated TUs, including Vega (http://vega.sanger.ac.uk/Homo_sapiens/), ENSEMBL (http://www.ensembl.org/), FANTOM2.0 (Okazaki et al. 2002; Numata et al. 2003), NCBI Acembly/AceView (http://www.ncbi.nlm.nih.gov/IEB/Research/Acembly/), NCBI (http://www.ncbi.nlm.nih.gov/mapview/), and FLJ transcripts (Ota et al. 2004). Genome coordinate overlaps between miRNAs and annotated TUs were classified according to the exonic or intronic nature of the miRNA. The FatiGo data mining tool (http://fatigo.bioinfocnio.es/; Al-Shahrour et al. 2004) was used to determine if any GO terms are significantly overrepre sented in microRNA host genes.
mRNA-Like Noncoding RNA Databases
A total of ∼10,000 mlncRNAs sequences were collected from the FANTOM Functional Annotation Database (http://fantom2.gsc.riken.go.jp/; Numata et al. 2003), the Noncoding RNAs Database (http://biobases.ibch.poznan.pl/ncRNA/; Erdmann et al. 2000), and the Full-Length Japan database (http://www.nedo.go.jp/bio-e/; Ota et al. 2004), and mapped to the genome using SSAHA. The positions of these ncRNAs were made accessible in the ENSEMBL genome graphical interface via the DAS protocol and analyzed for potential overlap with miRNAs in the human or mouse genome.
Identification of mRNA-Like Noncoding RNAs
miRNA host transcripts were designated as mlncRNAs in a process similar to that employed by the FANTOM Genome Exploration Research Group (Okazaki et al. 2002). To identify mlncRNA candidate miRNA hosts, we eliminated those ENSEMBL or NCBI Acembly AceView Transcripts overlapping with miRNAs with similarity to known protein sequences using BLASTX (E-value < 10-5) or those that showed homology to known motifs or domains in the Pfam database (Bateman et al. 2004).
miRNA Host Gene RT-PCR
Total RNA was prepared from 2-3-wk-old mice using the RNA RT-PCR Miniprep Kit (Stratagene). To safeguard against RT amplification of hnRNA (nuclear precursor RNA), an oligoDT primer was used in the first-strand cDNA synthesis step instead of gene-specific primers. In order to unambiguously distinguish spliced cDNA from genomic DNA contamination, specific primers were designed to amplify across introns of the predicted microRNA host transcript, and -RT controls reactions were performed. The primers used to detect each of the five microRNA transcription units by PCR were as follows: miR-9-3 host transcript, ENSMUSESTG00000007408, 5′-GTGGGCCAGGAGAAAAGAGAAAAG-3′ (forward) and 5′-CTCTGTGTTCATCCTGTGGCTTTG-3′ (reverse); miR-22 carrier transcript, 2210403K04Rik, 5′-CAGTGA TTTTGCTCCTCTGTCCAC-3′ (forward) and 5′-CAACTGAGC TACAACCCCAGTCAT-3′ (reverse); miR-137 carrier transcript, ENSMUESTG00000012545, 5′-CAGAGTCCGCATGAAGCAGAG CAA-3′ (forward) and 5′-GTCCTTGGCAACCGGGAGCTTTTA-3′ (reverse); miR-153 host transcript, ENSMUSG00000054701, 5′-CTGTGCTGACCTATGACCACTC-3′ (forward) and 5′-CAATCAG GACGTAGGTTCCACT-3′ (reverse); miR-219 host transcript, ENSMUESTG00000007085MBuild30, 5′-AGAGGCGCCCGCTT GGGTTCAG-3′ (forward) and 5′-TCATTCCCCAGCTTCTGGG TTT-3′ (reverse).
miRNA Host Gene Expression Pattern Curation
Gene expression pattern information was obtained from the primary literature using NCBI PubMed (http://www.ncbi.nlm.nih.gov:80/) as a source for articles. In cases where the host gene/TU had no primary literature citations, expression data were instead obtained from the RIKEN Expression Array Database (http://read.gsc.riken.go.jp/; Bono et al. 2003), the Gene Expression Atlas Database (http://expression.gnf.org/cgi-bin/index.cgi; Su et al. 2002), the NCBI Acembly/AceView Web site, or from SWISS-PROT (http://us.expasy.org/sprot/).
Acknowledgments
We thank Tony Cox and James Stalker for technical assistance in generating ENSEMBL DAS sources for genome annotation, Dr. Russell Grocock for useful discussions and technical help, and Drs. Jos Jonkers and Madhuri Warren for critical reading of the manuscript. A.R. is an NIH Postdoctoral Fellow. This work was funded by The Wellcome Trust.
Footnotes
-
[Supplemental material is available online at www.genome.org.]
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2722704. Article published online before print in September 2004.
-
↵1 Corresponding author. E-MAIL abradley{at}sanger.ac.uk; FAX 44-1223-494714.
-
- Accepted July 27, 2004.
- Received April 28, 2004.
- Cold Spring Harbor Laboratory Press








