
Allele-Specific Holliday Junction Formation: A New Mechanism of Allelic Discrimination for SNP Scoring
Abstract
We report here a new mechanism for allelic discrimination—allele-specific Holliday Junction formation. The Holliday Junction (HJ) is a unique DNA structure that can be formed in a sequence-nonspecific manner by routine PCR. To cause the PCR-based HJ formation to occur in an allele-specific manner, the PCR primers are manipulated such that an extra mismatch next to a SNP of interest is introduced between a target and a reference amplicon and a GC-clamp is added. Based on this new mechanism, novel SNP genotyping methods were developed, including a homogeneous fluorescence polarization (FP) competition assay that requires neither labeled primers/probes nor expensive enzymes/substrates. Using this novel genotyping technology, we were able to convert >95% of SNP sequences into genotyping assays that work well under a universal set of assay conditions and achieved 100% accuracy in clinical samples.
SNPs can be detected in either a sequence-specific or sequence-nonspecific way (Kowk 2001). Sequence-specific detection relies on four general mechanisms for allelic discrimination: allele-specific hybridization, allele-specific nucleotide incorporation, allele-specific oligonucleotide ligation, and allele-specific invasive cleavage (Kowk 2001). We describe here a fifth mechanism for allelic discrimination: allele-specific Holliday Junction (HJ) formation.
It has been shown by Panyutin and Hsieh (1993) that two partial duplexes will quickly form a four-stranded cruciform Holliday Junction if their tails are complementary enough to anneal to each other (Fig. 1A). The two partial duplexes that form the Holliday Junction will then undergo strand exchange through spontaneous branch migration, during which the energy loss due to the breaking of a base pair is coupled with the energy gain from the formation of a new base pair (Panyutin and Hsieh 1993). The strand exchange will go to completion and the Holliday Junction will quickly resolve into two duplexes when the sequences of the duplex portions of the two partial duplexes are the same (Panyutin and Hsieh 1993). However, when there is a difference between the duplex portions of the two partial duplexes, in the presence of Mg2+, the spontaneous branch migration will encounter an energy barrier at the mismatch position, leading to the formation of stable Holliday Junctions (Panyutin and Hsieh 1993). Based on Panyutin's discovery, Lishanski and coworkers developed a method for sequence-nonspecific mutation detection using PCR-amplified DNA fragments (Fig. 1B; Lishanski 2000; Lishanski et al. 2000). Like hybridization or heteroduplex formation, Holliday Junction formation is itself sequence-nonspecific. To make Holliday Junctions form in an allele-specific manner, it is necessary to make the target amplicons and the reference amplicons short enough so that the possibility for the existence of an additional SNP is negligibly small. However, when the forward and the reverse primers are separated only by one or a few bases, a single-base mismatch can no longer impede branch migration and cause the formation of detectable Holliday Junctions in the resultingshort amplicons. To overcome this problem, an extra single-base mismatch was introduced between the target and the reference amplicons at the vicinity of the SNP of interest. The introduced extra single-base mismatch is itself too weak to impede branch migration in short amplicons (<80 bp). However, when it is coupled with the SNP of interest, the combined double mismatch becomes strongenough to impede branch migration and cause the formation of stable Holliday Junctions. The introduction of an extra mismatch alone is enough to produce allele-specific Holliday Junction formation for short amplicons that are highly GC-rich. However, for average or AT-rich short amplicons, the addition of a 2–8-bp GC-clamp at the 5′ end of the amplicons is required for allele-specific Holliday structure formation. The extra singlebase mismatch and the 5′-GC-clamp are introduced through primer design.

Mismatch-induced Holliday Junction formation. (A) A single-base mismatch between two partial duplexes impedes branch migration and causes the stabilization of the Holliday Junction formed between the two partial duplexes through the annealing of their complementary tails. (B) Illustration of partial duplex and Holliday Junction formation by using PCR.
The Holliday Junction has physical and biochemical properties that are very different from those of duplex DNA or single-stranded DNA. These differences can be exploited in various ways for the detection of Holliday Junctions and the development of novel SNP-scoring methods. We demonstrate here two ways of detecting Holliday Junctions (HJ): a homogeneous, FP (fluorescence polarization) competition assay and a heterogeneous, gel electrophoresis assay. Through the successful development of genotyping assays for 108 SNPs randomly selected from the NCBI SNP database and 11 clinically relevant SNPs, we show that Holliday Junction-based genotyping provides a robust, very inexpensive technology for the scoring of a diverse set of SNPs or mutations with a high degree of accuracy.
RESULTS
Mismatch-Induced Holliday Junction Formation of PCR Amplicons Can Be Detected by Gel Electrophoresis
When a forward primer and two tailed reverse primers are used to amplify DNA samples, ∼50% of the amplicons will be partial duplexes (Fig. 1B). These partial duplexes will form Holliday Junctions and undergo spontaneous branch migration (Fig. 1B). When such PCR amplicons of one genotype are mixed with the amplicons of a different genotype at 1:1 ratio, it can be calculated that ∼6% of the total amplicons should form stable Holliday Junctions. On the other hand, when the amplicons of one genotype are mixed with the amplicons of the same genotype, no stable Holliday Junctions will form. In theory, the presence or absence of Holliday Junctions should be detectable on gels because Holliday Junctions have much lower mobility than duplex amplicons. However, such PCR-generated Holliday Junction bands were never detected on gels before when ethidium bromide was used to visualize the DNA bands. When more sensitive DNA dyes such as SybreGold and SybreGreen were used, we were able to detect them directly on gels (Fig. 2).
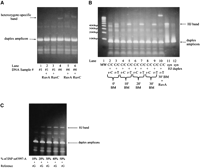
PCR-generated Holliday Junctions can be detected on gels. (A) A heterozygote-specific, lower-mobility band was the only band that was up-shifted by RuvA and RuvC. DNA sample #4 is a homozygote, and DNA sample #1 is a heterozygote. (B) After 10 min of branch migration (BM), the target amplicons of a C/C homozygote of SNP rs#1551570 formed a lower-mobility band with the T/T reference amplicons (r-T) but not with the C/C reference amplicons (r-C). This lower-mobility band was the only band that was up-shifted by RuvA, and it had the same mobility as the synthetic HJ designed to mimic the HJ formed between the C/C target amplicons and the r-T reference amplicons. (C) A PAGE gel showing the correlation between the number of Holliday Junctions and the allele frequency of SNP ss#3997 allele A (percent of SNP ss#3997-A).
PCR-generated Holliday Junction bands were first detected in our early studies when PCR amplicons were relatively long(>120 bp). Deriving PCR amplicons from a heterozygote is essentially the same as mixing the PCR amplicons of one genotype with the amplicons of a different genotype at 1:1 ratio. For each SNP tested, we detected a DNA band that was only present in PCR assays from heterozygotes. Figure 2A shows that this heterozygote-specific band moved more slowly than its respective duplex amplicons and only this band was up-shifted by RuvA and RuvC, two proteins that specifically bind to Holliday Junctions. These data strongly imply that the heterozygote-specific bands are, indeed, Holliday Junction bands. This was confirmed later by using synthetic Holliday Junctions that mimic the PCR-generated HJ in very short amplicons (Fig. 2B). Figure 2B shows that a DNA band of much lower mobility was present when the target amplicons from a C/C homozygote of SNP rs#1551570 were mixed with the T/T reference amplicons (r-T) but absent when mixed with the C/C reference amplicons (r-C; Fig. 2B, lanes 2–9). Moreover, this band was the only band up-shifted by RuvA (Fig. 2B, lane 10), and it had the same mobility as the synthetic HJ on gel (Fig. 2B, lane 11).
Allele-Specific Holliday Junction Formation Requires an Introduced Extra Mismatch and in Most Cases, the Addition of a GC-Clamp
When we tried to score SNPs through the detection of HJ formation in PCR amplicons that are 120 bp or longer, we found a preponderance of false-positive results that were subsequently proved to be caused by the presence of a second SNP in the PCR amplicons. To achieve allele-specific HJ formation, we designed PCR primers that were very close to the SNP to be interrogated, and the resulting amplicons were very short (<70 bp).
To find out if all types of SNPs (A/T, A/G, C/T, G/T, A/C, and G/C) can induce HJ formation in short amplicons, we generated all possible mismatches by using forward primers (Table 1A, SEQ ID NO 3–7) that contain different “wobbles” at the SNP ss#4129 (NCBI SNP database) and a 6-bp overlap with the reverse-tailed primers (Table 1B, T1-2 and T2-2, SEQ ID NO 31 and 32.) As a result, the wobbles overwrite the naturally occurring C in the genomic DNA template used (a C/C homozygote) and thus generate a series of short amplicons containing all possible SNPs at the same distance from both amplicon ends and in the same sequence environment. Each wobble-containing forward primer was used in combination with the (T1-2/T2-2, 1:1) reverse-tailed primer mix to generate a 67-bp amplicon. The extent to which various mismatches impede branch migration in these amplicons was judged by the intensity of respective HJ bands on 6% PAGE gels (Fig. 3A).
Forward Primers and SNP Sequences
Reverse-Tailed Primers
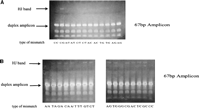
An extra mismatch is required to achieve allele-specific Holliday Junction formation. (A) Various mismatches inhibit branch migration to a different extent in 67-bp amplicons as judged by relative intensity of their HJ bands. (B) Effect of an extra mismatch (mm1) on Holliday Junction formation by various types of mismatches in very short (67-bp) amplicons.
Figure 3A shows that for the 67-bp amplicons, a single A/T, A/G, or A/C mismatch did not produce any detectable HJ at all—judging by the lack of HJ bands on gels and, later, by the lack of inhibition in FP competition assays (data not shown). A noticeable but weak HJ band was produced only by a single G/C mismatch, and the HJ band produced by a single T/G or C/T mismatch is barely visible.
Therefore, a single-base mismatch, A/T, A/G, or A/C, cannot induce detectable HJ formation in these short amplicons. One possible way to solve this problem is to introduce an extra single-base mismatch (e.g., A/T, A/G, or A/C) next to the SNP of interest. This extra mismatch would be, by itself, too weak to block branch migration but, in combination with the SNP, might be strong enough to block branch migration and cause detectable HJ formation. To test this, four target PCR amplicons, each with a predetermined genotype (A, T, C, or G) at SNP ss#4129, were produced by using the four forward primers (SEQ ID 13–16) that have a 4-bp overlap with the tailed reverse primers (T1-2 and T2-2, SEQ ID NO 31 and 32.) At the same time, four reference PCR amplicons, each with a predetermined genotype (A, T, C, or G) at SNP ss#4129 and an extra T→ A mutation (mm1) introduced at the base immediately before the SNP, were produced by using the four forward primers (SEQ ID 17–20) that all have a 4-bp overlap with the tailed reverse primers (T1-2 and T2-2, SEQ ID NO 31 and 32.) Each of the 4 target amplicons was then mixed at a 1:1 ratio with each of the four reference amplicons. All 16 possible mixtures were subjected to denaturation and branch migration. The formation of HJ in these 16 mixtures was then visualized on 6% PAGE gels (Fig. 3B.) Figure 3B shows that a single-base mismatch such as A/G, A/C, or A/T caused HJ formation in the 67-bp amplicons of SNP ss#4129 only after an extra mismatch mm1 (an A/T mismatch) was introduced before it.
To test whether the introduction of an extra mismatch can be used widely to achieve allele-specific HJ formation, we randomly selected ∼100 SNPs from the NCBI SNP database and tried to develop genotyping assays for them. We found that only a small fraction of SNPs was able to achieve allele-specific HJ formation after the introduction of an extra mismatch. Careful analysis and comparison of the sequences of these SNPs revealed that all the SNPs that had achieved allele-specific HJ formation after the introduction of an extra mismatch were either extremely GC-rich or had an overall average GC content with long stretches of GC (>5 bp of continuous G or C). In contrast, those that failed to achieve allele-specific HJ formation were either AT-rich or had an overall average GC content with long stretches of AT (>5 bp of continuous A or T). To mimic the sequence content of the SNPs that had achieved allele-specific HJ formation, a GC-clamp was added to each of the SNPs that had failed to do so. With this GC-clamp permutation, all of the SNPs were able to achieve allele-specific HJ formation after the introduction of an extra mismatch.
Figure 4 illustrates the development of an HJ-based genotyping assay for SNP ss#4130, an SNP with an A/T-rich sequence context. When target amplicons were amplified from genomic DNA sample #1 of the M08PDR panel using primer sets 4130F/(4130T1 + 4130T2) (Table 1A,B, SEQ ID 19, 37, and 38) and then mixed with each of the two reference amplicons amplified using primer sets 4130ref-C/(4130T1 + 4130T2) (Table 1A,B, SEQ ID 22, 37, and 38) and 4130ref-T/(4130T1 + 4130T2) (Table 1A,B, SEQ ID 23, 37, and 38), the introduced extra mismatch alone was not enough to achieve allele-specific Holliday Junction formation (Fig. 5, lanes 1,2) However, when an 8-bp or 7-bp GC-clamp was added to the two redesigned target forward primers 4130F2 and 4130F3, respectively, allele-specific Holliday Junction formation was achieved (Fig. 5, lanes 3–6).

General rules of primer design for the SNP genotyping assays based on allele-specific Holliday structure formation. (A) The primer designs for those GC-rich SNPs that do not require the addition of a GC-clamp. (B,C) Two alternative ways to design primers for the SNPs that require the addition of a GC-clamp.
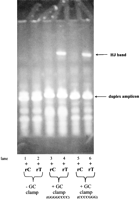
A GC-clampis required to achieve allele-specific Holliday Junction formation. In addition to an introduced extra mismatch, a GC-clampis required to achieve allele-specific Holliday Junction formation for a C/T SNP ss#4130 in sample #1 (a C/C homozygote) of the M24PDR panel from the Corriel Cell Repository.
We succeeded in developing gel-based genotyping assays for all the SNPs selected, including108 SNPs randomly picked from the NCBI SNP database and 11 clinically relevant SNPs. Figure 6A shows the data of a typical PAGE gel-based genotyping assay, whereas Figure 6B shows that of a typical agarose gel-based assay. The genotype of the genomic DNA sample was determined by detecting the formation of HJ between the target amplicons and the two reference amplicons. If the genomic DNA was a homozygote of one allele, it only formed Holliday Junctions with the reference amplicon of the other allele (Fig. 6A, samples 1, 3, 6, 7; Fig. 6B, samples 76–80). In contrast, if the genomic DNA was a heterozygote, it formed Holliday Junctions with both reference amplicons, and the HJ bands were about half of the intensity of those formed in homozygotes (Fig. 6A, samples 2, 4, 5; Fig. 6B, samples 70–75).
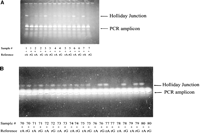
SNP genotyping by gel electrophoresis. (A) A PAGE gel showing Prothrombin (FII) G20210A genotyping of 7 of the 150 clinical samples obtained from the UC Davis Medical Center. (B) An agarose gel showing the genotyping of SNP ss#4181 for 11 DNA samples (#70–#80) from the Corriel Human Diversity Panel.
Holliday Junctions Can Be Detected Homogeneously by an FP Competition Assay
The Holliday Junction is an intermediate product during homologous recombination. Several proteins that specifically bind to Holliday Junctions have been cloned, expressed, and purified from several organisms (West 1997). One of them is RuvA, which has very high specificity for binding to Holliday Junctions. Its specificity for binding to Holliday Junctions is >1 × 104 higher than for binding to duplex DNA (West 1997). By using RuvA and a universal fluorescent tracer molecule, we were able to develop a homogeneous, FP competition assay for SNP scoring. In the assay, the presence and the number of Holliday Junctions can be determined through their competition with the fluorescent tracer molecules for binding to RuvA. The tracer is a small fluorescence-labeled synthetic Holliday Junction formed via the annealing of four 18-bp oligonucleotides with one of the 4 oligonucleotides labeled with fluorescein. Because of its small size and special shape, the free tracer has a low FP of ∼80 millipolarization units (MP). After binding to RuvA, its FP jumps to ∼400 MP. The big FP difference (>300 MP) between the free and bound tracer allows the development of a robust FP-based competition assay. As illustrated in Figure 7A, the more unlabeled HJs coexisting with the tracer, the fewer tracers will bind to RuvA and the lower the FP value. In this way, the amount of unlabeled HJs can be determined through the monitoring of the FP value of the tracers.
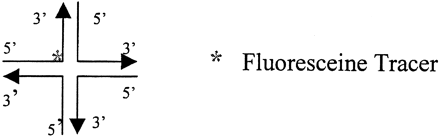
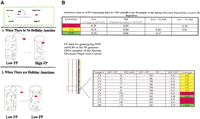
SNP genotyping by the homogeneous FP competition assay. (A) Illustration of Holliday Junction detection by the FP competition assay. (B) FP Data and statistical analysis for genotyping SNP ss#4188 in the 90 genomic DNA samples of the Human Diversity Panel from the Corriel Cell Repository.
Figure 7B shows the data of a typical FP genotyping assay: an HJ-based genotyping assay developed for a G/A SNP ss#4188 was tested against the 90 genomic DNA samples of the Corriel Human Diversity Panel. For each sample, the number of Holliday Junctions formed between the target amplicons and each of the two reference amplicons was determined by the two FP values of the tracer: FP(+refA) and FP(+refG). The difference between the two FP values was then calculated: ΔFP = FP(+refA) - FP(+refG). For an A/A homozygote such as sample #76, FP(+refA) is much higher than FP(+refG) and ΔFP = FP(+refA) - FP(+refG) = 190 mP. On the contrary, for a G/G homozygote such as sample #77, FP(+refA) is much lower than FP(+refG) and ΔFP = FP(+refA) - FP(+refG) = -175 mP. For a G/A heterozygote such as sample #70, the difference between FP(+refA) and FP(+refG) is much smaller and ΔFP = FP(+refA) - FP(+refG) = 41 mP. In our assay analysis, ΔFP is normalized by dividing by FP, and the ratio of ΔFP/FP is used as the final parameter for genotyping calling in our assays. The statistical analysis of the FP data (Fig. 7B) shows that our FP-based genotyping assay for SNP ss#4188 is a robust assay because the ΔFP/FP ratios of the three genotypes in the 90 samples are at least 3 standard deviations apart.
RuvA binds to all Holliday Junctions, including those that formed in an allele-specific manner or those that formed nonspecifically. One source for nonspecific Holliday Junction formation is the genomic DNA template used in PCR. The second source is truncated primers that exist in low-quality (salt-free) tailed reverse primers. These nonspecific Holliday Junctions do not interfere with the detection of the specific HJ band on gel, probably because they are a heterogeneous population with randomly distributed lengths. However, these nonspecific Holliday Junctions do bind to RuvA and interfere with the FP-based HJ detection. Therefore, the homogeneous FP competition assay has stricter requirements than the gel-based heterogeneous assay: First, the genomic DNA concentration used in the PCR should be 0.2 ng/μL or less. Second, the two tailed reverse primers usually need to be PAGE-purified, although desalted primers are always satisfactory for the three forward primers.
The only labeled oligonucleotide for the FP homogeneous assay is the 18-bp fluorescein-labeled oligonucleotide used to make the tracer. Because the tracer used in the assay is universal for all SNPs and the working concentration for the tracer is only 300–500 pM, the cost for the tracer is negligible. RuvA protein can be easily and inexpensively manufactured in large scale and can be stored in a refrigerator for more than 6 mo without detectable loss of activity.
We succeeded in developing genotyping assays for 108 SNPs randomly selected from the NCBI SNP database. These SNPs cover all possible mismatches (G/C, A/T, A/G, A/C, C/T, G/T) in various sequence contents (from extremely AT-rich to extremely GC-rich). Among the 108 SNP assays, 95% of them use the universal PCR/assay conditions outlined in Methods and ∼90% of them were designed with little optimization. Each assay was tested against the 24 genomic DNA samples of the Corriel M24RDP panel. Then 10 of the 108 SNPs were selected and tested against the 90 genomic DNA samples of the Corriel Human Diversity Panel. In both cases, the gel-based and the FP-based assay formats were used in parallel and the genotyping callings from using the two formats were found to have 100% correlation (one example can be seen by comparing Figs. 6B and 7A). To test the accuracy of our genotyping assays, 10 of the 108 SNPs were randomly selected, and the genotypes at these 10 SNP positions for each of the 24 genomic DNA samples of the Corriel M24RDP panel were determined by sequencing(Sequetech, Inc.) and compared with those determined by the Holliday Junction-based genotyping assays. There was 100% concordance between the Holliday Junction-based genotyping assay and the sequencing results.
To determine the sensitivity of the technology and its potential application to diagnostics, we developed genotyping assays for 11 clinically relevant mutations. All 11 SNP assays performed well under a set of universal PCR/assay conditions. Moreover, the genotyping assays for Factor V Leiden, the prothrombin G20210 mutation, and the two MTHFR polymorphisms were each tested blindly on 50 clinical samples (the samples were made anonymous and received an exemption from the Institutional Review Board) obtained from UC Davis Medical Center that had previously been tested using standard genotyping technologies. The DNA concentrations of these 150 clinical samples were unknown, and a 1000× dilution was applied universally for all of them for PCR for the FP-based assays. The allele-specific Holliday structure formation genotyping was found to have 100% accuracy in these samples as compared with the clinical results.
DISCUSSION
We describe here a new allelic discrimination mechanism— allele-specific Holliday Junction formation. Based on this mechanism, two SNP genotyping methods were developed: a gel-based heterogeneous assay and an FP-based homogeneous assay. Although the two methods rely on different detection modalities, the assay development for them proved to be interchangeable. In addition to the two detection methods illustrated here, it is foreseeable that a variety of other methods and instruments such as capillary electrophoresis, liquid chromatography, or FRET-based assays can also be used for the same purpose.
Like other SNP genotyping methods, the key to successful assay design for Holliday Junction-based SNP genotyping is the design of PCR primers. On average, by following the primer design rules outlined in Methods and illustrated in Figure 4, successful genotyping assays performed under a universal set of PCR/assay conditions can be developed for ∼90% of SNPs with little optimization. Successful genotyping assays can be developed for the remaining 10% of SNPs by redesigning primers or changing the PCR or branch migration conditions.
There are already many SNP genotyping methods available today, each equipped with its unique set of desirable attributes. For an example, some heterogeneous assays (such as RFLP) do not require expensive labeled primers but are labor-intensive and time-consuming. In contrast, homogeneous assays (such as the TaqMan or the READIT assay) are fast and can be easily automated but all require expensive labeled primers/probes or expensive enzymes/substrate. We developed here an FP-based, homogeneous genotyping assay that is fast, easily automatable, and, at the same time, does not require expensive labeled primers/probes or enzymes/substrates. In fact, it is the only homogeneous assay available today that uses universal assay conditions and requires neither labeled primers/probes nor expensive enzymes/substrates. However, our FP-based assay is not yet a close-tube assay like the Taqman assay. We are working on the development of a thermostable tracer and RuvA to make our assay a close-tube assay.
Not only can Holliday Junction-based genotyping assays be used for scoring SNPs on individual DNA samples, they can also be used to detect allele frequency directly in pooled DNA samples. As shown in Figure 6, the number of HJs formed in heterozygotes is about half of those formed in homozygotes. This is compatible with the fact that, in heterozygotes, only 50% of the target DNA amplicons can form HJs with each of the two reference amplicons. Therefore, the number of HJs formed between the target DNA amplicons and the reference DNA amplicons seems to reflect the allele frequency in the target DNA sample. Using DNA samples with known allele frequency at 10% increments (0%, 10%, 20%, 30%, 40%, and 50%), we were able to show a clear correlation between allele frequency and the relative amount of HJ formation (Fig. 2C).
Besides various single-plex genotyping assays that have already been or can be developed based on this new allelic discrimination mechanism, allele-specific Holliday Junction formation also has the potential to be used for the development of genotyping technologies capable of performing multiplex assays in parallel. For example, because the length of the amplicons and thus the size of the HJs formed between them can be arbitrarily determined through primer design, multiple SNPs can be genotyped in a single lane on PAGE gels or capillary electrophoresis if the Holliday Junctions formed for different SNPs are of different size and therefore have different mobility. A more drastic approach, however, will involve performing the allelic discrimination for multiple SNPs in parallel directly on genomic DNA followed by the isolation/purification of the allelic discrimination products—Holliday Junctions using RuvA. The identities of individual Holliday Junctions purified can then be elucidated, for an example, on a micro-array after amplification by PCR. Certainly, many technical obstacles need to be overcome before this potential can be applied to practice.
Finally, our FP-based competition assay also has the potential to be used as a homogeneous yet inexpensive method for the detection of gene expression or the detection of pathogens. As long as two partial duplexes have tails that are complementary to each other, a Holliday Junction will form between them regardless of the sequence homology between the duplex portions of the two partial duplexes (Fig. 1A). In fact, the less homology shared between the sequences of the duplex parts of the two partial duplexes, the more stable the Holliday Junction. When two tailed reverse primers are used to amplify a target DNA, part of the target amplicons will be partial duplexes (Fig. 1B). The partial duplex portion of the target amplicons can be easily transformed into Holliday Junctions by annealing with partial duplexes that have complementary tails but heterologous sequence at their duplex part. The resulting HJs can then be detected homogeneously using our FP-based competition assay. The target amplicons, in these cases, can derive from host RNA/genomic DNA or from the RNA/DNA of various pathogens such as HIV, HBV, or HCV.
METHODS
SNP Assay Design
We selected 91 SNPs from the batch of SHGC/AffymetrixChip/Seq-Genomic (from ss#4117 to ss#4210 except ss#4131, ss#4178, and ss#4208) and 17 SNPs from the batch of SHGC/Affymetrix Chip-Genomic (from ss#3985 to ss#4003 except ss#3989) from the NCBI SNP database. Among the four SNPs that were omitted for assay development, ss#4131 does not have the sequence information about its allele position, ss#4178 and ss#4208 have longrepetitive sequences, and ss#3989 was skipped because it had been extensively used before for PCR in our lab and might cause some contamination problem. Of the total SNPs, 11 were clinically relevant: 7 β-thalassemia mutations (-28, cd17, cd70/72, IVS-2 nt 654, cd27/28, cd41/42 and +40/+43), the Factor V Leiden mutation, the Prothrombin gene G20210A mutation, and two common MTHFR polymorphisms (MTHFR677 and MTHFR1298).
For each SNP, a total of five primers were designed for the development of a genotyping assay: three short (18–36 bp) forward primers and two long(40–50 bp) tailed reverse primers (Fig. 4.) For the three forward primers, one was designed to produce the target amplicons with the pair of the tailed reverse primers, whereas each of the other two forward primers was designed to produce reference amplicons with the same pair of tailed reverse primers. The two reference amplicons have known genotypes at the SNP of interest as well as an introduced extra mismatch next to the SNP of interest (Fig. 4.) In particular, each reference primer has at least a 4-bp-long, perfectly matched overlap with the tailed reverse primers so that the reference amplicons have a predetermined sequence (Fig. 4).
Depending on whether the extra mismatch is introduced at the base immediately before or after the SNP of interest, the tailed reverse primers begin at (or in other words, their 3′ ends terminate at) the base that is 1 base or 2 bases after the SNP of interest, respectively (Fig. 4). This rule for designing tailed reverse primers holds true unless it causes 3′-dimer or hairpin formation. The hairpin and 3′-dimer problems can be solved by the introduction of permutations of the primer at appropriate positions or by moving the 3′ ends of tailed primers farther away from the SNP of interest.
For the SNPs that are in a very GC-rich sequence environment, no GC-clamp is needed (Fig. 4A). For the SNPs that are in an AT-rich or average GC content environment, a 2–8-bp GC-clamp must be added to both the target and the reference amplicons to achieve allele-specific Holliday Junction formation. The GC-clamp can be added to the target and the reference amplicons by attaching it to all of the three forward primers (Fig. 4B.) In both cases, the target forward primers used to amplify the target DNA ends just before the SNP of interest (Fig. 4A,B).
Alternatively, the GC-clamps are added only to the target forward primer during primer design to avoid long reference forward primers (Fig. 4C.) In these cases, the GC-clamp is added to the reference amplicons in a second round PCR using the target forward primer as the forward primer and the amplicons produced by the reference forward primers as the template.
All primers were designed using the software Oligo 6 purchased from Molecular Biology Insights, Inc. The general rules for primer design are as follows: (1) the priming part of each primer—the part that actually hybridizes with the template— should have a Tm above 66°C. (2) The final Tm for each target forward primer after the addition of a 0–8-bp GC-clamp should be above 78°C. (3) If the base next to the SNP is G or C, a G A or C A mutation should be introduced there in the reference forward primers to introduce an A/G or A/C mismatch between the target and the reference amplicons. If the base next to the SNP is T, a T→ A mutation should be introduced there in the reference forward primers to introduce an A/T mismatch between the target and the reference amplicons. If the base next to the SNP is A, an A→ T mutation should be introduced there in the reference forward primers to introduce an A/T mismatch between the target and the reference amplicons.
Oligonucleotides
The oligonucleotides were purchased either from Operon Technologies or from Genset Corp.
PCR Amplification
PCR amplifications were carried out using a PTC-200 DNA Engine thermocycler (MJ Research Inc.). For genomic PCR, 45 PCR cycles were performed with 10 sec of denaturation at 94°C, 15 sec of reannealing at 58°C, and 45 sec of extension at 72°C. The cycling was preceded by a 10-min incubation at 95°C to activate AmpliTaq Gold DNA polymerase (Applied Biosystems). The reaction mixtures contained 0.2 ng/μL or less genomic DNA, 0.025 U/μL AmpliTaq Gold DNA polymerase, 200 μM each dNTP, 1 μM the salt-free forward primer, 250 nM each of the two reverse tailed primers if PAGE-purified, and 500 nM each of the two reverse tailed primers if salt-free, 10 mM Tris-HCl, (pH 8.3), 50 mM KCl, 2 mM MgCl2, and 200 μg/mL BSA.
The target amplicons and each of the reference amplicons were either amplified separately or amplified together in a single PCR reaction. When amplified together, the forward primer of the PCR reaction was a mixture of the target forward primer and one of the reference forward primers at a ratio of 4:1.
Branch Migration
Branch migration was carried out in the same PCR buffer using a PTC-200 DNA Engine thermocycler (MJ Research Inc.) at 65°C for 30 min following a 2-min denaturation step at 95°C. When the target and the reference amplicons were amplified together in a single PCR reaction (see above), branch migration was simply linked to the PCR in a single program. When the target and the reference amplicons were amplified separately, the target amplicons were mixed with each of the reference amplicons at a 1:1 ratio before branch migration was performed.
RuvA and RuvC Proteins
The 190 μM wild-type Escherichia coli RuvA and 35.5 μM wild-type E. coli RuvC in storage buffer (20 mM Tris-HCl at pH 7.5, 0.1 mM EDTA, 2 mM 2-mercaptoethanol, 200 mM Nacl, 50% glycerol) were purchased from H. Shinagawa at the University of Osaka in Japan and kept at -80°C. Aliquots of 50 μM RuvA diluted in PCR buffer (10 mM Tris-HCl at pH 8.3, 50 mM KCl, 4 mM MgCl2, and 200 μg/mL BSA) were made and kept at 4°C for at least 24 h before being used as the RuvA stock solution in experiments.
Band-Shift Analysis
For band-shift analysis, 5 μL of PCR/branch migration products was mixed with 1 μL of 0.25 μM freshly diluted RuvA or 1 μL of 0.5 μM freshly diluted RuvC before being mixed with 1 μL of 6× TBE Loading Buffer (Invitrogen Inc.) and loaded in a well of a 6% 12-well precast TBE gel (Invitrogen Inc.).
Tracer
A fluorescein-labeled synthetic Holliday Junction formed by the annealing of four 25 μM 18-bp oligonucleotides (HJ18-1, HJ18-2, HJ18-3, and HJ18-4) in PCR buffer at room temperature for 1 h. The fluorescein is attached to one of the four nucleotides at the center of the Holliday Junction. HJ18-1: GCGCATAGQCCGAATGGC (Q = fluorescein-dT); HJ18-2: GCCATTCGGTGAGCAGCG; HJ18-3: CGCTGCTCAGGATTGACG; HJ18-4: CGTCAATCCACTATGCGC.
FP Competition Assay Using Analyst AD
All the FP-based genotyping assays for the ∼120 SNPs were performed using both 96-well (LJL HE-96B) and 384-well (LJL HE-384B) plates on an Analyst AD fluorescence plate reader (Molecular Devices Inc.).
When LJL HE-96B plates were used, 10 μL of PCR/branch migration products was mixed, in each well, with 10 μL of 0.67 nM tracer before adding 4 μL of a 1:400 dilution of the 50 μM RuvA stock solution. After incubation at room temperature for 10 min, the fluorescence polarization of the samples was measured using an Analyst AD fluorescence plate reader (Molecular Devices Inc.).
When LJL HE-384B plates were used, 5 μL of PCR/branch migration products was mixed, in each well, with 3 μL of 1 nM tracer before adding 2 μL of a 1:400 dilution of the 50 μM RuvA stock solution. After incubation at room temperature for 10 min, the fluorescence polarization of the samples was measured using an Analyst AD fluorescence plate reader (Molecular Devices Inc.).
PAGE Gel Analysis
For PAGE gel analysis, 5 μL of PCR/branch migration products was mixed with 1 μL of 6× High Density Loading Buffer (Invitrogen Inc.) before being loaded in a well of a 6% 12-well precast TBE PAGE gel (Invitrogen Inc.). The gels were run in a matching SureLock Mini cell (Invitrogen Inc.) at 160 V for 30 min before being stained with SybreGold (Molecular Probes) and photographed with 667 films.
Agarose Gel Analysis
For agarose gel analysis, 5 μL of PCR/branch migration products was mixed with 1 μL of 6× High Density Loading Buffer (Invitrogen Inc.). Then 2.5 μL of the mixture was loaded in a well of a 3% 400-well agarose gel that was run in a 20 × 40cm SubMarine gel apparatus (CBS Scientific) purchased from VWR. The gels were run at 190 V for 55 min before being stained with SybreGold (Molecular Probes) and photographed with 667 films.
Synthetic Holliday Junctions
The synthetic Holliday Junctions that mimic the HJ formed between C/C target amplicons and T/T reference amplicons (r-T) of SNP rs#1551570 were made by the sequential annealing of four PAGE-purified 78-bp oligonucleotides (1551570#1, 1551570#2, 1551570#5, and 1551570#6). First, two partial duplexes were made by annealing1551570#1 to 1551570#2 and 1551570#5 to 1551570#6, respectively, in PCR buffer at room temperature for 30 min. The two partial duplexes were then mixed at a 1:1 ratio and underwent branch migration at 65°C for 30 min. 1551570#1: 5′-CTCACTGGCATCTCAGCTA TAGCAACAGGATCTTAGCCTTTAGAGTCTGTCGGACGCAGATTTGAGTTCTAACGGCCC-3′ 1551570#2: 5′-GGGCCGTTAGAACTCAAATCTGCGTCCGACAGACTCTAAAGGCTAAGATCCTGTTGCTCTGCTAGATCTCACAGTGGT-3′ 1551570#5: 5′-GGGCCGTTAGAACTCAAATCTGCGTCCGATTGACTCTAAAGGCTAAGATCCTGTTGCTATAGCTGAGATGCCAGTGAG-3′ 1551570#6: 5′-ACCACTGTGAGATCTAGCAGAGCAACAGGATCTTAGCCTTTAGAGTCAATCGGACGCAGATTTGAGTTCTAACGGCCC-3′.
Acknowledgments
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.997703.
-
↵3 Corresponding author. E-MAIL qinghong{at}freshgene.com; FAX (925) 681-4012.
-
- Accepted April 18, 2003.
- Received November 15, 2002.
- Cold Spring Harbor Laboratory Press








