
Analysis of mRNA With Microsomal Fractionation Using a SAGE-Based DNA Microarray System Facilitates Identification of the Genes Encoding Secretory Proteins
Abstract
In the regulation of host defense responses such as inflammation and immunity, the secretory proteins, including membrane proteins, play central roles. Although many secretory proteins have been identified by using methods such as differential display, random screening, or the signal sequence trap method, each method suffers from poor reproducibility, low sensitivity, or time-consuming or laborious work. Therefore, the strategy for facilitating the selection of the genes encoding the secretory proteins is desired. In this paper, we describe a system for isolating the genes encoding secretory proteins by analyzing mRNAs with microsomal fractionation on serial analysis of gene expression (SAGE)–based DNA microarray system. This system succeeded in discriminating the genes encoding secretory proteins from ones encoding nonsecretory proteins with 80% accuracy. We applied this system to human T lymphocytes. As a result, we were able to identify the genes that are not only encoding secretory proteins but also expressing selectively in a specific subset of T lymphocytes. The SAGE-based DNA microarray system is a promising system to identify the genes encoding specific secretory proteins.
Most of the secretory proteins—such as receptors, transporters, adhesion molecules, hormones, cytokines, and chemokines—play central roles in homeostatic and host defensive responses; therefore, identification of novel secretory proteins may lead to further clarification of molecular mechanisms of life phenomena and establishment of new therapies for human diseases. Many secretory proteins have been identified by using differential display (Liang and Pardee 1992), random screening, and the signal sequence trap method (Tashiro et al. 1993). However, those methods appeared unlikely to be suitable for the comprehensive identification of the genes encoding secretory proteins for which expression is dynamically regulated in response to particular external stimuli. This is because differential display suffers from poor reproducibility, random screening suffers from time-consuming and laborious work, and the signal sequence trap method suffers from low sensitivity.
The comprehensive search for expressed genes by serial analysis of gene expression (SAGE) reveals not only known but also novel transcripts present within RNA population studied (Velculescu et al. 1995). However, it is generally difficult to predict whether the gene encodes secretory or nonsecretory protein from the SAGE tag. We solved this obstacle by analyzing mRNA with subcellular fractionation by using DNA microarray (Diehn et al. 2000). Consequently, we were able to develop a SAGE-based DNA microarray system that is efficient in identifying the genes encoding novel secretory proteins (Fig 1).
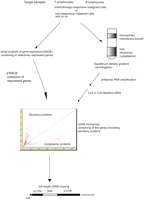
The strategy for identifying the genes encoding secretory proteins for which expression is dependent on the cell types or the cell states. In the present study, our strategy was applied to human T lymphocytes. First, SAGE was performed to reveal genes selectively expressed under specific T lymphocyte conditions such as activated Th1, activated Th2, or activated T lymphocytes. Second, the specific genes were collected by using 3′ RACE. Finally, by using a DNA microarray system, the genes encoding secretory proteins were identified by analyzing RNAs from the free ribosomal and the microsomal fraction after equilibrium density–gradient centrifugation.
In our previous paper, we constructed the expression profile of human activated Th1 lymphocytes and that of human activated Th2 lymphocytes by using SAGE technology (Nagai et al. 2000). In addition, we performed SAGE on human resting CD4+ T lymphocytes. We show here that the genes encoding secretory proteins in a specific subset of human T lymphocytes are identified efficiently by using the SAGE-based DNA microarray system.
RESULTS
Screening of the Genes Expressed Selectively in Specific Subsets of T Lymphocytes
In activated Th1-, activated Th2-, and resting CD4+ T lymphocyte SAGE libraries, a total of 32,219, 32,291, and 62,459 tags, respectively, were sequenced. To identify individual genes, the expressed genes were analyzed with the National Center for Biotechnology Information (NCBI) SAGE database (http://www.ncbi.nlm.nlh.gov/SAGE/). Those SAGE results permitted selecting the genes satisfying the following criteria: (1) the genes encode hypothetical proteins or ESTs on 2002.1; (2) the genes encode proteins expressed selectively in either activated Th1 or Th2 lymphocytes, or selectively induced by activated T lymphocytes; and (3) the genes correspond with the products amplified by 3′ RACE. The genes satisfying these criteria are listed in Table 1. Ten genes were selected from activated Th1 lymphocytes, one gene from activated Th2 lymphocytes, and 20 genes from activated T lymphocytes.
Selectively Expressed Genes With Uncharacterized Subcellular Localization From the SAGE Results
Isolation of Membrane-Bound Polysomes From Activated T Lymphocytes
We performed equilibrium density–gradient centrifugation to separate microsomes and free ribosomes from the peripheral blood mononuclear cells (PBMCs) stimulated with phorbol myristate acetate (PMA) and ionomycin (Mechler and Rabbitts 1981; Mueckler and Pitot 1981; Mechler 1987). The separation into the free and membrane-bound polysomes from the PBMCs was confirmed by reverse transciption–polymerase chain reaction (RT-PCR; Fig. 2A). We arbitrarily selected GATA3 and G3PDH for cytoplasmic proteins; CD48, CXCR4, and CCR7 for cell surface molecules; and IFNγ and MIP1β for secreted proteins. Figure 2A shows that cytoplasmic genes such as GATA3 and G3PDH are rich in the free ribosome fraction, and secreted and membrane-associated genes such as IFNγ, MIP1β, CD48, CXCR4, and CCR7 are rich in the microsome fraction.
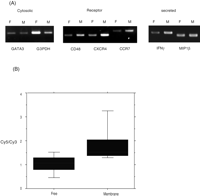
Distribution analysis of RNA encoding proteins with characterized subcellular localization. (A) RT-PCR was performed on total RNA isolated from the free ribosomal and the microsomal fraction. GATA3 and G3PDH were selected for cytoplasmic proteins; CD48, CXCR4, and CCR7 for receptors; and IFNγ and MIP1β for secreted proteins. F and M indicate the free ribosomal and the microsomal fraction, respectively. (B) Microarray analysis gave a Cy5/Cy3 ratio to each gene. The free column shows the box plot of Cy5/Cy3 ratios of 150 genes encoding cytoplasmic proteins; the membrane column shows the box plot of Cy5/Cy3 ratios of 229 genes encoding secretory proteins. Each box includes 60% of genes encoding the cytoplasmic or secretory proteins.
Identification of the Genes Encoding Secretory Proteins
Fluorescently labeled cDNA was synthesized from each fraction (Cy5-labeled cDNA for membrane-associated mRNA, and Cy3-labeled cDNA for cytosolic mRNA). The Cy3- and Cy5-labeled cDNAs were simultaneously hybridized to the genes on the microarray glass slide. Figure 2B shows the distribution of Cy5/Cy3 fluorescent intensity ratio of 379 genes for which subcellular localization has been established. The ratio of the fluorescent intensity of the genes encoding proteins of known subcellular localization is used as internal control. Eighty percent of genes encoding cytoplasmic proteins are <1.35 in Cy5/Cy3 ratio; on the other hand, 80% of genes encoding secretory proteins >1.37 in Cy5/Cy3 ratio.
Selection of the Novel Genes Encoding Secreted and Membrane-Associated Molecules
Among the genes in Table 1, nine genes had a Cy5/Cy3 ratio >1.35, suggesting that these genes encode secretory proteins (six cDNAs encoding hypothetical proteins and 3′ ESTs). Two genes are expressed selectively on activated Th1 lymphocytes, one gene on activated Th2 lymphocytes, and six genes on activated T lymphocytes. By using the algorithms such as SOSUI (http://sosui.proteome.bio.tuat.ac.jp) and PSORTII (http://psort.nibb.ac.jp/form2.html), their subcellular localization was predicted (Fig. 3). The hypothetical protein encoded by Hs.286131 gene, of which expression is selective in activated Th1 lymphocytes, has four transmembrane domains. Although the product of Hs.7718 gene selectively expressed in activated Th2 lymphocytes has no transmembrane domain in accordance with the algorithms, the ratio of its fluorescent intensity (Cy5/Cy3 = 1.73) was sufficiently high to be a secretory protein. This discrepancy was due to the incomplete information of the sequence of Hs.7718 cDNA in the public database. Although it is reported in the public database that the product of Hs.7718 gene comprised of 522 amino acids, the full-length cDNA of this gene turned out to encode a protein comprising 775 amino acids with a signal sequence at the N-terminal portion (Nielsen et al. 1997). Hs.349306 gene product has 13 predicted transmembrane domains, which suggests that this product is a seven-transmembrane receptor selectively induced in activated T lymphocytes. Hs.99486 gene product has a signal sequence and a transmembrane domain.

Predicted structures of six hypothetical proteins and RT-PCR analysis of their expression level. By using an algorithm such as SOSUI and PSORTII, transmembrane domains of the six hypothetical proteins were predicted on the basis of the presumed human ortholog. Sequences were obtained from the public database. In the column predicted structure, the black oval represents putative signal sequence; the black boxes, putative transmembrane domains. RT-PCR was performed on total RNA isolated to confirm SAGE results. Th1, Th2, act T, and res T indicate activated Th1 lymphocytes, activated Th2 lymphocytes, activated T lymphocytes, and resting T lymphocytes, respectively. The Cy5/Cy3 fluorescent ratio is a value that is given to each gene by the microarray analysis; candidates of secretory proteins have Cy5/Cy3 fluorescent ratios >1.35.
Full-Length cDNA Cloning of Hs.182285
To examine whether secretory proteins can be identified from the limited information of the EST of 3′-site, the full-length cDNA cloning of Hs.182285 gene was conducted. The result indicated that Hs.182285 cDNA encodes a novel protein comprising 167 amino acids, which has four predicted transmembrane domains, as shown in Figure 4.
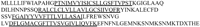
Amino acid sequence of Hs.182285 gene product. 5′ RACE analysis reveals the predicted structure of Hs.182285 gene product. Hs.182285 gene product comprises 167 amino acids. The four predicted transmembrane domains are shown by the underlines.
Analysis of In Vitro Translated Hs.7718 Gene Product
To assess the topology of Hs.7718 gene product relative to the microsomal vesicle membranes, proteinase K susceptibility studies was performed (Fig. 5A). The in vitro translated Hs.7718 gene product resolved at ∼106 kD on SDS-PAGE, which is 20 kD larger than the size calculated from amino acid sequence (Fig. 5A, the first lane). Hs.7718 gene product translated in the presence of microsome showed higher molecular weight even after treatment with proteinase K (Fig. 5A, the second lane), which is possibly due to N-glycosylation at amino acids 138–141 and amino acids 361–364. Upon disruption of the microsomal membrane, Hs.7718 gene product was completely degraded (the third lane). Together, these results indicate that Hs.7718 gene product is a secretory protein.
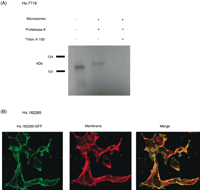
(A) Autoradiogram demonstrating the susceptibility of Hs.7718 gene product to proteinase K when translated in vitro in the presence of microsomal vesicles (microsomes). Equal aliquots of each translation reaction were exposed to proteinase K in the absence (-) or presence (+) of Triton X-100. (B) HEK293 cells overexpressing green fluorescent protein (GFP)–fused Hs.182285 gene product. HEK293 cells expressing GFP-fused Hs.182285 gene product (left) were fixed, and the membrane is labeled with DilC12 (center). Images were merged (right). When GFP-fused Hs.182285 gene product distribution is compared with the membrane labeling in red performed with DilC12, the GFP fluorescent signal was identical to the plasma membrane.
Hs.182285 Gene Product–Green Fluorescent Protein Distributed Through the Plasma Membrane
To directly visualize the intracellular distribution of Hs.182285 gene product, we expressed chimera consisting of Hs.182285 gene product fused to green fluorescent protein (GFP) in cultured HEK293 cells and examined the GFP fluorescence by laser-scanning confocal microscopy. The Hs.182285-GFP fused at the C terminus was dispersed throughout the plasma membrane (Fig. 5B). There is no difference in distribution between Hs.182285 gene product with GFP at the N terminus and at the C terminus. In addition, Hs.182285-GFP fusion protein mostly merged with membrane marker DilC12 (Fig. 5B). These results indicate that Hs.182285 gene product is indeed located at the plasma membrane.
DISCUSSION
Intercellular communication between secreted proteins and membrane receptors plays a central role in most fundamental biological processes. In addition, secretory proteins are the most probable therapeutic agents, or targets, for antagonistic or agonistic therapy. Therefore, the development of efficient systems for identifying the genes encoding secretory proteins has been desired.
So far, several strategies for identifying secretory proteins have been developed. One is the signal sequence trap method that takes advantage of a characteristic N-terminal signal peptide sequence. However, this strategy has difficulty in identifying novel specific secretory protein genes, because unequal representation of different messages makes the identification of weakly expressed genes difficult and because comparisons among the expression profiles of multiple samples cannot be performed. Another is a genome-wide screening method using DNA microarray (Diehn et al. 2000); DNA microarray analysis of mRNAs with subcellular fractionation reveals candidate genes encoding secretory proteins. This strategy cannot be applied to identify the genes encoding secretory proteins that are expressed in response to particular external stimuli because of the following reasons. One is that DNA microarray analysis is limited by the ability to analyze only previously isolated genes. Furthermore, it is usually difficult for laboratories to collect, verify, and manage thousands of genes for DNA microarray analysis, and many genes represented on the microarrays cannot be assessed because they are not expressed at a sufficient level. Another reason is that it is difficult to prepare sufficient materials in specific conditions such as Th1-polarized lymphocytes or Th2-polarized lymphocytes for the purpose of discriminating secretory proteins from nonsecretory proteins by equilibrium sediment–density centrifugation. Our strategy could be applied to identify the genes expressed in a limited amount of samples.
The striking feature of our system is the ability to isolate not only the genes that are encoding secretory proteins but also dynamically regulated proteins in response to particular external stimuli. In addition, our system can identify novel genes without interference of highly expressed genes. This is the fundamental limitation in identifying novel specific secretory protein genes. We have overcome this problem by analysis of mRNA with subcellular fractionation by using the SAGE-based DNA microarray system. In this study, we found that the SAGE-based DNA microarray system is very efficient in identifying the cDNAs encoding novel secretory proteins with selective expression in a specific subset of T lymphocytes, although the gene expression in human Th1 lymphocytes, human Th2 lymphocytes, and activated human T lymphocytes has been intensively analyzed. Our system is efficient in discovering new secretory proteins, but it is impossible to detect all secretory proteins. The reason is that some proteins translocate across the endoplasmic reticulum (ER) membrane by posttranslational modification (Deshaies et al. 1991; Panzner et al. 1995), and that some mitochondrial membrane proteins are synthesized on free polysomes, released into cytosol, and transported across the organelle membrane by a posttranslational mechanism.
We have succeeded in cloning the full-length cDNA of a novel gene (cluster ID, Hs.182285) for which the product can be a transmembrane protein induced in activated T lymphocytes. This molecule indeed exists at the plasma membrane (Fig. 5B), and has four predicted transmembrane domains and one WW domain (amino acids 8–34, also known as rsp5 or WWP) in accordance with PROSITE (http://www.expasy.ch/prosite/). The WW domain is one of the smallest, yet most versatile, protein–protein interaction modules. The ability of this simple domain to interact with a number of proline-containing ligands (Chen and Sudol 1995), which somewhat resembles SH3 (Src homology 3) domains, has resulted in a great deal of functional diversity. Although it is difficult to speculate the function of this molecule, this molecule may directly activate or stabilize T lymphocytes, because WW domains can be found in many signal transduction molecules.
In conclusion, the SAGE-based DNA microarray system described here facilitates identification of the genes encoding novel secretory proteins for which expression is regulated during biological processes in any type of cells or tissues. Because subcellular fractionation by equilibrium density–gradient centrifugation cannot discriminate perfectly secretory proteins from nonsecretory proteins, the microsome fraction could contain mRNAs for cytoplasmic proteins. However, the 3′-EST information revealed by SAGE suffices full-length cDNA cloning, and the subcellular localization of the molecules are predicted with considerable accuracy from the amino acid sequence by using the algorithms such as SOSUI or PSORTII. Application of our system to other cell types and conditions could lead to further identification of the genes encoding novel secretory proteins that remain veiled in the human genome (Velculescu et al. 1999; Sara et al. 2002).
METHODS
SAGE
The SAGE procedure was performed on mRNAs from human activated Th1 lymphocytes, activated Th2 lymphocytes, and resting CD4+ T lymphocytes as described (Nagai et al. 2000). Sequenced files were analyzed with the SAGE software, SAGE map (http://www.ncbi.nlm.nlh.gov/SAGE/), and NCBI's sequence search tool (advanced BLAST search; http://www.ncbi.nlm.nih.gov/BLAST/). After elimination of linker sequences and the repeated ditags, a total of 126,969 tags representing 32,219, 32,291, and 62,459 tags from activated Th1, activated Th2, and resting CD4+ T lymphocytes, respectively, were analyzed.
DNA Microarray Manufacture
We used 3′ rapid amplification of cDNA ends method (3′ RACE) to collect cDNAs for the microarray analysis, using the RACE kit (Clontech) according to the manufacture's instructions. The 3′ RACE products were cloned into the p-GEM T easy vector (Promega), and inserts of at least two independent Escherichia coli clones were sequenced by using a M13 forward primer. Sequence homology was confirmed by the advanced BLAST search. For the analysis, we used 463 cDNA clones consisting of 150 genes encoding cytoplasmic proteins, 229 genes encoding secretory proteins, 82 genes with uncharacterized subcellular localization, and two negative controls (λ [Takara] and tobacco chloroplast gene). Sequences of all genes were verified. PCR products prepared from these clones were spotted onto glass slides by using a robot with four printing tips (Kakengeneqs); length of the amplicon ranged from 300–1000 bp. To normalized carrying efficiencies of labeling and detection, a series of housekeeping genes (encoding β-actin, ribosomal protein L32, G3PDH, and thymocin β10) and negative control genes (λ and tobacco chloroplast gene) were spotted in each of the four rectangles of DNA spots. The λ and tobacco chloroplast gene that we used as negative controls have no sequence homology to any sequence in the human genome.
Subcellular Fractionation
We used equilibrium density–gradient centrifugation to separate the free ribosomes and the microsomes (Mechler and Rabbitts 1981; Mueckler and Pitot 1981; Mechler 1987). Briefly, PBMCs were cultured in RPMI1640 supplemented with 2 mM l-glutamine, 0.1 mM nonessential amino acids, 1 mM pyruvate, 5 × 10-5 mM 2-mercaptoethanol, and 10 % FCS containing 4 ng/mL of IL-2 (R&D Systems) for 14 d. The cells were expanded from 2 × 107 to 3 × 108 cells. The cultured PBMCs were further stimulated for 4 h with 50 ng/mL of PMA and 1 μg/mL of ionomycin (Sigma). The 3 × 108 PBMCs were treated with cycloheximide (50 μM; Sigma) for 10 min at 37°C and lysed hypotonically by using a ball-bearing homogenizer followed by elimination of the nuclei by centrifugating the homogenate at 2000g for 2 min at 4°C. The supernatant containing cytoplasmic extract was diluted with 2.5 M sucrose until the sucrose concentration reached 2.1 M and was layered on 2.5 M sucrose. Two successive layers of sucrose solutions, one with 1.98 M sucrose and the second with 1.3 M sucrose, were then layered over the sample. After the gradients were centrifuged for 15 h at 90,000g and 4 °C, we collected the free ribosomal fraction in load zone and the microsomal fraction between the 1.98 M and 1.3 M sucrose layers.
RNA Isolation and Antisense RNA Amplification
Total RNA was isolated from the membrane and the cytoplasmic fraction by using Trizol (Invitrogen). Poly (A)+ RNA was isolated from total RNA by using oligo-dT30 (Roche Diagnostic System) according to the manufacturer's protocol. Poly (A)+ RNA fractions recovered were amplified by using a linear T7-based antisense RNA (aRNA)-amplification method. One fifth of resultant aRNA sample was electrophoresed in 1% agarose gel, and the amount of aRNA was quantitatively estimated by ethidium bromide staining. Thus, the amount of aRNA to be used for the labeling procedure was adjusted.
Preparation of Fluorescence-Labeled cDNA and Microarray Hybridization
aRNA from the cytoplasmic fraction was labeled with the fluorescent dye Cy3; aRNA from the membrane fraction was labeled with the fluorescent dye Cy5. The labeled probes were purified on Microcon 30 columns (Millipore), and 10 μg of yeast transfer RNA, 4 μg of poly (dA), and 15 μg of Cot1 human DNA as blocking reagents were added to probes and concentrated to 12 μL. Then, 2.55 μL of 20× standard saline citrate (SSC) and 0.45 μL of 10% sodium dodecyl sulfate (SDS) were added, and a final volume of 15 μL was used as probe solution for hybridization on each cDNA-spotted slide. The slides were covered with glass coverslips and fixed in a hybridization cassette (TeleChem), and hybridization was performed for 12 h at 65 °C. Glass slides were then washed in 2× SSC and 0.03% SDS for 5 min, 1× SSC for 5 min, and 0.2× SSC for 5 min.
Image Analysis
Fluorescent images of hybridized microarrays were obtained by using a ScanArray 4000 (GSI Lumonics) and analyzed by using ScanAlyze 2 software (http://rana.lbl.gov/EisenSoftware.htm).
Semiquantitative RT-PCR
Each total RNA (200 ng) was treated with DNase (Roche), and RT cDNA, corresponding to 40 ng of total RNA, was amplified by PCR. Reaction mixtures were incubated in a DNA Thermal Cycler (Perkin-Elmer) for 25–30 cycles.
Explanation of Box Plot
In each column, the genes for which Cy5/Cy3 ratios are from the 20th to 80th percentile are included in the box, and the genes for which Cy5/Cy3 ratios are from the 10th to 90th percentile are between the top and bottom bar.

Full-Length cDNA Cloning of Hs.182285 cDNA
The human thymus marathon-ready cDNA (Clontech) was used to clone the 5′ end of Hs.182285 cDNA by 5′ RACE analysis. The first PCR amplification was performed with ccatc ctaatacgactcactatagggc (adapter primer 1) and aaaggggtc aatttgctctaatgtgtc (gene-specific primer 1): 32 cycles for 20 s at 94°C and 3 min at 72°C, five cycles for 20 s at 94°C and 3 min at 70°C, five cycles for 20 s at 94°C and 3 min at 68°C × 22 cycles. The second (nested) amplification was performed with actcactatagggctcgagcggc (adapter primer 2) and tgtcaaagacata ccacttacatgacgg (gene specific primer 2): 27 cycles for 20 s at 94°C and 32 min at 72°C and five cycles for 20 s at 94°C and 3 min at 68°C × 22 cycles. PCR fragments were analyzed by agarose gel electrophoresis and sequenced.
In Vitro Translation
The protein was synthesized from isolated plasmid Hs.7718 cDNA by using the TNT in vitro transcription/translation system (Promega) in the absence or the presence of canine pancreatic microsomal membranes (Promega). Synthesized protein was detected by incorporation of [35S] methionine (Amersham) as described by the manufacturer. Proteinase K treatment of translation reactions was performed at final concentration of 50 μg/mL for 1 h on ice in the presence or absence of 0.5% Triton X-100, followed by inactivation of the protease with 3 mM phenylmethylsulfonyl fluoride. The reactions were mixed with sample buffer and loaded onto SDS-PAGE (7.5%) gels. Molecular mass standards were obtained from Bio-Rad.
Hs.182285 Gene Product–GFP Construct and Transient Transfections
Hs.182285 gene product-GFP was obtained by PCR amplification of Hs.182285 open reading frame and cloned in the pEGFP-C1 and pEGFP-N2 vector (Clontech). HEK293 cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. pEGFP-C1/N2–Hs.182285 transfection was carried out by using LipofectAMINE Plus (Invitrogen) according to the instruction of the manufacturer.
Confocal-Scanning Laser Microscopy
Transfected cells were cultured on cover glass and fixed in 4% formaldehyde in PBS for 15 min, and the membrane was labeled by DilC12 (Molecular Probe Inc.). The specimens were examined with a laser-scanning confocal microscope (Olympus).
Acknowledgments
This work was supported by CREST/SORST and Grant-in-Aid for Scientific Research on Priority Areas (C) “Medical Genome Science” from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
Article published online before print in June 2003.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.709603.
-
↵3 Corresponding author. E-MAIL koujim{at}m.u-tokyo.ac.jp; FAX 81-3-5684-2297.
-
- Accepted April 3, 2003.
- Received August 14, 2002.
- Cold Spring Harbor Laboratory Press








