
Antisense Transcripts With FANTOM2 Clone Set and Their Implications for Gene Regulation
- Hidenori Kiyosawa1,2,4,
- Itaru Yamanaka1,
- Naoki Osato1,
- Shinji Kondo1,
- RIKEN GER Group1,
- GSLMembers3,5, and
- Yoshihide Hayashizaki1,2,3,6
- 1Laboratory for Genome Exploration Research Group, RIKEN Genomic Sciences Center (GSC), RIKEN Yokohama Institute, Suehiro-cho, Tsurumi-ku, Yokohama, Kanagawa 230-0045, Japan
- 2Division of Genomic Information Resource Exploration, Science of Biological Supramolecular Systems, Yokohama City University, Graduate School of Integrated Science, Suehiro-cho, Tsurumi-ku, Yokohama 230-0045, Japan
- 3Genome Science Laboratory, RIKEN, Hirosawa, Wako, Saitama 351-0198, Japan
Abstract
We have used the FANTOM2 mouse cDNA set (60,770 clones), public mRNA data, and mouse genome sequence data to identify 2481 pairs of sense–antisense transcripts and 899 further pairs of nonantisense bidirectional transcription based upon genomic mapping. The analysis greatly expands the number of known examples of sense–antisense transcript and nonantisense bidirectional transcription pairs in mammals. The FANTOM2 cDNA set appears to contain substantially large numbers of noncoding transcripts suitable for antisense transcript analysis. The average proportion of loci encoding sense–antisense transcript and nonantisense bidirectional transcription pairs on autosomes was 15.1 and 5.4%, respectively. Those on the X chromosome were 6.3 and 4.2%, respectively. Sense–antisense transcript pairs, rather than nonantisense bidirectional transcription pairs, may be less prevalent on the X chromosome, possibly due to X chromosome inactivation. Sense and antisense transcripts tended to be isolated from the same libraries, where nonantisense bidirectional transcription pairs were not apparently coregulated. The existence of large numbers of natural antisense transcripts implies that the regulation of gene expression by antisense transcripts is more common that previously recognized. The viewer showing mapping patterns of sense–antisense transcript pairs and nonantisense bidirectional transcription pairs on the genome and other related statistical data is available on our Web site.
The level of mRNA in a eukaryotic cell, and its translation into protein, can be controlled at many levels subsequent to transcription initiation. Because mRNA is single stranded, the presence of a complementary antisense strand may alter transcription, elongation, processing, location stability, and translation. Functional antisense RNA has been identified in bacteria (review by Wagner and Simons 1994) and also implicated in gene regulation and differentiation in several eukaryotic organisms (Terryn and Rouze 2000; Elmendorf et al. 2001), including mammals (review by Dolnick 1997; Vanhee-Brossollet and Vaquero 1998). Natural antisense transcripts usually arise via separate transcription initiation from the opposite DNA strand at the same genomic locus as the sense strand.
They may be coding or noncoding RNA (ncRNA) complementary to mature processed sense coding mRNA, or they may be complementary only to the primary unprocessed transcript, being contained solely within an intron or overlapping a 5′ UTR or 3′ UTR. They may or may not be spliced. There are now many examples of functional antisense transcripts in developmental gene regulation (review by Vanhee-Brossollet and Vaquero 1998) and imprinting (Jong et al. 1999b; Mitsuya et al. 1999; Hayward and Bonthron 2000; Chamberlain and Brannan 2001; Li et al. 2002; Sleutels et al. 2002), but the number of well-characterized antisense transcripts is still small (Dolnick 1997; Vanhee-Brossollet and Vaquero 1998). In each case, the natural antisense transcript was discovered in the course of studies on the sense RNA.
A genome-wide search for possible antisense transcripts was performed recently (Lehner et al. 2002), and 87 pairs of sense–antisense transcripts that originated from the same chromosomal locus were identified. This search focused mainly on sense–antisense mRNA containing open reading frames, whereas in many known functional examples at least one partner is nonprotein coding.
Another recent search for antisense transcripts has been done on an actual transcript sampling from cDNA libraries from the protozoan parasite Giardia lamblia (Elmendorf et al. 2001). Random sampling of 100 cDNA clones from the libraries revealed 23 clones that appeared noncoding, of which three were antisense transcripts complementary to the protein-coding mRNA.
The simultaneous availability of the draft genomic sequence (http://genome-archive.cse.ucsc.edu/) and large cDNA resources in the mouse permits the first global analysis of the frequency of genuine antisense transcription in a mammal. In this study, we have searched for pairs of sense–antisense transcripts mainly based upon the FANTOM2 cDNA sequence set, a product of the Mouse Gene Encyclopedia Project in the RIKEN Genomic Sciences Center (The FANTOM Consortium and The RIKEN Genome Exploration Research Group Phase I and II Team 2002; http://fantom2.gsc.riken.go.jp/db/). This set contains 60,770 full-length enriched mouse cDNA sequences derived from the libraries of various tissues at various developmental time points. We identified more than 2000 examples of sense–antisense transcript pairs, clearly indicating the prevalence of this mechanism and its potential importance in mammals.
RESULTS
Listing Sense–Antisense Pairs of cDNA Sequences
To list all of the antisense cDNA sequences, the entire set of FANTOM2 cDNA sequences and public mRNA sequences was mapped on the mouse genome sequence draft. The set of the cDNA sequence pairs that originated from the same locus but from opposite strand were selected. These pairs were classified according to the categories depending upon the nature of the overlap. The analysis includes pairs in which the cDNA sequences do not overlap, but they derive from the same locus. We call this type of pair “nonantisense bidirectional transcription pair” (categories 3, 4, and 5 in Fig. 1) in this article. In such cases, we infer that the transcription and/or processing of one member of the pair could interfere with that of the other. We call the pair that shares sequence complementarity with each other “sense–antisense transcript pair” (categories 1 and 2 in Fig. 1). The words “bidirectional transcription pair” (categories 1, 2, 3, 4, and 5 in Fig. 1) refer to both sense–antisense transcript pair and nonantisense bidirectional transcription pair. The categories and the number of the pairs in each category are summarized in Figure 1. The histogram showing the distribution of the size of overlaps between the pairs of sense–antisense transcript is presented in Figure 2. The number of the pairs mapped on each chromosome is shown in Table 1. The mapping pattern on each chromosome is also shown in Figure 3. One striking feature, which suggests that the mapping pattern of the sense–antisense transcript pairs is nonrandom, is that far fewer pairs were mapped on the X chromosome. The fewer pairs mapped on the X chromosome were specific to sense–antisense transcript pairs, and this phenomenon was not observed in the case of nonantisense bidirectional transcription pairs.
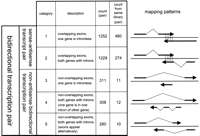
Classification of sense–antisense transcript and nonantisense bidirectional transcription pair patterns. Five categories of bidirectional transcription patterns are shown. The categories are classified according to the patterns of how the two transcripts are mapped on the genome sequences. The schematic examples from each category are shown next to each explanation. Total pair counts as well as pair counts that are from the same cDNA library sources for each category are presented.
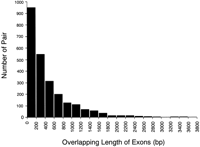
Histogram of overlapping length distribution. The distribution of sense–antisense transcript pairs for their overlapping length of exons is shown. The horizontal axis represents overlapping length (bp) of exons for each sense–antisense transcript pair. The vertical axis represents the number of pair for each bin.
Number of Bidirectional Transcription Pairs per Chromosome

Chromosome map of sense-antisense transcript and nonantisense bidirectional transcription pairs. All of the mapped positions of the bidirectional transcription pairs are schematically presented. The sense-antisense transcript and nonantisense bidirectional transcription pairs are presented in green on the left and in blue on the right, respectively. The red lines indicate the positions of the known imprinted genes. The pink areas are the regions ± 5 Mb of the known imprinted gene positions. Note that there are a few mapped positions of the sense-antisense transcripts on the X chromosome, compared with the autosomes. The names of known imprinted genes mapped in each pink region are: Wt1 (1); Nnat (2); Gnas, Gnasxl, Nespas (3); p73 (4); Sgce (5); Copg2, Mit1/Ib9 (6); Peg3/Pw1, Usp29, Zim1, Zim3, Zfp264 (7); Frat3, GABRA5, GABRB3, GABRG3, Magel2, Mkrn3/Zfp127, Ndn, Snrpn, Snurf, Ube3a (8); H19, Igf2, Igf2as, Ins2, Ipl/Tssc3, Kvlqt1, Mash2, Nap1l4, Obph1, p57KIP2/Cdkn1c, Slc22a1l, Tapa1/Cd81, Tssc4 (9); Rasgrf1 (10); Zac1 (11); Dcn (12); Meg1/Grb10 (13); U2af1-rs1 region1, U2af1-rs1 region2 (14); Dlk, Meg3/Gtl2 (15); Htr2a (16); Ata3 (17); Igf2r, Mas1, Slc22a2, Slc22a3 (18); Impact (19); Ins1 (20). Because the genes on the X chromosome are imprinted only on the paternal X chromosome in the extraembryonic tissues (reviewed by Latham 1996), we did not shade the X chromosome with pink color. Although the chromosomal locations of Ipw, Kvlqt1-as, Msuit, Peg1/Mest, and Pwcr1 are already known, these genes were not computationally mapped by BLAST program. Accordingly, these genes are not shown in Figure 3.
Overall Analysis of the Bidirectional Transcription Pairs
The actual mapping patterns of each bidirectional transcription pair on the genome sequences and accompanying data can be viewed at http://genome.gsc.riken.go.jp/m/antisense/viewer/. Examples of the data on this Web site are shown in Figure 4. The types of analyses performed are listed in the field (the most upper line) of Figure 4C. The cDNA sequence counts (Fig. 1; 2481 pairs) belonging to categories 1 and 2, the actual sense–antisense transcript pairs, are striking, considering the small number of sense–antisense gene pairs previously reported (Dolnic 1997; Vanhee-Brossollet and Vaquero 1998; Lehner et al. 2002). Many of the antisense transcripts, especially in category 2, were processed by removal of at least one intron, based upon mapping to the genome sequences. This is one supporting fact that these are genuine transcripts. To confirm that these are reproducibly transcribed, we performed a FASTA-search for these sense–antisense candidates against the EST database. At least one EST sequence hit was obtained in 2265 (category 1), 2246 (category 2), 518 (category 3), 542 (category 4), and 497 (category 5) cDNA sequences (please see our Web site for the exact number of EST support for each cDNA sequence; Fig. 4C). The histogram indicating the overall distribution of EST hits for the bidirectional transcription pairs is also presented in Figure 5.

Examples of antisense viewer. All data of the bidirectional transcription pairs can be viewed at http://genome.gsc.riken.go.jp/m/antisense/viewer/. (A) Entrance of the viewer. The data are either sorted by chromosome or category. By clicking “Annotation,” the FANTOM2 annotation of each cDNA (B) will be shown. By clicking chromosome numbers or category numbers, the pages for the description of the bidirectional transcription pairs will be shown (C). (B) Gene names by FANTOM annotation are shown for each sense–antisense cDNA pairs. (C) Data for each bidirectional transcription pair are shown. By clicking either “Image” or “Applet,” graphical mapping patterns of cDNA on the genome sequences (D) will be presented. (D) Mapping patterns of bidirectional transcription cDNA pairs on the genome. Forward and reverse directions on the chromosomes are represented in blue and red colors, respectively. The filled boxes represent the positions of exons. The black line represents GC content along the genome sequences. The yellow and green lines represent a ratio of the observed to expected CpG score, and a final CpG score, respectively (please see the Methods section for a detailed calculation of these scores). The positions of major promoter consensus sequences are shown underneath the positions of exons.
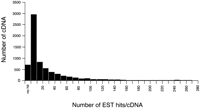
Histogram of number of EST hits distribution. The distribution of bidirectional transcription pair cDNA for the number of EST hits is shown. The horizontal axis represents the number of EST hits for each bidirectional transcription pair cDNA. The vertical axis represents the number of cDNA for each bin. We also analyzed distributions for sense–antisense transcript pairs and nonantisense bidirectional transcription pairs separately, but there was no significant difference between these distributions (data not shown).
To function in regulation of its complementary partner, a natural antisense transcript might be expected to be coexpressed, although the alternative would be that the expression is exclusive. To test these alternatives, we traced the library origin of each cDNA sequence. The FANTOM2 cDNA was fully sequenced for the representative clone after clustering with 3′ end sequences of over one million clones (phase I sequences; The RIKEN Genome Exploration Research Group Phase II Team and the FANTOM Consortium 2001; The FANTOM Consortium and The RIKEN Genome Exploration Research Group Phase I and II Team 2002). When we counted the number of the common libraries for sense and antisense cDNA, we took all the phase I sequences into account and used the library origin of phase I sequences homologous to sense–antisense sequences with at least 96% identity. The count of the common library source for each bidirectional transcription pair is shown on the Web (examples given in Fig. 4C). The lists of cDNA libraries that produced both members of bidirectional transcription pair were shown on the Web (http://genome.gsc.riken.go.jp/m/antisense/). There was no major apparent enrichment of bidirectional transcription pairs in any particular library. The numbers of the bidirectional transcription pairs expressed in the same library were 480, 274, 11, 12, and 10 pairs for the categories 1 through 5, respectively (Fig. 1). Thus, there is a significant bias that the sense–antisense transcript pairs tended to be isolated from the same library sources while nonantisense bidirectional transcription pairs did not.
We investigated CDS (protein coding sequence) in sequences of sense–antisense transcript pairs. Because we used potential CDS data of FANTOM2 collaboration (The FANTOM Consortium and The RIKEN Genome Exploration Research Group Phase I and II Team 2002), first we chose sense–antisense transcript pairs consisting of FANTOM2 sequences out of our bidirectional transcription pairs. There were 1924 sense–antisense transcript pairs that have more than 20 bp of exon overlapping region. Among them, 519 pairs had potential CDS (more than 300 bp) in both sense and antisense strands. In 1054 pairs, only one member of each pair had potential CDS (more than 300 bp). In 351 pairs, no members of each pair had potential CDS (more than 300 bp). Thus, antisense transcripts tended to be ncRNA.
The mapping pattern of sense–antisense transcript and nonantisense bidirectional transcription pairs on the genome can be viewed by clicking “image” or “applet,” given as an example at the right end of Figure 4C. The examples of the viewer is shown in Figure 4D. The 5′ and 3′ directions are represented by the blue and red colors, respectively. The green waveform indicates the position of a CpG island. Other consensus sequences found at the promoter region are also shown.
Known Sense–Antisense Transcript Pairs Found in Our Search
We searched the known sense–antisense transcript pairs in all categories of bidirectional transcription pair data. First, we chose 19 sense cDNA sequences of protein coding genes that have been reportedly accompanied by antisense transcripts, and searched for them among our bidirectional transcription pairs (see the Methods section for a list of these 19 genes). Seven (Hif1a, Mkrn2, Raf1, Ercc1, Cd3z, Thra, and Hoxa11) of the 19 genes had antisense sequences in our list of bidirectional transcription.
Sense–Antisense Transcript and Nonantisense Bidirectional Transcription Pairs Related to Known Imprinted Genes
As approximately 15% of the known imprinted genes accompany the antisense transcripts that are suspected to regulate imprinting of the sense gene (Reik and Walter 2001), we investigated the relationship between the imprinted regions on the chromosomes and the chromosomal positions of the cDNA sequences of all bidirectional transcription pairs. The positions of known imprinted genes and the chromosome regions within ±5 Mb are highlighted in Figure 3. The cDNA mapped in the pink regions (±5 Mb of known imprinted genes) is listed on the Web (http://genome.gsc.riken.go.jp/m/antisense/imprinted_genes/). There was no apparent relationship between the mapping positions of imprinted genes and the bidirectional transcription pairs. We also searched antisense sequences for the known imprinted genes in our bidirectional transcription pair sequences. Out of 58 imprinted genes (see the Methods section for the names of these 58 genes), 22 genes (Copg2, GABRA5, Gnas, Gnasxl, Htr2a, Igf2, Igf2r, Kvlqt1, Magel2, Mas1, Mit1/lb9, Ndn, Nespas, Nnat, Slc22a3, Tapa1/Cd81, U2af1-rs1 region1, U2af1-rs1 region2, Ube3a, Usp29, Zfp264, and Zim3) had bidirectional sequences.
DISCUSSION
This is the first systematic analysis of sense–antisense transcripts using a comprehensive full-length transcript sequence set. Major findings of this analysis are: (1) As many as 2481 pairs of sense–antisense transcript were identified; (2) there is a strong bias in the frequency of the mapping patterns of the sense–antisense transcript pairs (few only on the X chromosome); (3) cDNA clones of the sense–antisense transcript pairs tended to be isolated from the same cDNA library sources. These findings are made possible with the large-scale isolation of full-length cDNA from the RIKEN Mouse Gene Encyclopedia Project. (The RIKEN Genome Exploration Research Group Phase II Team and the FANTOM Consortium 2001; The FANTOM Consortium and The RIKEN Genome Exploration Research Group Phase I and II Team, 2002.) Although the mouse genome sequence draft is now available for extensive bioinformatic analyses (Mouse Genome Sequencing Consortium 2002), predicting noncoding antisense transcripts on the genome sequences by computer programs is almost impossible because virtually all the existing programs are designed to predict protein-coding regions (exons). Recently, trials to predict ncRNA were attempted using computational algorithms (Argaman et al. 2001; Carter et al. 2001; Rivas et al., 2001; Wassarman et al. 2001), but these attempts were possible only for ncRNA that shares structural similarities. The antisense RNA is expected to function through sequence complementarity to the sense strand. This type of ncRNA cannot be predicted by the methods based on RNA sequence or structural similarities. The most recent review of natural antisense transcripts listed 20 pairs of sense–antisense transcript in mammals (Vanhee-Brossollet and Vaquero 1998). The number we counted for sense–antisense transcript pair is 2481. The gene expression regulation by the natural antisense transcripts is now recognized as common in mammals (Dolnick 1997; Vanhee-Brossollet and Vaquero 1998), but this number is still much greater than previously envisaged.
Although the numbers of the cDNA sequences mapped on the X chromosome were relatively small compared to those on similar-length autosomes (see Table 1), the ratio of sense–antisense transcript pairs mapped on the X chromosome was particularly low. By contrast, the percentage of nonantisense bidirectional transcription pairs that do not generate complementary products, mapped on the X chromosome, was similar to those on other chromosomes. These differences in the numbers between sense–antisense transcript pairs and nonantisense bidirectional transcription pairs were also found in the library origins as mentioned in the Results section (Fig. 1), and indicate that there is a basic, biologic difference in the nature of the two types of transcription. Precisely, the fact that both cDNA of the sense–antisense transcript pair were isolated from the same cDNA library source does not necessarily mean that the pairs of the transcripts existed in the same single cell. Expression analyses such as RT-PCR with the RNA from a single cell, or RNA-FISH may be necessary to confirm the existence of both of the pair in a single cell. Yet the bias found in the cDNA library source differences between two types of bidirectional transcription pairs (Fig. 1) is still significant.
The small number of antisense transcripts on the X chromosome argues that the regulation of gene expression by antisense transcripts may have something to do with X chromosome inactivation. If the sense and antisense transcripts are expressed in a mono-allelic manner, each of which is expressed only from either the paternal or maternal chromosome, and if both sense and antisense transcripts are necessary for the regulation of those sense and antisense transcripts' expression, loci encoding sense–antisense transcript pairs may have been excluded on the X chromosome during evolution.
The actual biologic functions of these natural antisense transcripts in living organisms are hardly known, leaving a reasonable speculation that they form a double-stranded RNA (dsRNA) to downregulate the expression of sense RNA molecules. The dsRNA would prevent single-stranded mRNA from interacting with cellular components for normal gene expression. Alternatively, the resultant dsRNA could be a target for RNA interference (RNAi). The molecular mechanisms of RNAi began to be revealed (recent reviews by Hutvagner and Zamore 2002; Zamore 2002), and the target genes can be efficiently knocked out with dsRNA in model organisms such as worms and flies (Fire et al. 1998; Caplen et al. 2001; Elbashir et al. 2001), as well as mammalian cells (Brummelkamp et al. 2002; Paddison et al. 2002; Sui et al. 2002). However, the native biologic function and meaning of RNAi are still unknown, except for a few examples, such as RNAi-like, antiviral capability in plants (Kasschau and Carrington 1998) or degradation of Stellate mRNA by a RNAi-like mechanism in Drosophila (Aravin et al. 2001). Virtually all of the sense–antisense transcripts found in our analysis could be the target of a dsRNA-dependent RNAi mechanism, if both sense and antisense transcripts were produced in the same single cell and the concentration of the transcripts were high enough for the RNAi mechanism to proceed with its function. The abundance of such sense and antisense transcripts in living mice indicates that gene expression regulation by sense–antisense transcripts, possibly via a RNAi-like mechanism, might be fairly common.
The number of genes or transcripts on the genome has always been a hot area of scientific concern, especially in the genome era (Ewing and Green 2000; Liang et al. 2000; Roest Crollius et al. 2000). An estimation of genes from the human genome sequences is roughly between 30,000 and 40,000 (International Human Genome Sequencing Consortium 2001; Venter et al. 2001). However, these numbers are given based on the protein-coding RNA. Recently, the importance of ncRNA began to be widely accepted (review by Eddy 2001; Storz 2002). A subset of very small ncRNA exists among ncRNA, for example, in microRNA (miRNA), lin-4 (22 bp) (Lee et al. 1993) and in Caenorhabitis elegans, let-7 (21 bp) (Reinhart et al. 2000). The FANTOM cDNA set does not contain such small transcripts, but appears to contain substantial numbers of noncoding RNA (The FANTOM Consortium and The RIKEN Genome Exploration Research Group Phase I and II Team 2002), which is an excellent source for analyzing genome-wide, noncoding transcripts. The transcript number on the genome might be much higher because these noncoding RNAs also have important functions. A recent work analyzing actual transcriptional activity in the selected regions of chromosomes 21 and 22 revealed as much as an order of magnitude and more genomic sequences are being transcribed (Kapranov et al. 2002). As we presented in the Results section, the antisense transcripts are not always ncRNA but tend to be ncRNA. As one type of ncRNA, antisense transcripts may play essential roles in cellular gene expression regulation. Confirmation of the actual expression and function of these antisense transcripts will contribute to evaluation of the total number of genes on the genome.
METHODS
Mapping cDNA Sequences Onto the Mouse Genome Sequences
We used the mapping data calculated in the FANTOM2 collaboration. (The FANTOM Consortium and The RIKEN Genome Exploration Research Group Phase I and II Team 2002) In the data, the FANTOM2 cDNA sequence set (60,770 sequences), RefSeq mouse sequences (http://www.ncbi.nih.gov/RefSeq/) and GenBank mRNA sequences were attempted to map on the mouse genome sequence (MGSCv3) assembly (Mouse Genome Sequencing Consortium 2002). In this article, we refer to the region where cDNA sequences align with the genome sequences' “exon” expedient. We extracted sense–antisense transcript pairs and nonantisense bidirectional transcription pairs from the map data based on loci overlapping. We call the list of the extracted pairs as redundant version of our pair list. We eliminated redundancy with the following procedure: First, we split cDNA sequences into exon-overlapping and exon-nonoverlapping groups. In the exon-overlapping group, we made clusters based on exon overlapping. For each cluster, we chose a pair having the longest exon overlapping as a representative pair. In the exonnonoverlapping group, we made clusters based on loci overlapping. In this case, we chose a pair that has the longest loci overlapping as a representative pair for each cluster. Then we split the exon-overlapping group into two categories and the exon-nonoverlapping group into three categories. Definitions and the number of pairs of these five categories are as follows. Category 1—exons overlapping: one of the genes is intronless; 1252 pairs were found. Category 2—exons overlapping: both genes have introns; 1229 pairs were found. Category 3—no exons overlapping: one of the genes is intronless; 311 pairs were found. Category 4—no exons overlapping: both genes have introns. One of the genes is within one intron of the other gene; 308 pairs were found. Category 5—no exons overlapping: both genes have introns. Several exons of two genes appear alternately; 280 pairs were found.
Identification of CpG Islands
A CpG island is a region of vertebrate DNA with a high GC content and a high frequency of CpG. Three criteria for the CpG island are defined as follows (Gardiner-Garden and Frommer 1987):
-
“A region of DNA used for evaluation of its base content should be longer than 200 bp in length.” Our computer program with a window size of 200 bp met this condition. In Figure 4D, for all bases between 900 bp upstream of genes and 900 bp downstream, the following quantities were calculated by shifting the window one base pair per each calculation.
-
“GC content should be over 0.5.” GC content = [(number of G) + (number of C)]/(window size). In Figure 4D, a black line represents GC content.
-
“CpG score defined below should be over 0.6.” (observed CpG) = (number of CpG)/(window size)
(expected CpG) = [(number of C)/(window size)] * [(number of G)/(window size)]
(CpG score) = observed/expected = [(number of CpG) * (window size)]/[(number of C) * (number of G)].
CpG score is represented as a yellow line in Figure 4D. Because the threshold of the CpG score is 0.6, a final CpG island score is defined as the CpG score minus 0.6, and the criterion (3) is equivalent to a positive final CpG island score. In Figure 4D, the positive final CpG island score is indicated by a green line only in a region of DNA that satisfies the criterion (2). Therefore, the green line represents the existence of a CpG island in the DNA region, because the region satisfies all three criteria explained above.
Search for EST Sequences Homologous to Bidirectional Transcription Pairs
To confirm that sense–antisense candidates are derived from natural transcripts, we performed a FASTA search (Pearson and Lipman 1988) for these sense–antisense candidates against approximately one million EST sequences annotated as mouse or Mus musculus, and a 5′ end in the EST division of the GenBank database. The number of EST sequences that match a sense–antisense candidate with ≥94% identical with ≥80% overall length was counted for each candidate.
Library Origin of Bidirectional Transcription Pairs
To analyze the library origin of the sequences from bidirectional transcription, we used the library origin of not only the full-length sequences, but also the mouse end sequences highly homologous to the full-length sequences. After we masked repeat sequences in all the sequences from the redundant version of our pair list using RepeatMasker software (http://ftp.genome.washington.edu/RM/RepeatMasker.html), we searched the sequences against approximately 1.3 million 3′ end sequences using BLAST software (Altschul et al. 1990). The library origins of 3′ end sequences with ≥94% identity were added to the library origin of the matched sense or antisense sequences. Next, we counted the frequency of libraries in which both sense and antisense clones expressed simultaneously as follows. For each library, we counted the number of FANTOM clones expressed in the library and the number of sense and antisense clones expressed in the library, and then calculated the percentage of the number of the sense and antisense clones to the number of the FANTOM clones. We sorted the names of the library origins included in each category of bidirectional transcription pairs by their ratios and listed them (http://genome.gsc.riken.go.jp/m/antisense/). We also counted the numbers of sense and antisense clones expressed only in a unique library and listed them (http://genome.gsc.riken.go.jp/m/antisense/).
A List of Known Sense Transcripts With Antisense Transcripts and a Search of Antisense Transcripts
The following are known mammal cDNA sequences reported to accompany antisense transcripts selected from the recent literature and used in this study: Fgf2 (Knee et al. 1997), Hif1a (Thrash-Bingham and Tartof 1999), Tgfb2 (Coker et al. 1998), Tnfrsf17 (Hatzoglou et al. 2002), Mkrn2 (Gray et al. 2001), Hfe (Thenie et al. 2001), Klhl1 (Benzow and Koob 2002), Ucn (Shi et al. 2000), Wt1 (Moorwood et al. 1998), Tnni1 (Podlowski et al. 2002), Thra (Hastings et al. 2000), Hsp70.2 (Murashov and Wolgemuth 1996), Hoxd-3 (Bedford et al. 1995), Tyms (Dolnick 1993), CD3z (Lerner et al. 1993), N-myc (Krystal et al. 1990), Trp53 (Khochbin and Lawrence 1989), Ercc1 (van Duin et al. 1989), and Hoxa11 (Hsieh-Li et al. 1995). To identify the known sense–antisense transcripts in the bidirectional transcription pairs calculated in this article, we looked up the GenBank# or Refseq# of the known transcripts in the redundant version of our pair list. In addition, we performed a FASTA search (Pearson and Lipman 1988) for the known sense–antisense genes against bidirectional transcription pair sequences. The sequences ≥96% identical with ≥80% overall length were selected.
A List of Known Imprinted Genes and a Search for Antisense Transcripts of Known Imprinted Genes
Symbols used as known imprinted gene references are as follows; Ata3 (Mizuno et al. 2002), Copg2 (Lee et al. 2000), Dcn (Mizuno et al. 2002), Dlk (Takada et al. 2000), Frat3 (Chai J-H et al. 2001), Gabra5 (Knoll et al. 1993), Gabrb3 (Wagstaff et al. 1991), Gabrg3 (Greger et al. 1995), Gnas (Williamson et al. 1996), Gnasxl (Peters et al. 1999), H19 (Leighton et al. 1995), Htr2a (Kato et al. 1998), Igf2 (De Chiara et al. 1991), Igf2as (Moore et al. 1997), Igf2r (Barlow et al. 1991), Impact (Hagiwara et al. 1997), Ins1 (Giddings et al. 1994), Ins2 (Leighton et al. 1995), Ipl/Tssc3 (Qian et al. 1997), Ipw (Wevrick and Francke 1997), Kvlqt1 (Gould and Pfeifer 1998), Kvlqt1-as (Smilinich et al. 1999), Magel2 (Boccaccio et al. 1999), Mas1 (Villar and Pedersen 1994), Mash2 (Guillemot et al. 1995), Meg1/Grb10 (Miyoshi et al. 1998), Meg3/Gtl2 (Miyoshi et al. 2000), Mit1/lb9 (Lee et al. 2000), Mkrn3/Zfp127 (Jong et al. 1999a), Msuit (Onyango et al. 2000), Nap1l4 (Paulsen et al. 1998), Ndn (MacDonald and Wevrick 1997), Nespas (Wroe et al. 2000), Nnat (Kagitani et al. 1997), Obph1 (Engemann et al. 2000), p57KIP2/Cdkn1c (Hatada and Mukai 1995), p73 (Kaghad et al. 1997), Peg1/Mest (Kaneko-Ishino et al. 1995), Peg3/Pw1 (Kaneko-Ishino et al. 1995), Pwcr1 (de Los Santos et al. 2000), Rasgrf1 (Plass et al. 1996), Sgce (Piras et al. 2000), Slc22a1l (Cooper et al. 1998), Slc22a2 (Zwart et al. 2001), Slc22a3 (Zwart et al. 2001), Snrpn (Leff et al. 1992), Snurf (Gray et al. 1999), Tapa1/Cd81 (Andria et al. 1991), Tssc4 (Paulsen et al. 2000), U2af1-rs1 region1 (Nabetani et al. 1997), U2af1-rs1 region2 (Nabetani et al. 1997), Ube3a (Albrecht et al. 1997), Usp29 (Kim et al. 2000), Wt1 (Rainier et al. 1993), Zac1 (Piras et al. 2000), Zfp264 (Kim et al. 2001), Zim1 (Kim et al. 1999), and Zim3 (Kim et al. 2001). These genes were chosen based on the data at http://www.mgu.har.mrc.ac.uk/imprinting/imprinting.html and http://www.geneimprint.com/. To search for antisense transcripts of known imprinted genes, we looked up the GenBank# or Refseq# of the known imprinted genes in the redundant version of our pair list. In addition, we performed a FASTA search (Pearson and Lipman 1988) for the known imprinted genes against bidirectional transcription pair sequences. The sequences ≥96% identical over ≥80% overall length were selected.
Acknowledgments
We thank A. Hasegawa for Web interface. This study was supported by a Research Grant for the RIKEN Genome Exploration Research Project from the Ministry of Education, Culture, Sports, Science and Technology of the Japanese Government to Y.H.
Footnotes
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.982903.
-
↵4 Present address: RIKEN Tsukuba Institute, BioResource Center (BRC), Tsukuba, Ibaraki, 305-0074, Japan.
-
↵5 Takahiro Arakawa, Piero Carninci, and Jun Kawai.
-
↵6 Corresponding author. E-MAIL yosihide{at}gsc.riken.go.jp; FAX 45-5039216.
-
- Accepted February 25, 2003.
- Received December 3, 2002.
- Cold Spring Harbor Laboratory Press








