
Genome Function and Nuclear Architecture: From Gene Expression to Nanoscience
- Timothy P. O'Brien1,9,
- Carol J. Bult1,
- Christoph Cremer2,
- Michael Grunze2,
- Barbara B. Knowles1,
- Jörg Langowski3,
- James McNally4,
- Thoru Pederson5,
- Joan C. Politz5,
- Ana Pombo6,
- Günter Schmahl7,
- Joachim P. Spatz2, and
- Roel van Driel8
- 1 The Jackson Laboratory, Bar Harbor, Maine 04609, USA
- 2 University of Heidelberg, D-69117 Heidelberg, Germany
- 3 German Cancer Research Center, 69120 Heidelberg, Germany
- 4 National Cancer Institute, Bethesda, Maryland 20892, USA
- 5 University of Massachusetts Medical School, Worcester, Massachusetts 01605, USA
- 6 MRC Clinical Sciences Centre, London W12 ONN, UK
- 7 University of Goettingen, 37070 Goettingen, Germany
- 8 University of Amsterdam, 1018 TV Amsterdam, The Netherlands
Abstract
Biophysical, chemical, and nanoscience approaches to the study of nuclear structure and activity have been developing recently and hold considerable promise. A selection of fundamental problems in genome organization and function are reviewed and discussed in the context of these new perspectives and approaches. Advancing these concepts will require coordinated networks of physicists, chemists, and materials scientists collaborating with cell, developmental, and genome biologists.
The Human Genome Project has laid the foundation for a wide variety of new studies and initiatives (Lander et al. 2001; Venter et al. 2001). The recent release of an annotated draft assembly of the mouse genome has added a profound evolutionary window as well as an experimental basis for enhancing the dissection of mammalian gene function (Mouse Genome Sequencing Consortium 2002). Despite the ongoing successes associated with the analysis of the human, mouse, and other model organism genomes, our understanding of the functions of genomic sequences is still quite limited. Progress depends on the classification of each gene product and an appreciation of gene activities as networks of genetic and biophysical interactions. Additional insights will be afforded by determining how physiological, biochemical, and genomic regulatory networks function as an integrated system to direct biological activities (Hartwell et al. 1999; Tyson et al. 2001; Davidson et al. 2002). A key aspect of understanding the systematic output of genetic information is the realization that the genome is spatially organized within the nucleus, and that this organization represents a critical dimension of genome function.
Describing genome organization in the nucleus as a function of cell type or physiology poses profound scientific challenges that require a combination of experimental and theoretical approaches. To convey this interdisciplinary perspective, we provide an overview on recent progress in nuclear organization and genomics research. There have been numerous focused reviews addressing these topics, and these are cited throughout the text. Specific concepts are advanced to discuss how molecular, cell biological, genetic, and computational biology approaches can be extended by the disciplines of physics, chemical biology, materials science, and nanoscience to relate genome organization and nuclear architecture with development and disease.
Integrating Genome Organization With Nuclear Structure
The contents of the nucleus are segregated into functional compartments (Pombo et al. 2000; Dundr and Misteli 2001; Spector 2001; Pederson 2002). These include the nucleolus, splicing-factor compartments (interchromatin granule clusters), Cajal bodies, promyelocytic leukemia bodies, replication and transcription factories, and a growing list of assemblies that await further analysis (Fig. 1). These compartments contain populations of molecules that are potentially being stored, cleared, recycled, and moved or are engaged in active processes, such as transcription and chromosome maintenance (Lewis and Tollervey 2000). The assembly of these compartments is closely connected to their role in processing the genetic information contained in the genomic sequence.
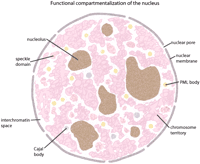
Major components of the nucleus are shown as indicated. For simplicity, only a few prominent nuclear assemblies are shown, although a number of others have been identified.
Chromosomes and Genomic Sequences Form Nuclear Compartments
Understanding relationships between genome organization and nuclear structure and activity will require linking the assembly and maintenance of various compartments with defined genomic sequences. The nucleolus provides a compelling example, in which sequence and function intersect in the organization of a nuclear substructure. In the nucleolus, the machineries needed for ribosome assembly are associated with the ribosomal RNA genes, the nucleolar organizers. In human cells, the nucleolar organizer regions are located on five chromosomes and each contain ∼80 copies of a ∼43-Kb ribosomal RNA gene tandem repeat (Sullivan et al. 2001). Thus, in diploid cells, multiple genomic regions sharing a common feature, ∼3 Mb of rDNA gene sequences, serve as the nucleation point for the self-organization of the most prominent compartment within the nucleus.
It is now well recognized that chromosomes form distinct substructures arranged in defined positions in the nuclear volume during cell cycle progression, including discernable chromosome territories in the interphase nucleus (Cremer and Cremer 2001). These territories and subcompartments within and between them are potentially arranged in activity-dependent and cell type-specific positions (Parada and Misteli 2002; Tanabe et al. 2002). The differential arrangement of transcriptionally favorable versus less permissive chromatin domains suggests that sequence composition is connected to the formation of nuclear compartments that correlate with the expression profile of specific cell types. There are many indications that the transcription level of a chromosomal region influences its organization within the nucleus. Transcriptionally active loci loop out from compact chromatin domains (Cmarko et al. 1999; Verschure et al. 1999), sometimes up to distances in the submicrometer range (Volpi et al. 2000; Mahy et al. 2002). Intranuclear position is linked to gene activity. For instance, there is a close relationship between the association of loci with pericentromeric heterochromatin and the cell differentiation-dependent epigenetic silencing of genes (Fisher 2002). A consideration of the possibilities that intranuclear position determines transcriptional activity or that transcriptional activity determines intranuclear position illustrates the need for advancing approaches that will provide a greater appreciation of the potential interdependence between position and activity.
Interestingly, genes are not distributed randomly on chromosomes. Loci that are transcribed at high rates, including many housekeeping genes, form large clusters on several chromosomes separated by domains that contain genes that are expressed at relatively low levels (Caron et al. 2001; Lercher et al. 2002). It is likely that this clustered arrangement of genes on the linear chromosome has consequences for the spatial arrangement of chromatin in the interphase nucleus.
Nuclear Components and Structures Are Highly Dynamic
The assembly of architecturally stable macromolecular complexes, domains, and compartments within the nucleus requires multiple levels of interactions involving DNA and a variety of molecules. However, these assemblies are comprised of dynamic components (Misteli 2001b), in which associations between proteins, and between proteins and DNA, at times display a remarkably high rate of exchange (Fig. 2). One important dimension of these dynamic associations is functional coupling, in which some molecules remain and others are exchanged as the assemblies perform sequential functions. Thus, an important consideration is the fraction of each molecular species that is distributed among different compartments and the extent of functional coupling between processes, macromolecular structures, and compartments (e.g., Maniatis and Reed 2002).
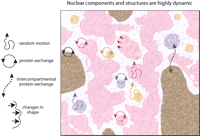
Most nuclear structures exhibit random, constrained motion. In some cases (e.g., PML or Cajal bodies) instances of energy-dependent or directed motion have been observed. Compartments may also expand to move toward interactive sites (e.g., speckles advance toward a chromosome territory) as they change shape, and thus, relative position in the nucleus. Proteins found in specific compartments typically reside only on the order of seconds to minutes within that compartment. After release, proteins migrate through the interchromatin space and associate with the same type of compartment elsewhere. However, some proteins also exhibit exchange between different types of compartments (dotted purple arrow).
In addition to exchange of molecules within or between compartments, the compartments themselves can move within the nucleus. Chromosomal domains display a degree of mobility. These movements are associated with changes in chromatin organization that are potentially linked with activity. For instance, transcriptional activation of genes is preceded by a considerable decondensation of chromatin (Tumbar et al. 1999; Volpi et al. 2000; Muller et al. 2001). Interphase chromatin has also been shown to be dynamic, showing a topologically constrained type of diffusion, allowing chromatin domains to move more or less randomly over distances of a few tenths of a micrometer (Gasser 2002). Interactions between chromosomes and nuclear structures are proposed to impose local and long-range constraints on motion (Marshall et al. 1997; Vazquez et al. 2001). Interestingly, chromatin in heterochromatin domains appears to move less than euchromatic chromatin (Chubb et al. 2002). This dynamic behavior of chromatin is likely to be important for the regulation of gene expression. It would allow chromatin domains that lie far apart in the linear DNA to interact. For example, such physical contacts over distances of many kilo-bases are a key element in the regulation of β-globin gene expression by the locus control region (Carter et al. 2002).
Mobility of compartments may reflect transient interactions with chromosomal sites. For example, a recent study of Cajal body movements throughout the nucleus showed an intermittent, ATP-dependent binding to chromatin that revealed itself as a paradoxical increase in mobility when ATP production was inhibited (Platani et al. 2002). The emerging picture is that various complexes within the nucleus are undergoing constant assembly and disassembly, as well as non-directed movement versus possibly active transport, interspersed with transient fixed locations.
The dynamic environment also suggests that self-organization plays a significant role in establishing nuclear architecture. Thus, steady-state dynamics influence the assembly of supramolecular structures whose components are in constant flux, owing to functional interactions both within the assemblies and with other macromolecules (Pederson 2002). Such an environment offers the level of plasticity needed for a complex biological system to be responsive to environmental and developmental stimuli, creating the opportunity to modulate and shift the balance of components and the dimension of the interactions, resulting in structural transitions that correlate with dynamic states of nuclear activity (Misteli 2001a).
Gene Expression and Organizing an Active Genome Within the Nucleus
Genetic information, including genes, regulatory sequences, and structural elements is arranged along chromosomes that are packaged within the nucleus. The nucleus has evolved to manage this information to achieve the differential gene expression profiles associated with the function of unique cell types. A complete appreciation of the global regulation of gene expression will depend on defining relationships between the linear content of each chromosome, chromatin structure, and the functional organization of chromosomes in the nucleus.
Hierarchical Organization of the Genome
Complex structures, such as folded proteins or chromatin, are often described in terms of ordered levels of organization (Woodcock and Dimitrov 2001; Horn and Peterson 2002). Discriminating between levels of complexity is useful for conceptually and experimentally framing the connection between genomic sequences and the functional organization of the genome. The nucleotide sequence represents the first level. At this level, DNA can be defined as an encrypted polymer containing signals that control the properties of its assembled form beyond that of a uniform fiber. Chromosome conformation is likely to be shaped by the spatial arrangement in the linear sequence of the genes, and regulatory and structural features needed to prepare the chromosome for processes such as transcription, replication, and segregation during the cell cycle. These features include centromeric, telomeric, and repetitive DNA as well as promoter proximal elements that control the activity of individual genes and locus control regions, and insulators that regulate the activity of large chromosomal domains. Patterns emerging from the analysis of genomic regions include long stretches of gene-poor sequence and the arrangement of genes and modular regulatory elements as clusters that are cooperatively positioned along the chromosomes. Genomic pattern analysis is at an early stage and has already revealed interesting results. For example, a recent study of the transcriptional activity across human chromosomes 21 and 22 revealed a surprising extent of expression associated with genomic sequences beyond those features annotated as predicted genes and exons (Kapranov et al. 2002). Thus, even at the primary sequence level, there is still much to learn about the relationships between sequence content and activity.
A second level of organization encompasses the direct interactions between DNA and molecules in the nucleus. DNA sequences show time-dependent associations with a variety of molecules. These associations are defined by patterns contained within the genomic sequence and epigenetic covalent modifications of chromatin-bound proteins, the concentrations and binding affinities of various molecules in the nucleus, and the physical properties intrinsic to the nuclear environment that govern their interactions. The molecules involved range from proteins that coat or package DNA to sequence-specific nucleic acid binding proteins. The epigenetic modifications of chromatin proteins include methylation, acetylation, phosphorylation, and ubiquitination of histone tails, all of which have an impact on transcription-factor accessibility, and hence, on gene expression. These modifications are proposed to constitute a histone code that influences the long-term and short-term regulation of transcriptional activity (Berger and Felsenfeld 2001; Schreiber and Bernstein 2002; Turner 2002). In addition to molecules that package DNA, there are also sets of proteins that interact with specific sequences to control gene expression. The dissection of insulator and boundary regulatory elements in chromatin provides a specific example of the importance of these interactions and how the arrangement of genomic features contributes to the functional design of a locus (Bell et al. 2001).
The third level of organization takes into account the three-dimensional topology of the genome within the cell nucleus. It is considered to involve, among other factors, the tethering of specific sites in the chromosomes to regions of the nuclear envelope (Cremer and Cremer 2001), the protein-facilitated juxtapositioning of DNA regions within a chromosome to form a stable bridge between distant sequences from which loops project (Woodcock and Dimitrov 2001), and the possible interaction of certain DNA regions with presently ill-defined nonchromatin elements of intranuclear structure (Pederson 2002). Recently, there has been increasing evidence for both actin and myosin within the nucleus (Pederson and Aebi 2002), adding another dimension to the unsolved problem of intranuclear structure. Although less well understood than the aforementioned primary and secondary levels, this third level can be thought of as the culmination of the hierarchy of interactions that functionally link the linear genomic sequence and the way it is folded and compartmentalized inside the cell nucleus (Fig. 3).
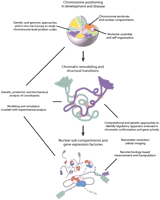
The information contained in the genomic DNA sequence is packaged within the nucleus. Multilevel hierarchical interactions are theorized to modulate the spatial arrangement and packaging of DNA in order to decode and process this information. These multilevel interactions are envisioned to exist on several scales to influence the relative position of chromosomes and the formation of the variety of supramolecular assemblies associated with dynamic nuclear compartments and chromatin domains. (Top) Individual chromosomes represented as different colored domains occupy definable territories within the interphase nuclear volume. Organizational principles that influence the location and potential nonrandom positioning of chromosomes relative to each other in the interphase nucleus are proposed to reflect an interplay between chromosome composition and sequence-based, protein-mediated associations with the nuclear envelope, nuclear pore complexes, and other chromosomes, compartments, and factors in the nuclear milieu. In the chromosome territory, DNA is likely to be organized in a variety of condensed and decondensed structural conformations. (Middle) Chromatin remodeling and scaffolding complexes (light red ovals) are proposed to generate domains within and between chromosome territories that contain extended chromatin fibers (green and blue loops). The extension of the chromatin fiber potentially provides an opportunity for further interactions between nuclear factors and loci leading to the assembly of various nuclear compartments. For example, positioning a gene cluster away from heterochromatin may favor the formation of a gene expression factory in which conditions are more permissive for RNA production and processing. Further remodeling of the chromatin provides transcriptional accessory proteins with increased access to the regulatory apparatus for individual or sets of genes. (Bottom) Transcriptional accessory proteins (red and purple clusters) interact with regulatory elements and the basal transcriptional machinery (light blue oval) to promote or repress the expression of a gene. Global as well as local modulations in chromosome organization and the packaging of DNA are needed to achieve cell-type specific profiles of gene expression.
The Spatial Organization of the Genome in the Nucleus Controls Gene Expression
There is mounting evidence that the position of a gene within a chromosome and within the three-dimensional space of the nucleus influences its activity (Fig. 4). Additionally, there is increasing recognition that the regulatory sequences that control expression also affect the spatial position of a gene and its association with transcriptionally inactive or permissive chromatin domains (Francastel et al. 2000). Thus, local and higher-order interactions that result in the differential arrangement of genes in the nucleus have the potential to make a substantial contribution to the establishment of global patterns of cell type-specific gene expression.
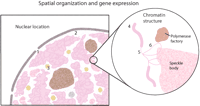
Two levels of spatial organization may affect gene expression. In the first level (left), the association of gene loci with a nuclear pore (1), the nuclear periphery (2), or with specific nuclear bodies (3) could affect gene expression. At the second level (right), the compaction and organization of chromatin may influence gene activity. Decondensation from a thicker (4) to a thinner fiber (5) could affect transcription. In addition, outlooping (6) of the chromatin fiber may occur and also be involved in gene expression. Speckle domains and polymerase factories are associated with these active regions.
A connection between the intranuclear position of a gene and its expression supports the notion that fundamental processes, such as cellular differentiation during embryonic development, are dependent on the regulation of dynamic changes in nuclear architecture. This idea also underscores the importance of maintaining a stable genomic architecture in order to sustain the differentiated state and normal function of the cell. For example, many cancers are characterized by translocations, duplications, or deletions that disrupt gene function or chromosome structure leading to overall genomic instability (Chakraborty et al. 2001), and such rearrangements also typify a number of other human diseases (Stankiewicz and Lupski 2002). It has often been assumed that diseases arising from gene rearrangements are a direct consequence of the locally altered DNA sequence per se and this is certainly true in a number of clearly demonstrated cases. However, it is also probable that by altering the longer-range organization of a chromosomal region within the nucleus, and thus changing its interactivity with other chromosomes or factors, such rearrangements, can, at least in some cases, influence the expression of genes in trans some distance away. This puts into perspective the importance of understanding how genetic perturbations lead to the global reorganization of nuclear architecture, chromatin structure, and widespread changes in gene expression. Such additional insights will be essential for elucidating the molecular and genomic basis of development and disease.
To understand genome architecture, it will be critical to identify sequence features that are connected to chromosome organization and the regulation of gene expression, such as insulators (Burgess-Beusse et al. 2002)and promoter-proximal tethering elements (Calhoun et al. 2002). Comparison between human and mouse genomic sequences reveals thousands of conserved noncoding sequence blocks that potentially perform biological functions. These conserved blocks conceivably contain clusters of novel regulatory elements that control networks of gene expression. These sequence elements need to be classified and their spatial arrangement carefully mapped to search for patterns that underlie the functional composition and organization of chromosomes. Such patterns could direct the formation and maintenance of specific chromosomal conformations associated with different levels of gene expression. However, when one attempts to relate our current knowledge of gene expression on the basis of models of gene regulatory networks to the three-dimensional organization of the genome in the nucleus, it is clear that we presently lack a way to conceptualize, much less confirm, such a correspondence map. A poignant demonstration of this epistemological conundrum can be immediately grasped by inspection of a systems biology-type diagram of a set of regulatory interactions involved in establishing the expression state of a relatively small number of key genes that play a role in endomesodermal specification during the early development of the sea urchin embryo (Fig. 5; see Davidson et al. 2002). Thus, a long-term goal that will require an interdisciplinary approach is to acquire sufficient information to be able to overlay a systems representation of gene regulation and cell-state specification onto a nanometer scale topography map of nuclear organization.
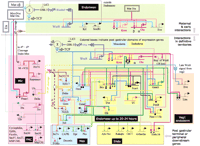
Hierarchical network of gene regulation involved in the specification of endoderm and mesoderm during early sea urchin development (Reprinted with permission from Davidson et al. 2002. Copyright 2002 American Association for the Advancement of Science.). Beginning with the detailed analysis of the cis-regulatory elements flanking a single gene involved in this process, endo16, as well as various trans-acting factors involved in the regulation of endo16, a network of genes and their levels of mRNA expression were studied. Regulatory interactions were determined by antisense oligo inhibition, injection of mRNAs into the embryo, and other perturbation methods. The network depicted in the diagram is based on modeling the various empirical parameters, including the experimental effects in embryos, using a computational environment on the basis of principles used to model mixed analog-digital circuits in the microelectronics industry. For details, see Brown et al. (2002). This envisioned network is but one example of how systems biology is conceptually impacting the field of gene expression. This figure also conveys how challenging it would be at the present time to relate the depicted gene regulatory circuits in relation to nuclear organization.
Multidisciplinary Approaches and Emerging Technologies to Address Genome Organization and Function
Studies in genetics, genomics, and cell biology have provided a foundation from which to build our understanding of genome biology. Extending this knowledge will require merging these approaches with additional disciplines and new technologies (see Fig. 3). Interactions between components need to be appreciated according to fundamental principles of physics and chemistry. High-resolution methods for obtaining quantitative measurements will be necessary to define relationships. The integration of this information will rely on computational tools and modeling to reveal how the nucleus and DNA work together on behalf of biological systems.
Assigning Functions to Genes and Additional Sequence Features in the Genome
Genetic studies have allowed genes to be identified, placed in interactive pathways, and organized into linkage groups along chromosomes, yielding an early view of chromosome content, organization, and gene function. Mutations, whether spontaneous, induced, or engineered are at the core of these studies. Classical and advanced approaches to generate point mutations, small deletions, or large chromosomal alterations can be extended to assign function to both coding and noncoding sequences across the genome. Gene-targeting technologies can be used to precisely modify or remove specific sequences, and have evolved to permit engineering large chromosomal rearrangements (Mills and Bradley 2001). Finally, mutagenesis and genetic screens can be used to survey large numbers of genomes under specific conditions in order to isolate mutations associated with a desired phenotype or cellular response. These approaches will permit functional classification of genes and sequence elements positioned across the genome (Nadeau et al. 2001).
Efforts in genomic analysis have generated numerous resources, from sequence-based markers for genetic mapping to EST libraries, large insert vectors (YACs and BACs), and clone-based physical maps. BAC clones arrayed across the genome can serve as FISH probes for spectral karyotyping and chromosomal localization studies (Liyanage et al. 1996). YAC and BAC clones have also been used as large insert transgenes (from ∼100 kb to >1 Mb), broadening our understanding of the regulatory apparatus acting on a gene. Improved techniques in clone manipulation promise to create increased flexibility for the use of BACs as transgenes or targeting vectors (Copeland et al. 2001).
Computational and comparative sequence analysis using genomic data from multiple species will continue to identify large sets of conserved sequences that will warrant classification and functional analysis (Hardison 2000; Meisler 2001). Advanced approaches in sequence annotation will be needed to diagram a locus, defining gene exons, local and distant regulatory elements, and structural features that contribute to its functional composition. These studies must be extended to describe sequence elements responsible for interactions between linked as well as unlinked loci to provide insight into how the organization within chromosomes and interactions between chromosomes influence the functional architecture of the nucleus.
Understanding the structural elements of a chromosome, such as the centromere, telomere, and replication origins, represents another important goal. These studies will lead to improved technologies for the construction of artificial chromosomes (Willard 2000). This opens possibilities for experimental analysis through the construction of designer chromosomes, for example, that are built around specific sequence elements predicted to be involved in chromosome behavior or regulation of gene expression. Such tools will enhance our ability to explore spatially dependent interactions between genomic sequence features on the same chromosome, as well as study how sequence composition influences interactions between chromosomes in the nuclear environment.
Studying Nuclear Compartments and Dynamics
Nuclear compartments and their molecular composition have been of major interest in the study of nuclear function (Spector 2001). Approaches in cell biology, structural biology, and proteomics will advance efforts to define the entire complement of molecules in the nucleus and provide classifications on the basis of their potential for functional interactions. A small number of nuclear bodies has been biochemically isolated and characterized for their protein content using mass spectroscopy analysis; these include spliceosomes (Neubauer et al. 1998; Zhou et al. 2002), interchromatin granule clusters (Mintz et al. 1999), and nucleoli (Andersen et al. 2002; Scherl et al. 2002). The results provide a first step toward a comprehensive appreciation of their biological function. These proteomics approaches, which are rapidly undergoing improvements in sensitivity and throughput, will have to be extended to other nuclear components and the biochemical composition data integrated with in situ studies.
Using a range of techniques, DNA, RNA, and protein have been localized in fixed samples after indirect immunolabeling or alternative detection, or in vivo using GFP or other fluorescent tags. Technical developments are needed in these areas to allow single molecule detection at the highest resolution, extending what can currently be obtained using the electron microscope (Weiss 1999). Smaller probes, such as aptamers, must be developed. The goal for in situ studies is to achieve a resolution in the dimension range of ∼10 nm, the size of the nucleosome.
Recent developments have allowed the in vivo visualization of gene activity primarily using ectopically expressed multicopy transgenes or tandemly repeated genes integrated at particular chromosomal sites (Tsukamoto et al. 2000; Muller et al. 2001). Due to current limitations of sensitivity and resolution, it is still difficult to visualize the expression of a single gene in its chromosomal context in mammalian cells. There have been encouraging initial reports on single protein tracking in the nucleus (Goulian and Simon 2000; Kues et al. 2001). This is an area in which investments in further developments are likely to be highly rewarding. High resolution, low-light level microscopic techniques discussed below must be improved to allow identification of single components.
In vivo results from fluorescence recovery after photo-bleaching (FRAP)and fluorescence correlation spectroscopy indicate that the protein and RNA molecules studied to date are highly mobile relative to the more limited movements associated with chromosomes (Politz and Pederson 2000; Misteli 2001b; Pederson 2001a,b; Vazquez et al. 2001; Chubb et al. 2002; Hediger et al. 2002). However, it is not well understood how a pool of mobile molecules and various chromosome configurations may give rise to relatively stable nuclear compartments. New approaches must be developed to address the problem concerning the mass transport properties of the nuclear interior. For example, how are disassembled components recycled, perhaps by local tethering or alternatively by complete release followed by diffusion-limited rebinding. We are very far from knowing basic parameters that are key to understanding nuclear dynamics and genome biology, such as the specific intranuclear concentrations of various components of a complex, their various on- and off-rates, and the fluid viscosity of the nuclear region in question.
Structural Analysis at the Molecular Level and Below
Detailed knowledge about biochemical processes occurring locally in the nucleus is a prerequisite for a comprehensive understanding of genome function. The nucleus is comprised of a complex ionic aqueous mixture of small molecules and macromolecules interacting via electrostatic, van der Waals, and hydrophobic and hydrophilic interactions, and forces due to macromolecular crowding and supramolecular organization. A comprehensive and predictive description of these interactions must build upon describing selected interactions between molecules or polymers. This will require knowledge of the chemical composition and properties of the intranuclear solvent phase, as well as the kinetics and structural tethering or freedom of the reactants. The transient nature of associations between molecules and how this influences the stability and flexibility of complexes and nuclear compartments must be described. It will be important to understand how structural transitions result from alterations in the balance and distribution of nuclear components or changes in intranuclear solvent properties. The thermodynamic and kinetic properties of component interactions must be known in order to define how these affect the collective behavior of the system. These structural transitions should be correlated with changes in patterns of gene expression that accompany both the global and local spatial reorganization of chromosomes and chromatin domains.
Living, fixed, or cryogenic biological specimens can be studied using x-ray microscopes. Vitrification or very short exposure times are needed to avoid cytological changes in living specimens caused by lethal-dose exposure during high-resolution imaging. Exposure times considerably less than 1 sec can be obtained with the new x-ray microscope, BESSY II, in Berlin. In the future, the most elegant approach to preserve the structure of living cells will be to take images within one micropulse of a free-electron laser.
The combination of a resolution range of a few nanometers, high penetrating power, analytical sensitivity, and compatibility with wet specimens allows x-ray microscopy and scanning x-ray microscopy of whole cells (Schmahl et al. 1980; Kirz et al. 1995). Applications include x-ray microscopic visualization of specific proteins (Vogt et al. 2000)and x-ray microscopic tomography of whole cells (Weiss et al. 2000) using cryo x-ray microscopy (Schneider 1998). The spatial resolution of 20 nm reached up to now is much better than that of current conventional light microscopes. In addition, x-ray microscopy and spectroscopy have already been successfully and routinely applied in environmental research, colloid physics, and materials science for morphological studies, as well as to study the elemental and molecular composition of interfaces and bulk materials.
Spectroscopic techniques can be applied to determine the distribution of atomic elements and low molecular weight components. Similar methods have reported on the nanoscale organization of structures comprised of nucleic acids and proteins on the bais of the phosphorous and nitrogen emission spectra (Hendzel et al. 1999). Elemental and molecular specificity and high spatial resolution can be achieved by soft x-ray absorption in combination with x-ray emission spectroscopy, using synchrotron radiation, which provides tunable x-rays of much higher brilliance than conventional x-ray sources. With the advance of sensitive x-ray detectors and improved spectrometers, it appears feasible that x-ray emission spectroscopy will be applied to study biological material in vivo.
Light Microscopy In Vivo at Nanometer Resolution
Cell type-specific profiles of chromosome position and the higher order organization of chromatin are likely to be linked to cell function. However, the relative positioning of chromosome territories and subchromosomal domains within and between these territories is poorly understood. Moreover, little is known about the potential relationships between the three-dimensional spatial organization of the chromosome territory and the sequence composition of the chromosome or the condensation status of a genomic segment in relation to gene activity. Already, there are exciting developments for methods to monitor the expression of sets of genes in the nucleus providing temporal and spatial resolution (Levsky et al. 2002). A long-term goal is to develop a positional code that defines the three-dimensional location of loci within the nucleus in relation to their activity. This will permit the construction of conformation maps of the genome within the nucleus as a function of discrete morphogenetic states during development and physiological states in adult cell lineages and terminally differentiated cells. In parallel, positional genomic information can be correlated with pathological states. It will be important to appreciate possible relationships between the repositioning of the genome and the onset of genome instability, for example, in the context of tumor progression.
To further our understanding of chromosome biology and nuclear function, it is essential to develop techniques that allow the measurement of structures inside the living cell with a spatial resolution down to the scale of ∼10 nm. This is becoming possible by use of new far-field light optical approaches, such as (1)4Pi microscopy (Egner et al. 2002), (2) point-spread-function (PSF)engineering by stimulated emission depletion (STED)(Klar et al. 2000), and (3)spectral precision distance microscopy (SPDM)and related methods (Esa et al. 2000; Lacoste et al. 2000). Presently, these approaches provide an optical/topological resolution down to the 30 to 100-nm range. An even better topological resolution limit and precision range, providing relative positions and distances, is anticipated for newly developed methods of spatially modulated illumination (SMI)far-field light microscopy. Experimental distance measurements in the direction of the optical axis down to a few nanometers, with a precision in the 1-nm range, have already been realized (Albrecht et al. 2002). Furthermore, SMI nanosizing approaches allow measurement of the diameter of fluorescent targets down to a few tens of nanometers. Improvements in light microscopy using these new approaches hold the promise of meeting the need to detect molecular complexes and components.
Nanotechnology and the Living Cell
Nanotechnology holds great promise for the analysis of complex processes inside living cells. It is anticipated to provide new tools to study the responses of different naturally occurring and genetically altered cell types and extend the approaches for monitoring cell behavior and activity in embryos, differentiated tissues, and organs as well as physiological systems (Bruchez Jr. et al. 1998). In addition to biological sensors that will be able to measure single molecule behavior, nanodevices are presently being developed that can be used to relocate various components inside the cell nucleus. This will allow different regions in the nucleus to be probed and manipulated to study various processes, such as their permissiveness for transcription. This will likely open direct approaches for investigating structure-function relationships by perturbing the local organization of the genome and determining its effect on function. A most promising method for nanomanipulation in living cells is the use of magnetic nanoparticles that are microinjected into the nucleus of living cells (Bausch et al. 1999). Such particles can be functionalized by the covalent attachment of selected molecules, for example, specific proteins (Abdelghani-Jacquin et al. 2001). Recently developed magnetic tweezers, in combination with high-resolution microscopy, would allow one to move such nanoparticles at will inside living cells, thereby changing local genome structure. Nanoprobes in the nucleus could be used to monitor changes in chromosome arrangement associated with changes in gene expression.
Cell-cell and cell-substrate interactions are important elements in the control of cellular function. Precisely engineered nanostructured substrates or three-dimensional environments for cells and cell assemblies are expected to become an important research tool (Chen et al. 1997; Kantlehner et al. 1999; Maheshwari et al. 2000; Spatz et al. 2000). These can be used to apply defined mechanical or chemical stimuli to cells and to study the biochemical, physical, and structural changes that occur in the nucleus using microscopic methods and integrated biosensors (Kao et al. 1999).
Quantitative Modeling of Highly Complex Biological Systems
Theoretical approaches to analyze complex datasets will be required to correlate dynamic genome architecture with the functional status of the cell in relation to development and disease. A validation that a more complete understanding of genome function in relation to nuclear structure has been attained would be the development of integrative and highly predictive models. Such models will rely on our understanding of fundamental principles of genome structure and function at the molecular level. To reach this point, it will be essential to integrate biological processes and interactions with observations at the molecular level and the mesoscopic level of organization (Laughlin et al. 2000). Relationships between nuclear architecture and activity will need to be described on several levels, from the self-assembly of nanoscale structures, to the self-organization of macromolecular complexes and nuclear compartments. Three-dimensional organization and structure need to be defined precisely, yet also account for the dynamics of nuclear components. It will also be critical to conceive and build models that incorporate the inherent plasticity of genomic structure-function relationships with careful attention as to which structural parameters are critical for function.
To understand molecular interactions inside the nucleus, requires an account of the complex liquid and tethered reactant environment, which is likely to affect the mechanical, elastic, and reactive properties of the macromolecules. Theoretical methods in physical chemistry are capable of describing chemical reactions that take place in a well-defined, rigid environment of a frozen solvent. The nature of quantum mechanics allows one to compute the products of some reactions given a configuration for a molecule and its local environment. This implies that environmental contributions to molecular conformation and supramolecular structure of individual entities can be described adequately at the level of the harmonic oscillator approximation, and can be compared with single molecule mechanical experiments using existing tools of theoretical chemistry and statistical mechanics (Kreuzer and Grunze 2001). These single molecule models can then provide the input for molecular dynamics and grand canonical Monte Carlo simulations to describe two body interactions in the intracellular environment. Studies on a larger structural scale can be initiated using Brownian motion models to simulate the positional mobility and spatial interactions involving chromatin regions and entire chromosome territories. For the time being, it will be challenging to describe the multiplicity of correlated parallel and serial reactions and transformations occurring in a living cell, but it appears within reach to model well-defined reaction steps and then apply the tools of computational biology to analyze complex biochemical reaction networks (Regev et al. 2001).
Extending modeling approaches will provide insights into nuclear phenomena such as the sequential turning on and off of networks of coordinately expressed genes. Such events have features that lend themselves to the same kind of hierarchical switching algorithms used in the electronics industry. An application of this type of modeling to address embryonic development is shown in Figure 5. The experimental parameters are from an analysis of the expression and regulation of a set of genes involved in cell specification during early development (Davidson et al. 2002). Moreover, recent studies have revealed a surprising degree of stochastic noise in gene regulatory events (Fedoroff and Fontana 2002) and in transcription events at the single cell level in a population of cells (Levsky et al. 2002), suggesting that a larger repertoire of creative computational approaches may be needed for the analysis of gene expression than might have been assumed initially.
A far-reaching goal is to develop a model that is able to account for major aspects of genome structure and function. Ultimately, the desire is to understand nuclear structure and genome biology in a manner comparable with present efforts to understand concepts in protein folding, namely, in terms of the free energy landscape and the chemical potential of all the constituents (Cheung et al. 2002). In general, investigations will be guided by developing quantitative models, albeit primitive in the beginning, and these models will provide insights for experimental design, resulting in an iterative planning matrix evolving from modeling and experimental testing in biological systems. This effort should yield a quantitative model of the nucleus that can be combined with models that describe additional paradigms of cellular activity. Such a systems biology-based, integrative cell model should eventually allow the simulation and engineering of cell behavior on a predictive basis.
Conclusions
During the last three centuries, theoretical modeling, in combination with experimental quantitative knowledge, has been the driving force of the technological revolution. The same approach is now beginning to be applied in the life sciences. Dramatic advances in microscopy, biophysics, and the nanosciences allied with genomic and biological sciences, provide a platform for understanding in vivo genome structure and function. To advance this effort, investigators from diverse disciplines have gathered to discuss ideas and needed directions, including the establishment of the Genome Architecture Consortium (GAC). The GAC seeks to provide a forum that will encourage international, multi-disciplinary research collaborations. As a first step, we have established an annual meeting that brings together researchers with expertise in several diverse, yet complementary fields (Politz and Pombo 2002; Politz et al. 2003). The objective is for individuals with distinct backgrounds to develop both a common language and an appreciation for the possibilities and the limitations of the different disciplines that can be applied to understand how the nucleus unlocks the information contained in the genomic sequence.
Acknowledgments
We thank Dr. Peter Schmidt for helpful discussions in theoretical physical chemistry and Drs. Tom Misteli and Lindsay Shopland for helpful discussions on nuclear organization. We thank Jennifer Smith and Sarah Williamson for help with figure preparation. We appreciate the valuable discussions and comments on the manuscript from many colleagues in our laboratories, at our institutions, and those who attended the 2001 and 2002 Nanostructural Genomics meetings held at The Jackson Laboratory. The 2001 Genomics Meets Nanoscience meeting was supported in part by NIH grant HG/GM02450, DOE grant DE-FG02-01ER63206, and the Office of Naval Research grant 01PR12804; the 2002 Advances in Nanostructural Genomics II meeting was supported in part by NIH grant HG02669.
Footnotes
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.946403. Article published online before print in May 2003.
-
↵9 Corresponding author. E-MAIL tpo{at}jax.org; FAX (207) 288-6073.
- Cold Spring Harbor Laboratory Press








