
Metallochaperones and Metal-Transporting ATPases: A Comparative Analysis of Sequences and Structures
- Fabio Arnesano1,
- Lucia Banci1,
- Ivano Bertini1,3,
- Simone Ciofi-Baffoni1,
- Elena Molteni1,
- David L. Huffman2, and
- Thomas V. O'Halloran2
- 1Magnetic Resonance Center CERM and Department of Chemistry, University of Florence, Via Luigi Sacconi 6, 50019, Sesto Fiorentino, Florence, Italy; 2Department of Chemistry and the Department of Biochemistry, Molecular Biology, and Cell Biology, Northwestern University, Evanston, Illinois 60208, USA
Abstract
A comparative structural genomic analysis of a new class of metal-trafficking proteins can provide insights into the intracellular chemistry of reactive cofactors such as copper and zinc. Starting from the sequences of the metallochaperone Atx1 and from the first soluble domain of the copper-transporting ATPase Ccc2, both from yeast, a search on the available genomes was performed using a homology criterion and a metal-binding motif x‘-x"-C-x‴-x⁗-C. By limiting ourselves to 20% identity with any of the proteins found, several soluble copper-transport proteins were identified, as well as soluble domains of membrane-bound ATPases. Structural models were calculated using high-resolution solution structures as templates, and the models were validated using statistical and energy criteria. Residue conservation and substitution have been interpreted and discussed in terms of structure–function relationship. The potential energy surfaces have been analyzed in terms of protein–protein interactions. We find that metallochaperones and their physiological partner ATPases from several phylogenetic kingdoms recognize one another, via an interplay of electrostatics, hydrogen bonding, and hydrophobic interactions, in a manner that precisely orients the metal-binding side chains for rapid metal transfer between otherwise tight binding sites. Finally, other putative metal-transport proteins are mentioned that have low homology and/or a different metal-binding consensus motif and that appear to use similar structures for recognition and transfer. This analysis highlights the wealth and the complexity of the field.
Copper, an essential trace metal, is used as a cofactor in a variety of redox proteins. In eukaryotes, copper-dependent metalloenzymes are found in multiple cellular locations including the cytosol, mitochondria, and cell surface (Linder 1991). Excess copper, however, is highly toxic to most organisms (Linder 1991; Vulpe and Packman 1995). For this reason, the transport and sequestration of copper must be tightly controlled (Rosenzweig 2001; Pena et al. 1999; Huffman and O’Halloran 2001).
A class of metal ion receptor proteins, called metallochaperones, have been identified, which deliver copper to specific intracellular targets (O'Halloran and Culotta 2000). Recent studies indicate that a main difficulty is faced by metallochaperones as they deliver copper to the correct intracellular destination: The cytoplasm has a significant thermodynamic overcapacity for copper chelation (Rae et al. 1999). This has led to the suggestion that metallochaperones act like enzymes to lower the activation barrier for copper transfer to specific partners (Huffman and O'Halloran 2000).
Copper chaperones were first isolated in the baker's yeastSaccharomyces cerevisiae (Lin and Culotta 1995; Pufahl et al. 1997), and functional homologs have been noted in humans (Klomp et al. 1997), sheep (Lockhart and Mercer 2000), mice (Nishihara et al. 1998),Arabidopsis thaliana (Himelblau et al. 1998), andCaenorhabditis elegans (Wakabayashi et al. 1998). A prototypical copper chaperone, Atx1, conducts Cu(I) in the cytoplasm and transfers this cargo directly to a specific partner protein (Pufahl et al. 1997; Huffman and O'Halloran 2000). Both the yeast Atx1 and the human Atx1 homolog (HAH1 or ATOX1) specifically traffic copper to the secretory pathway for incorporation into copper enzymes destined for the cell surface or extracellular milieu (Klomp et al. 1997; Pufahl et al. 1997; Hung et al. 1998). A homologous protein CopZ also binds copper, and variations are known in Enterococcus hirae (Odermatt and Solioz 1995) and Bacillus subtilis (Banci et al. 2001a).
A class of integral membrane proteins that transport heavy metals across cellular membranes provided the earliest insights into the biology of copper trafficking (Vulpe et al. 1993; Bull and Cox 1994;Solioz and Vulpe 1996). Members of this family, referred to as P-type or CPx-type ATPases, have been described, mainly by gene isolation, in a variety of bacteria, yeasts, nematodes, and mammals, including humans (Pena et al. 1999; Rensing et al. 2000; Fu et al. 1995; Vulpe et al. 1993; Bull et al. 1993; Lockhart et al. 2000; Tanzi et al. 1993;Yamaguchi et al. 1993; Chelly et al. 1993; Mercer et al. 1993). P-type ATPases are ubiquitous membrane proteins and have been classified into five groups according to ion specificity (Axelsen and Palmgren 1998). A growing subfamily of metal-transporting P-type ATPases is involved in homeostasis of heavy metal ions. This subfamily can be divided into two subgroups. The first is involved in the transfer of monovalent metal ions and includes Cu(I)- and Ag(I)-transporting proteins, such as the human Menkes (Vulpe et al. 1993; Chelly et al. 1993; Mercer et al. 1993) and Wilson (Bull et al. 1993; Lockhart et al. 2000; Tanzi et al. 1993; Yamaguchi et al. 1993) disease-related proteins and homologs fromS. cerevisiae (Yuan et al. 1995; Fu et al. 1995), E. hirae (Odermatt et al. 1993), Synechococcus (Phung et al. 1994), and Helicobacter pylori (Melchers et al. 1998). Members of the second subgroup of soft metal P-type ATPases transport divalent metal ions including Zn(II), Cd(II), and Pb(II). This subgroup includes ZntA from Escherichia coli and CadA from Staphylococcus aureus plasmid pI258 (Rensing et al. 1999). Recently, a related gene conferring Co(II) resistance was identified in SynechocystisPCC 6803 (Rutherford et al. 1999).
The cytoplasmic N-terminus of the copper P-type ATPases contains one or more metal-binding motifs (Bull and Cox 1994). These N-terminal domains and the copper chaperones and copper-transporting proteins are mainly characterized by M-x"-C-x‴-x⁗-C motif, also found in other proteins proposed to bind or transport the heavy metals (Solioz and Vulpe 1996;Silver et al. 1989), such as the bacterial periplasmic mercury-binding protein MerP (Sahlman and Jonsson 1992). Sequence similarities with metallochaperones indicate that each heavy-metal binding domain of ATPases comprises ∼70 residues (Pufahl et al. 1997).
In humans, the Menkes ATPase containing six N-terminal metal-binding motifs (Vulpe et al. 1993; Lutsenko et al. 1997; Chelly et al. 1993;Mercer et al. 1993) is located in the trans-Golgi network (Yamaguchi et al. 1996) and is believed to translocate copper, donated by HAH1 copper chaperone, across intracellular membranes into the secretory pathway. Similarly, in the yeast S. cerevisiae, the target of copper delivery by the chaperone Atx1 is a P-type copper-transporting ATPase, called Ccc2, confined to a late Golgi compartment (Pufahl et al. 1997; Yuan et al. 1997; Huffman and O'Halloran 2000).
Solution structures of the Cu(I)-bound and reduced apo forms of both Atx1 (Arnesano et al. 2001b) and the first soluble domain of Ccc2 (Ccc2a hereafter) (Banci et al. 2001b) have been recently solved. The solution structures of MerP (Steele and Opella 1997), of the fourth domain of the Menkes transporter (MNK4 hereafter) (Gitschier et al. 1998), and CopZ, in the reduced apo form from E. hirae (Wimmer et al. 1999) and the Cu(I)-bound form from B. subtilis (Banci et al. 2001a), are also available. The crystal structures were solved for the oxidized apo and Hg(II) forms of Atx1 (Rosenzweig et al. 1999). Both the MerP solution structure and the Atx1 crystal structure contained a Hg(II) ion with a linear coordination geometry, whereas the solution structure of MNK4 contained the Ag(I) ion. All these structures share a classical “ferredoxin-like” β1–α1–β2–β3–α2–β4 fold (Hubbard et al. 1997) in which the secondary structure elements, four β-strands and two α-helices, are connected by loop regions.
The aim of the present study is to browse the available genomes of all types of organisms and analyze them according to the specific consensus sequence for metal binding, as well as sequence homologies, with the ultimate goal of locating metallochaperones, metal-transporting proteins, and ATPases. The location of these sequences, their alignment, and the structural modeling allow us to identify the conserved amino acid segments and their role in the structural properties. Then, from this comparison, hints for the relation between structural properties and biological function are discussed.
The proteins analyzed here are all proposed to be metal-transport proteins. Some of them transport the metal through the membrane (P-type ATPases); others are small soluble proteins. Of the latter, those that carry the copper ion to a specific target protein are called copper chaperones, such as Atx1. The others, such as CopZ and MerP, are soluble metal-transport proteins involved in detoxification and, as shown below, fall into distinct structural classes.
RESULTS
Pattern-Based Sequence Analysis of Metallochaperones and Other Metal Transporters
Starting from the 73 amino acid sequence of Atx1 from yeast S. cerevisiae and using the consensus motif x‘-x"-C-x‴-x⁗-C and the amino acid chain length to restrict the search, and through a few cycles of the procedure described in the Methods section, the sequences of 57 proteins (including the template Atx1) were selected whose sequence alignment is shown in Figure1. Sequences from 12 eukaryotic organisms are grouped together in a green box. Pairwise residue identity within this subclass is 46% ± 19%. The most evolved organisms (Homo sapiens, Rattus norvegicus,Mus musculus, Ovis aries, and Canis familiaris) show a higher sequence similarity. All the eukaryotic organisms possess the metal-binding motif M-x"-C-x⁗-G-C. The proteins of this subclass contain a large number of Lys residues. The alignment highlights four positions (25, 29, 70, 73) where positively charged residues (Lys and Arg) are conserved. In particular, a Lys residue is always present at positions 70 and 73. The NMR structure of the Cu(I) form of yeast S. cerevisiae Atx1 (sequence 1) is available (Arnesano et al. 2001b).
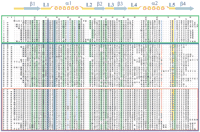
Sequence alignment of the Atx1 amino acid sequence from yeast S. cerevisiae with the metallochaperone homologs located in the present research. At the top, amino acid numbering is reported including gaps (first line) and according to the sequence of Atx1 (second line). The two metal-binding cysteines are shaded in blue. Positions where hydrophobic residues are conserved are shaded in green. Key positive (Arg and Lys) and negative (Glu and Asp) residues are indicated in blue and in red, respectively. The boxes include eukaryotic metallochaperone sequences (green), and bacterial, CopZ-like and MerP-like, sequences (blue). The red box includes the subgroup of MerP-like sequences. The secondary structure elements reported above the alignment are referred to Atx1. In column a we report sequence identity to Atx1 (sequence 1); in column b we report identity to sequence 13 and sequence 36 for CopZ-like and MerP-like sequences, respectively. Eukaryotic metallochaperones: 1 gi‖6730164 Saccharomyces cerevisiae(Atx1); 2 gi‖7492713 Schizosaccharomyces pombe;3 gi‖11290108 Oryza sativa; 4 gi‖11290106 Glycine max; 5 gi‖15228869 Arabidopsis thaliana;6 gi‖1945365 Homo sapiens; 7 gi‖6013208Canis familiaris; 8 gi‖7531050 Ovis aries;9 gi‖7531046 Mus musculus; 10 gi‖7531044Rattus norvegicus; 11 gi‖4165309 Caenorhabditis elegans; 12 gi‖7296474 Drosophila melanogaster;Bacterial and archaeal soluble metal transporters: CopZ-like: 13 gi‖5107576 Enterococcus hirae (CopZ); 14gi‖7429061 Bacillus subtilis (CopZ); 15 gi‖1477773Helicobacter pylori, strain A68; 16 gi‖9789743H. pylori, strain J99; 17 gi‖3121871Helicobacter felis; 18 gi‖7471711 Deinococcus radiodurans; 19 gi‖7462405 Thermotoga maritima;20 gi‖11346720 Campylobacter jejuni; 21gi‖13357760 Ureaplasma urealyticum; 22 gi‖8894834Streptomyces coelicolor; 23 gi‖9965436Streptococcus mutans; 24 gi‖13622769Streptococcus pyogenes; 25 gi‖11353981Neisseria meningitidis, serogroup A strain Z2491; 26gi‖11353792 N. meningitidis, serogroup B strain MC58;27 gi‖14600385 Aeropyrum pernix; 28gi‖14195314 Haemophilus influenzae; 29 gi‖12722322Pasteurella multocida; 30 gi‖12723757Lactococcus lactis; 31 gi‖12725117 L. lactis II; 32 gi‖10580283 Halobacterium sp.; 33gi‖10173169 Bacillus halodurans; 34 gi‖11349934Pseudomonas aeruginosa; 35 gi‖8388756Pseudomonas syringae; MerP-like: 36 gi‖127010Shigella flexneri (MerP); 37 gi‖2944141Pseudomonas stutzeri I; 38 gi‖2947088 P. stutzeri II; 39 gi‖2498542 Alcaligenes sp.;40 gi‖127008 P. aeruginosa, plasmid pVS1; 41gi‖2498544 Pseudomonas fluorescens; 42 gi‖4572444Sphingomonas paucimobilis; 43 gi‖127009Serratia marcescens; 44 gi‖4572382 E. coli;45 gi‖2498541 Acinetobacter calcoaceticus;46 gi‖2498543 Enterobacter cloacae; 47gi‖2052180 Pseudomonas sp., strain KHP41; 48gi‖2159997 Pseudomonas sp., strain K-62; 49gi‖6689527 Xanthomonas campestris; 50 gi‖2498545Shewanella putrefaciens; 51 gi‖2935549Pseudomonas alcaligenes; 52 gi‖2765117Thiobacillus sp.; 53 gi‖14195504 H. influenzae; 54 gi‖12721567 P. multocida;55 gi‖10640685 Thermoplasma acidophilum; 56gi‖14324466 Thermoplasma volcanium; 57 gi‖11496143Pseudoalteromonas haloplanktis.
A slightly larger sequence variability (34% ± 18% pairwise residue identity) is found within the subclass of bacterial proteins (grouped in a blue box in Fig. 1), comprising 45 sequences. In this case, only the pattern C-x‴-x⁗-C is conserved, with position x‘ being occupied by a Met, an Ile, a Leu, or a Val residue. However, within this class some proteins were grouped (red sub-box in Fig. 1) whose primary sequence is highly conserved, with pairwise residue identity of 56% ± 25%. As the first member of this subclass of organisms, the sequence of MerP from Shigella flexneri (sequence 28) was considered. This is a mercury detoxification protein (Foster 1987;O’Halloran 1993; Lund and Brown 1987; Sahlman and Jonsson 1992) whose Hg(II)-bound NMR structure is available (Steele and Opella 1997). There are 21 sequences belonging to this subclass. All of them are members of the widespread mercury detoxification operons (Liebert et al. 2000).
The sequence variability within the remaining bacterial metallochaperone homologs is larger (27% ± 11% pairwise residue identity). This subclass comprises 23 sequences. Fifteen of them contain a His residue at positions x" or x⁗ of the metal-binding motif. In two cases (sequences 20 and 25), His is replaced by Asn, which, however, can be considered His-like. The representative sequence of this subclass is that of CopZ, a protein found in E. hiraeand B. subtilis, which is proposed to act as a copper transporter (Odermatt et al. 1992; Odermatt et al. 1993; Odermatt and Solioz 1995; Cobine et al. 1999; Banci et al. 2001a). The NMR structures of the Cu(I)-bound form of CopZ from B. subtilis(sequence 14) (Banci et al. 2001a) and the apo form of CopZ fromE. hirae (sequence 13) (Wimmer et al. 1999) are available.
Figure 2A schematically shows amino acid variability among metal ion transporters in the protein frame of yeast Atx1. It can be noticed that sequence conservation increases when approaching the metal-binding site.
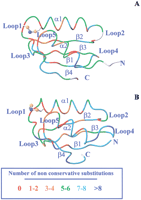
Amino acid variability among metallochaperone homolog sequences (A) and among metal-transporting ATPases (B). Color code is reported in the bottom panel. Positions with gaps in more than 50% of the aligned sequences are colored in white. The structures of yeast Cu-Atx1 (Arnesano et al. 2001b) (PDB ID 1FD8) and of the 1st domain of Cu-Ccc2 (Banci et al. 2001b) (PDB ID1FVS) are used.
Gram-positive bacteria show a different organization of the cell, which in particular lacks periplasm. This indicates that periplasmic metal resistance proteins, such as MerP, are absent. Although CopZ is in the cytoplasm, only a relatively small number of organisms show proteins having sequence similarity with the CopZ sequence. It can be inferred that other regulation mechanisms are effective in copper homeostasis (O'Halloran and Culotta 2000; Rosen 1999; Silver and Ji 1994).
To date, the complete genomes of 48 organisms (9 archaea, 35 bacteria, and 4 eukaryotes) have been sequenced. (Protein sequences reported in the present paper are those available in the GenBank in March 2001. Complete genomes are available at the following Internet site:http://www.ncbi.nlm.nih.gov/PMGifs/Genomes/org.html.) Under the present search conditions, small soluble metal-transport proteins were found only in 21 fully sequenced genomes of bacteria and archaea. For the other organisms with known complete genomes, it is probable that different proteins with low sequence homology and different metal-binding motifs are involved in metal homeostasis. On the other hand, Atx1-like metallochaperone proteins were found in all eukaryotes with complete genomes.
Pattern Analysis of Metal-Transporting ATPases
Metal-binding domains of large membrane proteins were searched, starting from the 72 amino acid sequence of the first soluble domain of the copper-transporting ATPase Ccc2 from yeast S. cerevisiae(Ccc2a) and using the x‘-x"-C-x‴-x⁗-C consensus motif. The search detected 116 sequences corresponding to single metal-binding domains. The sequence alignment is shown in Figure3. A total of 63 domains belonging to 16 different proteins was found in 12 eukaryotic organisms and 53 domains of 43 proteins in 36 bacteria and archaea. Copper-transporting ATPases were found in all fully sequenced eukaryotic genomes and in 24 bacterial and archaeal complete genomes. The number of metal-binding motifs in metal-transporting ATPases increases with evolution, ranging from one to two in bacteria and up to six in eukaryotes. In humans, two copper ATPases are present. They are called the Menkes and Wilson proteins and each contains six soluble domains. On the basis of sequence homology, both proteins were identified in the mouse and the rat, whereas only one copper-transporting ATPase was found in the sheep (Wilson-like) and the chinese hamster (Menkes-like). They all possess six domains, with the exception of the Wilson proteins of the mouse and the rat, in which only five are present. When six domains are present, a higher homology is found between corresponding domains of different organisms, with respect to homology within domains of the same organism. This also holds for proteins with only five metal-binding domains, taking into account that, on the basis of sequence homology, the fourth is missing.

Sequence alignment of the Ccc2a amino acid sequence from yeast S. cerevisiae with the heavy metal-binding domains located in the present research. At the top, amino acid numbering is reported including gaps (first line) and according to the sequence of Ccc2a (second line). The two metal-binding cysteines are shaded in blue. Key negative (Glu and Asp) and positive (Arg and Lys) residues are indicated in red and blue, respectively. Positions where hydrophobic residues are conserved are highlighted in green. The two boxes include eukaryotic (green) and bacterial (blue) sequences. The secondary structure elements reported above the alignment are referred to Ccc2a. In column a we report sequence identity to Ccc2a; in column b the length of interdomain linkers for multidomain proteins. 1 gi‖6320475 S. cerevisiae,1st domain (Ccc2a); 2 S. cerevisiae, 2nd domain (Ccc2b); 3 gi‖7492267 Schizosaccharomyces pombe;4 gi‖8745541 Candida albicans, 1st domain; 5 C. albicans, 2nd domain; 6 gi‖6633848 A. thaliana I, 1st domain; 7 A. thaliana I, 2nd domain; 8 gi‖12229667 A. thaliana II, 1st domain;9 A. thaliana II, 2nd domain; 10 gi‖1351993 human Menkes protein, 1st domain (MNK1); 11 human Menkes protein, 2nd domain (MNK2); 12 human Menkes protein, 3rd domain (MNK3); 13 human Menkes protein, 4th domain (MNK4); 14human Menkes protein, 5th domain (MNK5); 15 human Menkes protein, 6th domain (MNK6); 16 gi‖14758520 human Wilson protein, 1st domain; 17 human Wilson protein, 2nd domain;18 human Wilson protein, 3rd domain; 19 human Wilson protein, 4th domain; 20 human Wilson protein, 5th domain;21 human Wilson protein, 6th domain; 22 gi‖2739170O. aries, 1st domain; 23 O. aries, 2nd domain; 24 O. aries, 3rd domain; 25 O. aries, 4th domain; 26 O. aries, 5th domain;27 O. aries, 6th domain; 28 gi‖2440287M. musculus I, 1st domain; 29 M. musculus I, 2nd domain; 30 M. musculus I, 3rd domain; 31 M. musculus I, 4th domain; 32 M. musculus I, 5th domain; 33 M. musculus I, 6th domain; 34gi‖12229577 M. musculus II, 1st domain; 35 M. musculus II, 2nd domain; 36 M. musculus II, 3rd domain; 37 M. musculus II, 4th domain; 38 M. musculus II, 5th domain; 39 gi‖1351992Cricetulus griseus, 1st domain; 40 C. griseus, 2nd domain; 41 C. griseus, 3rd domain;42 C. griseus, 4th domain; 43 C. griseus, 5th domain; 44 C. griseus, 6th domain;45 gi‖12229551 R. norvegicus I, 1st domain;46 R. norvegicus I, 2nd domain; 47 R. norvegicus I, 3rd domain; 48 R. norvegicus I, 4th domain; 49 R. norvegicus I, 5th domain; 50 R. norvegicus I, 6th domain; 51 gi‖6006293 R. norvegicus II, 1st domain; 52 R. norvegicus II, 2nd domain; 53 R. norvegicus II, 3rd domain;54 R. norvegicus II, 4th domain; 55 R. norvegicus II, 5th domain; 56 gi‖7428301 C. elegans, 1st domain; 57 C. elegans, 2nd domain;58 C. elegans, 3rd domain; 59 gi‖7292707D. melanogaster, 1st domain; 60 D. melanogaster, 2nd domain; 61 D. melanogaster, 3rd domain; 62 D. melanogaster, 4th domain; 63gi‖10122140 Cryptosporidium parvum; 64 gi‖7531047B. subtilis, 1st domain (CopAa); 65 B. subtilis, 2nd domain (CopAb); 66 gi‖15283450 Aquifex aeolicus; 67 gi‖1354935 Escherichia coli, 1st domain; 68 E. coli, 2nd domain; 69gi‖2493017 Haemophilus influenzae; 70 gi‖8894835Streptomyces coelicolor I; 71 gi‖6689151 S. coelicolor II; 72 gi‖6714779 S. coelicolor III;73 gi‖2493001 Synechocystis I; 74gi‖7428304 Synechocystis II; 75 gi‖11267419Campylobacter jejuni; 76 gi‖7471220 Deinococcus radiodurans, 1st domain; 77 D. radiodurans, 2nd domain; 78 gi‖416665 E. hirae (CopA); 79gi‖2493003 H. pylori, strain 26695; 80 gi‖3121870H. felis; 81 gi‖13432119 Mycobacterium leprae I; 82 gi‖13637888 M. leprae II; 83gi‖1706183 Mycobacterium tuberculosis I; 84gi‖1706184 M. tuberculosis II; 85 gi‖11267401Neisseria meningitidis, serogroup A strain Z2491; 86gi‖11267405 N. meningitidis, serogroup B strain MC58;87 gi‖1353678 Proteus mirabilis, 1st domain;88 P. mirabilis, 2nd domain; 89 gi‖7531049Sinorhizobium meliloti, 1st domain; 90 S. meliloti, 2nd domain; 91 gi‖584792 Synechococcus I; 92 gi‖584820 Synechococcus II; 93gi‖7436397 Thermotoga maritima; 94 gi‖14601418Aeropyrum pernix; 95 gi‖11498084 Archaeoglobus fulgidus; 96 gi‖7428308 Methanobacterium thermoautotrophicum, 1st domain; 97 M. thermoautotrophicum, 2nd domain; 98 gi‖8388793Pseudomonas syringae, 1st domain; 99 P. syringae, 2nd domain; 100 gi‖9965435 S. mutans; 101 gi‖13622770 S. pyogenes;102 gi‖10173170 B. halodurans, 1st domain;103 B. halodurans, 2nd domain; 104gi‖11351437 P. aeruginosa, 1st domain; 105 P. aeruginosa, 2nd domain; 106 gi‖12723759 L. lactis; 107 gi‖10580282 Halobacterium sp., strain NRC-1, 1st domain; 108 Halobacterium sp., strain NRC-1, 2nd domain; 109 gi‖12722323 P. multocida; 110 gi‖120199 Sinorhizobium meliloti (fixI); 111 gi‖3122077 Rhizobium leguminosarum; 112 gi‖3122094 Bradyrhizobium japonicum; 113 gi‖13475529 Mesorhizobium loti;114 gi‖2338745 Rhodobacter capsulatus;115 gi‖7271788 Rhodobacter sphaeroides; 116gi‖13422766 Caulobacter crescentus.
The number of residues in the interdomain stretches is reported in column b of Figure 3. The interdomain sequences show a large variability in length. For instance, the two domains of yeast Ccc2 are joined immediately by a very short linker, as are the last two domains of the Wilson and Menkes proteins. In contrast, the other interdomain sequences of the latter proteins possess 10 to 90 residues. The exceptions are the mouse and rat Wilson ATPases in which, after the third domain, a stretch of approximately 150 residues is found that lacks the metal-binding motif. These interdomain regions may provide other structural elements or modulate motions of the individual metal-binding domains in concert with other elements of the ATPase.
If the six domains of the Menkes protein are aligned for the same organism, invariably domain 3 has the lowest homology with the other domains of the same protein. However, they are very similar among themselves in the various organisms (65% average identity). In particular, three key positions that discriminate domain 3 from the other domains can be identified from inspection of the sequence alignment shown in Figure 3. Position 13 (corresponding to x" in the consensus pattern) is always His in domain 3, whereas the other domains have a Thr. Positions 76 and 77 are two Pro or a Ser and a Pro in domain 3, whereas the other domains have two adjacent Gly and Phe. The calculated instability index (II) (Guruprasad et al. 1990) predicts a lower stability for domain 3 (II = 75 ± 8) compared with the others (II = 50 ± 8).
The Met residue present in the consensus motif is almost always conserved. In addition, a Gly residue precedes this Met in all eukaryotes and archaea and in almost all bacteria. A Phe (mainly in eukaryotes) or a Tyr (mainly in bacteria and archaea) residue is often present at position 77. Some negatively charged residues are partially conserved: A Glu is often found at position 22 in both bacteria and eukaryotes, with an Asp or a Glu at positions 72, 74, and 78, mainly in eukaryotes.
Almost all sequences aligned in Figure 3 have residue identity higher than 20% with respect to the Ccc2a sequence (Fig. 3, column a) with an average value of 29% for eukaryotes and 25% for bacteria and archaea.
In bacteria, the first domain of a metal-transporting ATPase fromB. subtilis (sequence 64) has the highest percentage of residue identity to the sequence of Ccc2a (35%). The average residue identity of all other bacterial sequences to sequence 64 from B. subtilis is 38% ± 8%. This sequence has 44% identity with CopA (sequence 78), a protein from E. hirae that is known to act as a copper-transporting ATPase (Odermatt et al. 1993). The protein CopZ has been postulated to act as an activator of gene transcription of the cop operon. Its action depends on the availability of copper, thus controlling the expression of CopA (Odermatt and Solioz 1995; Cobine et al. 1999). An interaction between CopZ and the ATPase, CopA, has not been shown so far.
Within the class of bacterial ATPases, some proteins can be identified (sequences 110–116 in Fig. 3) whose primary sequence is highly conserved one with the others, with pairwise residue identity of 41% ± 8%. These ATPases are found in organisms belonging to the group of Rhizobiaceae, Rhodobacter, andCaulobacter; all of them are members of the Fix operon (Nellen-Anthamatten et al. 1998; Cabanes et al. 2000; Delgado et al. 1998). This operon also includes cytochrome c oxidase (CCO), which requires copper to function, and the ATPases called FixI, which serve as copper pumps in the activation of CCO. FixI ATPases have an average residue identity of 20% with Ccc2a, and their soluble part is predicted to have the same βαββαβ fold.
The residue variability along the whole family of ATPase soluble domains is shown in Figure 2B.
Homology Modeling of Metallochaperones and Other Metal Transporters
First, we compared the structure of Cu(I)-Atx1 from S. cerevisiae (Arnesano et al. 2001b) (sequence 1 in Fig. 1) with the other available structures of soluble metal transporters, the apo-CopZ (Wimmer et al. 1999) from E. hirae (sequence 13), the Cu(I)-CopZ (Banci et al. 2001a) from B. subtilis (sequence 14), and the Hg(II)-MerP (Steele and Opella 1997) from Sh. flexneri (sequence 36). The protein fold is similar in all cases. Overall backbone RMSD values of 2.3 Å and 2.2 Å were found between Cu-Atx1 and Cu-CopZ and between Cu-Atx1 and apo-CopZ, respectively. The RMSD between Cu-Atx1 and Hg-MerP is 2.5 Å. The bacterial proteins apo-CopZ and Hg-MerP are quite similar to each other with a backbone RMSD value of 1.6 Å. When comparing Cu-CopZ from B. subtilisand the apo-CopZ from E. hirae, an RMSD value of 2.2 Å is found, which drops to 1.7 Å when residues close to the metal site (loop 1 and helix α1) are excluded, that is, the ones most affected by metal binding. The largest differences between Cu-Atx1 and bacterial metal transporters involve loops 2 and 4, which are the regions experiencing the largest variations in residue type, as well as insertions and deletions. Other differences involve helix α1, in particular at its N-terminus close to the metal site, and strand β4, which is shorter in MerP and CopZ than in Atx1.
For the sequences whose structures are not available, the PHD method (Rost and Sander 1993, 1994) has been used to predict secondary structural elements. Variations in the primary structure produce small changes in length and position of secondary structural elements, whereas the global fold is conserved. The proteins of unknown structure have then been modeled using Cu(I)-Atx1, apo- and Cu(I)-CopZ, and Hg(II)-MerP as templates.
An overlay of the structures for the superfamily of 57 metal transporters is reported in Figure 4A, in which three colors were used according to sequence classification. It can be clearly seen that structural differences between the three subgroups are correlated with those between the experimentally determined structures used as templates and are in agreement with sequence homology. The pairwise backbone RMSD between the modeled structures (calculated for the common stretches 7–12, 14–31, 37–44, 46–54, 59–67, 71–76) is 1.5 ± 0.7 Å. Higher deviations are found for regions in which residues are not conserved and insertions/deletions are found in the various sequences (Fig. 2A). Loop 2 has a variable length in the various sequences, as indicated by gaps in the alignment and, for this reason, different loop conformations are obtained from the modeling. Large changes are also observed in strand β4 and helix α1, as expected on the basis of the above considerations concerning the template structures. The metal ion position is well maintained among eukaryotic metallochaperone models, whereas it is more variable in bacterial metal transporters, reflecting a larger sequence variability in these latter sequences for residues in the metal-binding motif and its vicinity.
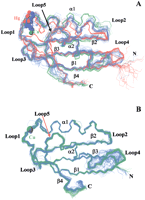
(A) Overlay of the super family of 57 metallochaperone homolog structures; three colors are used according to sequence classification of Fig. 1: Atx1-like (green), CopZ-like (blue), and MerP-like (red). The copper ion is shown as a green or a blue sphere for Atx1-like and CopZ-like structures, respectively; for MerP-like structures the mercury ion is shown in red. (B) Overlay of the super family of 116 structures, each corresponding to one soluble domain of a metal-transporting ATPase; two colors are used to distinguish between eukaryotes (green) and prokaryotes, including archaea (blue). The copper ion is shown as a sphere.
Homology Modeling of Metal-Transporting ATPases
The only available structures for a subdomain of metal-transporting ATPases are solution structures of the copper-bound 1stdomain of Ccc2 (Banci et al. 2001b) from S. cerevisiae(sequence 1 in Fig. 3) and of the silver-bound 4th domain of the human Menkes protein (Gitschier et al. 1998) (sequence 13), both belonging to eukaryotes. The residue identity between the two is 30% and the backbone RMSD between the two structures is 0.8 Å. The solution structures were used as templates for the modeling. All the eukaryote sequences show a large sequence homology with Ccc2a (29% ± 4% average identity). The third domains of Menkes and Wilson proteins show the lowest amino acid identity (22% ± 2%) with Ccc2a, but they have higher sequence identity with MNK4 (39% ± 2%). Concerning bacteria and archaea sequences, the use of these two templates is justified by a relatively large amino acid identity, as discussed above.
An overlay of the superfamily of 116 structures, each corresponding to one soluble domain of a metal-transporting ATPase, is reported in Figure 4B, in which two colors were used to distinguish between eukaryotes (green) and prokaryotes (blue), including archaea. All the structures are well superimposed with an average pairwise backbone RMSD of 0.4 ± 0.2 Å (calculated for the common stretches 12–32, 40–52, 67–72, 74–79). The most variable region is loop 4, which can be traced to the amino acid variability among ATPases reported in Figure2B in the protein frame of yeast Ccc2a. The third domain of the Menkes and Wilson proteins is well modeled, with an average RMSD of 0.8 Å to Ccc2a and 0.3 Å to MNK4, in agreement with residue identity.
Model Assessment: Energetics, Secondary Structure, and Hydrophobic Core Analysis
In the reported alignments (Figs. 1, 3), we included sequences having low residue identity with the members used as templates for the modeling but higher than 20% with at least one member of the family. A significant sequence homology is not necessary for two proteins to have a common fold (Orengo et al. 1994). Indeed, the sequences we have considered are correlated with each other, as there exists a path in sequence space such that every sequence can be reached from every other sequence. In this sense, a common fold requires a significant level of conservation of amino acid classes rather than of individual amino acids (Babajide et al. 1997). Also in low homology sequences reported here, key residues are conservatively substituted.
Even when the overall fold is the same, if sequence identity with template structures is lower than 30%, the following errors can affect the model: 1) the region is aligned correctly but is distorted or shifted as a rigid body relative to the correct structure (e.g., loops, helices); and 2) the region is modeled incorrectly because it does not have an equivalent segment in any of the templates (e.g., loops). RMSD to the template structure does not reflect these errors. In fact, modeling is strictly dependent on the structures chosen as templates. There are several criteria to evaluate a structural model and back-check for the choice of the templates (Sali 1995). A good model should have a low energy, according to a molecular mechanics force field, even if this is not sufficient (Novotny et al. 1984; Novotny et al. 1988). Thus, structural features have been analyzed from high-resolution protein structures, and related parameters have been used as strong indicators of errors in the model. Such features include packing, creation of a hydrophobic core, residue and atomic solvent accessibility, spatial distribution of charged groups, distribution of atom–atom distances, and main chain hydrogen bonding (Sali 1995 and references therein). In addition, programs based on the analysis of known protein structures provide stereochemical tests.
The quality of the structures was evaluated through Ramachandran plots obtained using the program PROCHECK (Laskowski et al. 1993). The latter program also analyzes the secondary structure elements. In addition, all models were inspected with the programPROSA (Sippl 1993), which allows the identification of poor models or regions in models with unsatisfactory interactions with the rest of the protein. The program checks if poor packing of the structure is present.
The results of the Ramachandran plot analysis are reported in Table1 for metallochaperones and soluble metal transporters, and in Table 2 for ATPase domain structures. The secondary structure elements, as analyzed byPROCHECK, particularly the two helices, are well conserved both in position and in length (Fig. 5), all structures sharing the β1–α1–β2–β3–α2–β4fold. The length of the last strand β4, however, shows a large variability. In particular, this strand is generally shorter in bacterial metal transporters than in eukaryotic metallochaperones and, in some cases, is totally absent. This reflects differences also observed in the template structures. In fact, in CopZ from E. hirae and B. subtilis and in MerP from Sh. flexneri, strand β4 has a reduced length with respect to Atx1 from S. cerevisiae. Hydrogen-bonded turns are often found close to the N- or C-termini of regular structural elements.
Structural Analysis of the Family of Metallochaperone and Metal Transporter Modeled Structures as it Results from the Ramachandran Plots
Structural Analysis of the Family of ATPase Metal Binding Domain Modeled Structures as it Results from the Ramachandran Plots
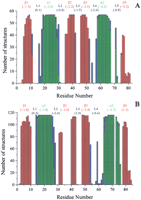
Secondary structure of structural models of metallochaperones and soluble metal transporters (A) and of ATPase domains (B). β-strands are indicated in red, α-helices in green, turns in blue. For each secondary structure element the pair/surface combined energy is reported in brackets as obtained from thePROSA analysis.
PROSA-combined energy (Methods section) has been reported for each secondary structure element on the top of plots of Figure 5. It can be noted that, generally, regular secondary structure elements, that is, α helices and β strands, have lower energy than loop regions. An exception is represented by strand β4 that, however, is less conserved as discussed above. On the other hand, loop 4 in chaperone and soluble metal-transport protein models and loop 2 in ATPase models have low energy. These energies were converted into Z-scores (Sippl 1993; Babajide et al. 1997), which can be considered as a measure for the quality of the modeling: A lower Z-score corresponds to a more favorable potential energy associated with the structure under examination. The Z-scores obtained for chaperone and soluble metal-transport protein structures are in the range −4.9 to −8.9, with a mean value of −6.6. The values for ATPase structures are in the range of −3.2 to −7.6, with a mean value of −6.1. These Z-scores are in a range expected on the basis of the amino acid sequence and of its length (∼70 a.a.) (Sippl 1993). In summary, the PROSAanalysis for the family of models confirms their consistency, as the range of observed Z-scores is quite narrow over all the structures. Inasmuch as this set includes experimental structures, this assessment implies that the overall quality of the models is comparable to that of the available experimental structures.
The hydrophobic core of chaperones, soluble metal-transport proteins, and ATPases are shown in Figure 6. In metallochaperones and other soluble metal transporters, 12 residues are buried in the majority of the structures (>70%), and 10 additional residues are buried in 30%–70%. In ATPases, 12 residues are buried in more than 70% structures and 10 additional residues are buried in 30%–70%. Buried residues are generally highly conserved in metallochaperones, soluble metal-transport proteins, and ATPase sequences (marked with * in Fig. 6). It is interesting to note that all these classes of proteins have a buried residue in corresponding positions of the amino acid sequences, consistent with a common βαββαβ fold. The most buried regions are strands β1 and β3 and part of helices α1 and α2. Residues in loops 2 and 4, in which amino acid variability is very high, are the most exposed. The second metal-binding cysteine of the motif (in helix α1) is partially exposed in metallochaperones and small soluble metal transporters, whereas it is buried in ATPase domains. At position x‘ of the consensus pattern, a Met often occurs, which is sometimes replaced by hydrophobic residues like Ile, Val, or Leu. These occupy a unique site in loop 1 that has a low solvent accessibility. In ATPases, the side chain of this Met is completely buried in almost all structures and penetrates into the core. Loop 1 and loop 5 regions are considerably more polar in the metallochaperones.
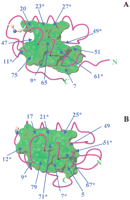
Hydrophobic core of structural models of metallochaperones and soluble metal transporters (A) and of ATPase soluble domains (B), defined as the van der Waals surface formed by atoms of residues that are buried in more than 70% structures. These residues are indicated in the figure. Conserved hydrophobic amino acids are marked with *.
Tables 3 and 4report the contacts conserved in more than 70% of the modeled proteins and involving buried residues. Nearly all amino acids involved in these contacts are conserved or conservatively substituted in the majority of the protein sequences that we have analyzed.
Contacts Involving Buried Residues (boldface) Conserved in More Than 70% of Metallochaperone and Metal Transporter Model Structures
Contacts Involving Buried Residues (in bold) Conserved in More Than 70% of Metal-transporting ATPase Model Structures
Other Potential Metal-Binding Domains
The search in the GenBank by using the x‘-x"-C-x‴-x⁗-C consensus motif led us to other proteins of different sequence length and/or low homology with the input sequences Atx1 and Ccc2. These significant differences prevented reliable structure modeling and are summarized below.
Some organisms possess other putative copper-transporting ATPases, in addition to those already reported in the alignment of Figure 3. InPseudomonas aeruginosa, Neisseria meningitidis,Campylobacter jejuni, H. pylori, and Vibrio cholerae the N-terminal soluble part of these ATPases encompasses ∼150 residues and contains three metal-binding motifs: CxHC, CxGC, and CxAC. Similarly, the archaeon Halobacterium sp. has an ATPase with three metal-binding motifs, CTLC, CRGC, and CATC.
The N-terminal domain of CCS, the copper chaperone for superoxide dismutase (SOD) (Culotta et al. 1997), is a structural homolog of Atx1 (Lamb et al. 1999), and it shares the same consensus motif for metal binding.
Two proteins of the mercury resistance (mer) operon, the mercuric reductase MerA and the inner membrane protein MerC, also contain a CxxC metal-binding motif (Miller 1999; Liebert et al. 2000). The N-terminal domain of MerA is aligned with the Atx1 sequence, with low homology (17% ± 2% residue identity), and with MerP (33% ± 3% residue identity).
Finally, we found two subgroups of ATPase sequences showing similarity to the E. coli zinc-transporting ZntA (Rensing et al. 1997) (sequences 1–12 in Fig. 7) or to theS. aureus cadmium-transporting CadA (Nucifora et al. 1989) (sequences 13–19 in Fig. 7). It was shown that ZntA and CadA also display Pb(II) translocating activity (Rensing et al. 1998). The average residue identity with respect to yeast Ccc2a and MNK4 is 19% and 20%, respectively, and the secondary structure prediction for the soluble domains of these Zn/Cd/Pb ATPases indicates the βαββαβ fold common to the copper transporters. It can be inferred that the metal-binding specificity for divalent cations does not reside in the overall fold but in sequence variation in the proximity of the metal-binding motif. Given the lack of experimental structures loaded with one of the divalent cations for which these proteins are specific, modeling was not pursued; however, some observations can be made on the basis of their sequence alignment (Fig 7).

Sequence alignment of zinc- and cadmium-transporting ATPases. At the top, amino acid numbering is reported including gaps. The two metal-binding cysteines are shaded in blue. Some conserved negative (Glu and Asp) and positive (Arg and Lys) residues are indicated in red and blue, respectively. Positions where hydrophobic residues are conserved are highlighted in green. In column a we report sequence identity to Ccc2a from S. cerevisiae. 1gi‖586655 E. coli, strain K-12 (ZntA); 2gi‖15803981 E. coli, strain EDL-933; 3gi‖15641046 Vibrio cholerae; 4 gi‖2624376 P. mirabilis; 5 gi‖3123078 Synechocystis; 6gi‖15807741 D. radiodurans; 7 gi‖7436386 B. subtilis; 8 gi‖14521140 Pyrococcus abyssi;9 gi‖15789464 Halobacterium sp., strain NRC-1;10 gi‖10720043 H. felis; 11 gi‖15611794H. pylori, strain J99; 12 gi‖2493007 H. pylori, strain 26695; 13 gi‖79893 Staphylococcus aureus (CadA); 14 gi‖14020985 S. aureus II, 1st domain; 15 S. aureus II, 2nd domain; 16gi‖231677 Bacillus firmus; 17 gi‖15616598Bacillus halodurans; 18 gi‖3121832 Listeria monocytogenes; 19 gi‖9789448 L. lactis.
These putative zinc- and cadmium-transporting ATPases display a different pattern of amino acid residues at the variable positions in the metal-binding motif. In particular, the presence of negatively charged residues near the metal-binding site may facilitate the binding of a divalent cation. For instance, the conserved Asp residue of the ZntA-like sequences is found in the metal-binding consensus motif x‘DCx‴x⁗C (x‘ = M, L) and can provide a metal-binding carboxylate group. In CadA-like sequences, the conserved residues are x⁗ = N with x‘ = F, L. Cadmium has a preference for nitrogen and oxygen donor atoms and tetrahedral or octahedral geometries (Cotton and Wilkinson 1990). Among the aligned CadA-like sequences there are no conserved His residues. On the contrary, Asn residues are conserved in various positions and in the consensus motif and may provide a donor atom. Conserved Glu residues may also coordinate the metal.
DISCUSSION
Metallochaperones, soluble metal-transport proteins, and the P-type ATPases share a common structural fold, yet salient differences in their structures correlate with their roles in the mechanism of metal trafficking. The conservation of key residues among the families provides a strong basis for modeling using as templates the available high-resolution solution structures. The solution structures of Atx1 and Ccc2a in both Cu-bound and Cu-free forms (Arnesano et al. 2001b;Banci et al. 2001b) reveal features important for rapid and reversible transfer between physiological partners. A structural model for the Atx1-Ccc2a complex has also been determined on the basis of experimental data (Arnesano et al. 2001a). This model provides information on the factors determining the specificity of the interaction. A structural genomics approach applied to two classes of proteins that are the partners in the copper transfer process allows us to explore and model features relevant to metal trafficking across different phyla.
Hydrophobic Core
The clustering of buried residues is structurally important for stabilizing the tertiary fold of these proteins, and it represents a support for the metal-binding region, that is, loops 1 and 5, which are partially exposed to the solvent. Conserved contacts involving buried residues are listed in Tables 3 and 4.
An important difference in the classes of proteins is found for residue at position 73 of the global alignment (residue 65 in loop 5 of yeast Atx1) in eukaryotic metallochaperones, with respect to the corresponding amino acid in the ATPase domains and the small soluble metal-transport proteins. In eukaryotic metallochaperones, this residue is invariably a Lys whose positively charged side chain is very close to the copper site and points toward the solvent (Fig.8A). In ATPase domains, as well as in bacterial metal-transport proteins, the corresponding position in loop 5 (number 77 and 73, respectively) is occupied by a Phe or a Tyr, with the exception of the third domain of Menkes and Wilson proteins and few other cases. Conserved contacts are found between the aromatic ring of this Phe/Tyr, the side chain of the conserved Met in loop 1, and the second metal binding Cys (in helix α1; Fig. 8B). Compared with Ccc2a (Banci et al. 2001b) and MNK4 (Gitschier et al. 1998), larger structural changes are observed in the Atx1 metal-binding region (loop 1 and loop 5) upon copper release (Arnesano et al. 2001b). Those studies indicate that key structure and functional roles of the conserved Lys in metallochaperones, relative to the Phe/Tyr, may account for this difference.
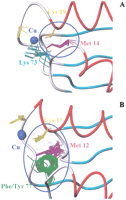
Conserved contacts between residues in the vicinity of the metal-binding site in eukaryotic metallochaperones (A) and ATPases (B).
The stabilization of the metal-binding site in Cu-Atx1, with respect to the apo form, is mainly attributable to Cu(I) coordination by the sulfur atoms of the cysteines. The positive side chain of Lys 65 in loop 5 leads to neutralization of the overall −1 charge of the Cu(I) bis-thiolate center, thus stabilizing the loop after metal binding and protecting the sulfur atom of the second Cys from solvent (Arnesano et al. 2001b). In the apo form, the first turn at the N-terminus of helix α1 folds back and loop 5 moves away from the copper site as Lys 65 is no longer attracted by the neutral protonated cysteines. The peptide dipoles of this turn also create a more positive potential that stabilizes the negative charge in the Cu(I)-loaded protein.
This trigger mechanism is absent in Ccc2a, in which a Phe replaces the Lys in loop 5. The apo form of the protein in the metal site is preorganized to receive the copper ion (Banci et al. 2001b).
The apparent flexibility of the peptide fold in the metal-binding region, observed in the metallochaperone (i.e., yeast apoAtx1) but not in the ATPase domain (i.e., yeast apoCcc2a), is supported by model analysis for other eukaryotic proteins (Fig. 8A,B). This feature of Atx1 may facilitate metal release to the target. These differences in the proximity of the metal-binding site between metallochaperones and ATPases can be related to their different functional roles in the copper-trafficking pathway.
The picture is somehow different in bacteria, wherein neither soluble metal transporters nor ATPase metal-binding domains contain a Lys in loop 5, and a Tyr or a Phe is found instead. In particular, in almost all bacterial ATPase domains this key position is occupied by a Tyr, at variance with eukaryotes, where a Phe is frequently found.
In the third metal-binding domain of Menkes and Wilson proteins, a Pro is invariably found in loop 5 in place of Phe/Tyr. This does not affect the conformation of the loop in modeled structures but may be relevant to the dynamic properties of these domains. Furthermore, the amino acid sequences of the third domain display less similarity to the other domains, and the computed instability index of the third domain, based on its primary structure, indicates lower stability. This can be either important for a cooperative metal binding of different domains or for relocation of the ATPases to the plasma membrane (Cobine et al. 2000), probably induced by conformational changes.
The Metal-Binding Site
The metal-binding motif conserved in metallochaperones, soluble metal-transport proteins, and metal-transporting ATPases is formed by the sequence x‘-x"-C-x‴-x⁗-C that has been used as a search criterion. Residue x‘ is highly conserved, being a Met in 91% of the proteins found. This side chain is not directly involved in metal ion coordination because it points toward the hydrophobic core of the protein and is involved in hydrophobic interactions, which may stabilize the folding of the metal-binding loop. Position x" shows only a few types of amino acids, usually Ser, Thr, or His. In ATPase domains from eukaryotes, a Thr residue is always at this position except for the third domain of the Menkes and Wilson proteins, where a His residue is found. Residue x‴ denotes a truly variable position, whereas x⁗ is quite conserved only within some subgroups. Residue x⁗ is always a Gly in eukaryotic metallochaperones. It is often a His or a Gly in CopZ-like proteins or an Ala or Thr in MerP-like proteins. It is Ser or Ala in eukaryotic ATPases. The large variability observed at position x‴ indicates that, in the extreme case, this residue may be deleted or an additional residue may be inserted, producing different consensus motifs for metal binding, CxC and CxxxC, respectively, as observed in other metal tranporters such as SilP, a silver ATPase fromSalmonella typhimurium (Gupta et al. 1999) containing CxxxC, or in CopY from E. hirae containing two conserved CxC motifs (Odermatt and Solioz 1995).
Other factors, in addition to the identity of residues in the metal-binding motif, could determine metal selectivity and its influence on protein–protein interactions, as also addressed by experimental works (Lutsenko et al. 1997; Larin et al. 1999; Veglia et al. 2000). One of them could be the size of the binding pocket, which on its turn can be controlled by the tightness of the turn of the binding loop or by electrostatics. Regarding the latter, differences in charge distribution in the vicinity of the metal-binding site among the various subfamilies of structural models are observed. The different pattern of charged residues in MerP-like structures with respect to eukaryotic metallochaperones and CopZ-like metal tranporters may be related to the different charge and type of coordination, for example, of Hg(II) with respect to Cu(I).
The nature of the residues in the turn of the metal-binding loop affects their interactions with the solvent and with other protein residues and can contribute to the turn tightness. Highly conserved residues are present in loop 3, which in almost all the structures are in contact with the residues following the second Cys of the metal-binding motif. A conformational change upon Hg(II) binding has been observed for a Phe residue in loop 3 in MerP from Sh. flexneri (Steele and Opella 1997).
Another important aspect to be considered is the surface interactions that determine the tightness of the turn of the metal-binding loop. Position 42 in metallochaperones and soluble metal transporters and position 44 in soluble domains of ATPases, both located in loop 3, are occupied by a Leu or a Phe in 86% of the sequences and are exposed to the solvent. In almost all structures, these residues are in contact with the amino acid immediately following the second Cys in the metal-binding motif (position 20 in metallochaperones and metal transporters; position 18 in ATPases). It can be proposed that these corresponding positions may be involved in controlling the stability of the metal-binding loop.
Surface Potentials and Protein–Protein Interactions
Residues on the surface of the structural models are less conserved than those forming the hydrophobic core. This is related to the specific function and interaction partner of the protein.
In metallochaperones, soluble metal-transport proteins, and ATPase domains we found different surface charge distributions, even within eukaryotic and bacterial subgroups. Figure9 shows the surface potentials of a metallochaperone (Atx1 from S. cerevisiae), a soluble metal-transport protein (CopZ from B. subtilis), and two ATPase-soluble domains (Ccc2a from S. cerevisiae and CopAa from B. subtilis).
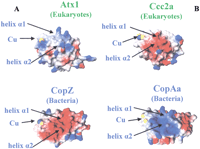
Surface potentials of S. cerevisiae Atx1 and B. subtilis CopZ structures (A). Surface potentials ofS. cerevisiae Ccc2a and B. subtilis CopAa structures (B).
In metallochaperones there are two conserved negative regions, corresponding to strand β2 and the N-terminal end of helix α2, and two conserved positive regions, helix α1 and the C-terminal part of helix α2 with loop 5. In particular, positions 25 and 29 (helix α1) and positions 67 and 73 (helix α2 and loop5) are occupied by Lys or Arg residues. On the contrary, in eukaryotic ATPases, several Glu and Asp residues, conserved in positions 22, 72, 74, and 78 with few exceptions, generate a negatively charged face on the protein surface in the proximity of the copper-binding region (Fig. 9). With respect to MerP-like proteins, little is known about the structure of putative partner proteins, but it is interesting to note that the negative regions are more scattered on the surface with respect to Atx1-like structures, whereas positive patches are present on helices α1 and α2, in analogy with metallochaperone sequences.
At variance with eukaryotic metallochaperones, CopZ-like models (bacteria) show a large variability of charged residues. Interestingly, the calculated isoelectric points (pI’s) show that the CopZ-like sequences (pI's of 4–5) are more acidic than the Atx1-like chaperones (pI's of 7–8).
It has been suggested that electrostatic forces play a crucial role in the interaction of the two yeast proteins Atx1 and Ccc2a (Portnoy et al. 1999; Wernimont et al. 2000). A model for the complex has been proposed based on experimental data (Arnesano et al. 2001a). Interactions occur on a protein–protein interface that includes the C-x-x-C metal-binding motif. In the model of the complex between Atx1 and Ccc2a, loop 1 and the N-terminus of helix α1 of one protein are in contact with loop 5 and the C-terminus of helix α2 of the partner, and the two helices α1 are tilted ∼45° to allow close contact between the two metal-binding regions of the two proteins (Arnesano et al. 2001a). The two pairs of cysteines are facing each other to facilitate a low barrier metal transfer through a series of two- and three-coordinate metal-bridged intermediates (Pufahl et al. 1997;Wernimont et al. 2000). The regions at the interface of the two molecules contain a large number of oppositely charged residues (Fig.9). For instance, an electrostatic interaction is possible between Asp 65 (loop 5) of Ccc2a and Lys 24 (helix α1) of Atx1, which are both conservatively substituted in homologous eukaryote sequences. This complementary charge distribution in metallochaperones and ATPases is essentially conserved in eukaryotes, strongly supporting a similar mechanism of interaction between physiological partners.
On this basis, some considerations can be made on multidomain ATPases in which differences in surface charge distribution are observed in distinct metal-binding domains. This indicates that metallochaperones may preferentially interact with some of these ATPase domains. Moreover, an interaction can be predicted among different domains of the same ATPase because they all share the same βαββαβ fold and, in some cases, they have surface potential complementarity, in full analogy with the case of Atx1 and Ccc2a.
In Figure 10 the surface potentials of the six metal-binding domains of the Menkes protein are reported. The structures are shown with the same orientation to show the charged helices α1 and α2 and loops 1 and 5, located at the protein–protein interface in the Atx1-Ccc2a complex. Surface potentials of domains 1 and 2 are similar to those of Ccc2a and Atx1, respectively, and they are likely to interact electrostatically. Another possible interaction can be predicted between domains 4 and 5. A change in the tertiary structure interactions, favored by interdomain flexible stretches, can cause cooperative metal binding signaling the ATPase of increased copper levels. This mechanism may permit ATPase translocation from the trans-Golgi network to the plasma membrane (Cobine et al. 2000).

Surface potentials of the six metal-binding domains of the Menkes protein.
In bacterial metallochaperone homologs and ATPase sequences, although large amino acid variability prevents us from identifying key surface residues from sequence analysis alone, the comparative modeling allows us to highlight complementary charge distributions on CopZ-like and CopA-like proteins from several different organisms, indicating that the two proteins can be partners in vivo. In fact, when changes occur in the surface electrostatic features between proteins from different organisms, they are compensated by coevolutionary changes on the surface of putative partner proteins. For example, in CopZ from B. subtilis (sequence 14), a negatively charged face is formed by Glu and Asp residues belonging to loop 1 and helices α1 and α2. In the CopA-like ATPase from the same organism (sequence 64), Arg and Lys residues form a positively charged face. This indicates an interaction mechanism between CopZ and CopA similar to that observed in yeast proteins, Atx1 and Ccc2 (Arnesano et al. 2001a), but with reversed charges (Fig. 9). A partnership between these proteins should be tested experimentally.
In E. hirae, CopZ is proposed to interact in vivo with the copper responsive repressor, CopY (Odermatt and Solioz 1995; Cobine et al. 1999), whose structure is unknown. Alternative partners have not yet been identified. In CopZ from E. hirae, two opposite charge distributions have been observed on the two faces of the protein and it has been speculated that one face could interact with a copper donor and the other could support recognition of the copper acceptor, such as CopY (Wimmer et al. 1999). However, we found CopY homologs only in E. hirae, Streptococcus mutans, Streptococcus pyogenes, Lactococcus lactis, and Lactobacillus sakei. Interestingly, the computed pI values for CopZ sequences from these organisms (with the exception of L. sakei, for which none of the available genome sequences are CopZ-like) are larger (average pI = 8.1) than those of other CopZ-like proteins lacking CopY (average pI = 4.6). It can be proposed that conserved negative regions identified in the CopY repressor from E. hirae,S. mutans, S. pyogenes, and L. lactis may be needed for the interaction with basic regions on the surface of the corresponding CopZ or, alternatively, that CopZ-like soluble metal transporters have another physiological target.
CONCLUSIONS
This comparative structural genomic survey of new classes of proteins involved in metal transport provides insights into how metal ions are transported within the cells.
Sequence homology and the metal-binding consensus motif x‘-x"-C-x‴-x⁗-C were used to locate protein sequences starting from two copper-binding proteins from yeast, the metallochaperone Atx1 and a soluble domain of Ccc2 ATPase, known to interact in vivo (Huffman and O’Halloran 2000). Inspection of the genomic databases reveals a significant number of homologs, which, based on sequence analysis and on the conservation of key residues, are readily segregated into a minimum of four groups: the Atx1-like metallochaperones, CopZ-like proteins, MerP-like proteins, and the domains of the P-type ATPases.
The most extensive structural group, the Atx1-like metallochaperones, function by protecting their essential metal ion cargo from adventitious binding agents in the cellular milieu but releasing the metal in a facile way to partner proteins (Pufahl et al. 1997). In this way, cells can overcome their chelation capacity that otherwise maintains an extremely low availability of metal cofactors and efficiently traffic them to intracellular destinations. Additional functions may emerge as well. The partnerships of the CopZ proteins are not yet clear, with both regulatory and transporter proteins as proposed targets. Two members of this subfamily have been characterized at the genetic and structural level to date (Wimmer et al. 1999; Banci et al. 2001a). The other members of the CopZ-like family may or may not correspond to copper-specific operons as some may provide tolerance or resistance to other types of metals and could even be involved in metal uptake.
Both Atx1 and CopZ share significant sequence similarities with a periplasmic mercury resistance protein, MerP, which interacts with a partner protein in the detoxification operon to import Hg(II) (Steele and Opella 1997). These proteins also share the same metal-binding motif and fold with the soluble domains of the physiological targets of Atx1, the copper-transporting P-type ATPases.
Some sequences, whose experimental structures were available, were used as templates for the modeling. Through the analysis of 3D-modeled structures, we were able to assign different functional roles to conserved amino acids: Some residues forming the hydrophobic core are conserved in all the classes in accordance with a common fold; charged surface residues are more conserved among eukaryotic sequences and more variable in bacteria and archaea.
Within each homology group, a series of distinguishing hallmarks of mechanistic importance can be discerned. In the Atx1-like subfamily we find a conserved metal-binding loop-helix junction and, on the helical face, a pattern of arginines and lysines, one of which folds over to protect the metal-binding site. Neither of these features is seen in the other subfamilies. In the family of P-type ATPases, the partners of Atx1, we find a more rigid hydrophobic core, complementary acidic electrostatic surfaces, and conserved hydrophobic patches.
Some observed sequence and structure variations between eukaryotic and bacterial proteins can be related to a different functional role and/or metal specificity. This will be difficult to assess until the functions and metal specificity are better established for CopZ and other members of this subfamily.
Alternatively, all these proteins may share similar functions but have different surface decoration that is related to different partnerships at the different phylogenetic levels. The presence of soluble metal transporters and soluble domains of ATPases in eukaryotes (as well as in bacteria and archaea), their common βαββαβ fold, and the conserved CxxC metal-binding motif, located in the loop between strand β1 and helix α1, indicate a common interaction mechanism similar to that observed between Atx1 and Ccc2a in yeast (Arnesano et al. 2001a). A partnership is proposed between CopZ-like and CopA-like proteins in bacteria. Furthermore, an interaction between individual metal-binding domains of ATPases can be predicted on the basis of surface potential analysis.
It will be interesting to see how the functions of these small proteins and domains that emerge from this genomic survey compare with the mechanistic and functional attributes of the initially characterized members. This survey can serve as a starting point for tests of the anticipated partner proteins or of the nature of physiological metal cargo, that is, a toxic nonessential metal such as Cd(II) or an essential metal such as Zn(II).
METHODS
Sequence Searching
Metallochaperones, small soluble metal-transport proteins, and ATPase sequences were searched for in the GenBank Databases (CDS translations + PDB + SwissProt + PIR + PRF) using sequence similarity criteria. This was accomplished by starting from sequences of proteins of known function (the copper chaperone Atx1 and the first soluble domain of the copper-transporting ATPase Ccc2 from yeastS. cerevisiae) and performing a PHI-BLAST(Altschul et al. 1997) search with a threshold of 20% sequence identity (http://www.ncbi.nlm.nih.gov/BLAST/phiblast.html) using the sequence length as search criterion and the x‘-x"-C-x‴-x⁗-C metal-binding motif. In the first search x‘ was set to M. On the ensemble of sequences, having residue identity above 20% with the reference sequences, we analyzed the conservation of x‘-x⁗ residues. Then, we performed a further PHI-BLAST search restricted to recurring residues at positions x, not imposing M in the first position of the metal-binding motif (x‘). Finally, the ensemble was further extended using as starting sequences each sequence of the ensemble. In the case of ATPases, in which more than one metal-binding motif was present at the N-terminal end, the sequences were subdivided into individual metal-binding domains. Each domain was aligned, neglecting the interdomain sequences and the membrane spanning portion. Sequence alignments were performed with CLUSTALW (Thompson et al. 1994).
Structural Prediction and Homology Modeling
Secondary structure prediction was performed through the PHD Method (Rost and Sander 1993, 1994) (http://pbil.ibcp.fr), which uses evolutionary information in the form of multiple sequence alignments that are used as input in place of single sequences.
A weight value of instability, as resulted from the statistical analysis of proteins of different stability, is assigned to
each of the 400 different pairs of amino acids (DIWV) (Guruprasad et al. 1990). Using these weight values it is possible to compute an instability index (II) (http://www.expasy.ch/tools/protparam.html) which is defined as: where: L is the sequence length,DIWV(x[i]x[i + 1])is the instability weight value for the dipeptide starting in position i. A protein whose instability index is smaller than
40 is predicted as stable, a value above 40 predicts that the protein may be unstable.
where: L is the sequence length,DIWV(x[i]x[i + 1])is the instability weight value for the dipeptide starting in position i. A protein whose instability index is smaller than
40 is predicted as stable, a value above 40 predicts that the protein may be unstable.
Structures were modeled with the program MODELLER (version 4.0) (Sali and Blundell 1993), using as reference the available solution structures: Cu(I)-Atx1 from S. cerevisiae (PDB ID1FD8); Cu(I)-CopZ from B. subtilis (PDB ID 1K0V); apo-CopZ from E. hirae (PDB ID 1CPZ); Hg(II)-MerP from Sh. flexneri (PDB ID 1AFJ); Cu(I)-Ccc2a, the first domain of Ccc2, fromS. cerevisiae (PDB ID 1FVS); and Ag(I)-MNK4, the fourth domain of the human Menkes protein (PDB ID 2AW0).
Amino acid residues of all metallochaperone homolog and ATPase sequences have been numbered on the basis of the corresponding alignment including gaps.
The program PROSA II (version 3.0, 1994) (Sippl 1993) for protein structure analysis was used to test the consistency and validity of the models. It provides an adimensional energy term per residue that is a combination of pair interaction energies and surface energies. The surface term is used to model the energetic features of solvent-protein interactions as it takes into account solvent exposure (Babajide et al. 1997). These energies are then converted into Z-scores using a polyprotein (Sippl 1993; Babajide et al. 1997) through the standard “hide and seek” procedure of the program. They are correlated with the difference in potential energy and calculated using mean field potentials between the input structure and other randomly assigned folds for its amino acid sequence. They can be considered as a measure for the quality of the modeling: a lower Z-score corresponds to a more favorable potential energy associated with the structure under examination.
Secondary structure and Ramachandran plot analyses were performed withPROCHECK (Laskowski et al. 1993). Solvent accessibility and surface potentials for individual residues were evaluated withMOLMOL (version 2.6) (Koradi et al. 1996). Two residues were assumed to be in contact if at least five pairs of atoms were closer than 4 Å. Buried residues were defined as those having a solvent accessibility lower than 25 Å2. For most residues, this value indicates that 90% or more of the surface is buried (Rodionov and Blundell 1998).
WEB SITE REFERENCES
http://pbil.ibcp.fr, the PHD Method, which uses evolutionary information in the form of multiple sequence alignments that are used as input in place of single sequences.
http://www.expasy.ch/tools/protparam.html, instability index.
http://www.ncbi.nlm.nih.gov/BLAST/phiblast.html, PHI-BLASTsearch.
http://www.ncbi.nlm.nih.gov/PMGifs/Genomes/org.html, for complete genomes.
Acknowledgments
We thank Professor Anthony G. Wedd for careful reading of the manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 Present address: CERM and Department of Chemistry, University of Florence, Via L. Sacconi 6, Sesto Fiorentino, Florence, Italy 50019.
-
3Corresponding author.
-
E-MAIL bertini{at}cerm.unifi.it; FAX 39-055-4574271.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.196802.
-
- Received May 17, 2001.
- Accepted November 30, 2001.
- Cold Spring Harbor Laboratory Press








