
Positional Candidate Cloning of a QTL in Dairy Cattle: Identification of a Missense Mutation in the Bovine DGAT1 Gene with Major Effect on Milk Yield and Composition
Abstract
We recently mapped a quantitative trait locus (QTL) with a major effect on milk composition—particularly fat content—to the centromeric end of bovine chromosome 14. We subsequently exploited linkage disequilibrium to refine the map position of this QTL to a 3-cM chromosome interval bounded by microsatellite markers BULGE13and BULGE09. We herein report the positional candidate cloning of this QTL, involving (1) the construction of a BAC contig spanning the corresponding marker interval, (2) the demonstration that a very strong candidate gene, acylCoA:diacylglycerol acyltransferase (DGAT1), maps to that contig, and (3) the identification of a nonconservative K232A substitution in the DGAT1 gene with a major effect on milk fat content and other milk characteristics.
[The sequence data described in this paper have been submitted to the GenBank data library under accession number AY065621.]
The majority of economically important traits in livestock are complex, continuously distributed phenotypes, which are influenced by multiple polygenes located at quantitative trait loci (QTL) dispersed across the genome. Spectacular advances in production efficiency have been realized following the implementation of sophisticated selection strategies founded on quantitative genetics theory. One of the strengths of these biometrical selection strategies is that they obviate the need for a detailed understanding of the genes upon which they act.
There is, however, great interest in gaining better knowledge of the molecular architecture of complex quantitative traits. This could indeed lead to new insights in the evolutionary forces undergone by natural and domestic populations, as well as the molecular physiology of the phenotypes of interest, and is expected to generate new opportunities for more effective “marker-assisted breeding.”
With the development of comprehensive marker maps for several species, it has become possible to map QTL influencing a number of medically and agronomically important traits. The picture that emerges is that of an exponential distribution of QTL effects: a few loci with moderate to large effects are amenable to mapping, while the remaining of the genetic variation remains elusive. Even for mappable QTL, however, the actual identity of the gene(s) and polymorphism(s) responsible for the QTL effect has so far remained unknown, with a few exceptions in plants and model organisms (Andersson 2001; Flint and Mott 2001; Mackay 2001;Mauricio 2001).
In this paper, we present very strong evidence for the first positional cloning of a QTL in an outbred mammal. This QTL, which has a major effect on milk yield and composition in dairy cattle, was previously mapped to a 3-cM interval on the telomeric end of bovine chromosome 14 (Coppieters et al. 1998; Heyen et al. 1999; Riquet et al. 1999; Looft et al. 2001; Farnir et al. 2002). We herein report the identification of a very strong positional candidate (DGAT1: acylCoA:diacylglycerol acyltransferase 1) in this interval, and the detection of a nonconservative K232A substitution in it that most likely causes the BTA14 QTL effect.
RESULTS
Construction of a BAC Contig Spanning the BULGE13-BULGE09Interval
To clone the gene(s) responsible for the observed QTL effect, we constructed a BAC contig spanning the BULGE13-BULGE09 marker interval containing the QTL. We accomplished this by screening a bovine BAC library (Warren et al. 2000) by filter hybridization with the microsatellite markers available for proximal BTA14q, as well as human cDNA clones known to map to the orthologous chromosome region in the human, that is, HSA8q23-tel (Riquet et al. 1999). The ends of the isolated BACs were sequenced, and sequence tagged sites (STSs) were developed from the corresponding sequences and mapped onto a bovine × hamster whole genome radiation hybrid panel (Womack et al. 1997). If the corresponding STSs were indeed mapping to proximal BTA14q as expected, they were tested on all other BACs available in the region of interest. This STS content mapping approach lead to the BAC contig shown in Figure 1.
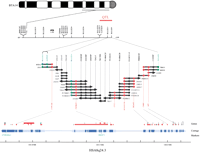
(See figure on page 224.) Generation of a BAC contig spanning the BULGE13-BULGE09 interval. The most likely position of the QTL (Farnir et al. 2002) is shown as a red bar on the FISH-anchored linkage map of proximal BTA14q. The BACs composing the contigs spanning the BULGE13-BULGE09 interval are shown as a series of black horizontal lines. The dots on each BAC indicate their individual STS content: black dots correspond to STSs derived from BAC ends, green dots to microsatellite markers, and red dots to gene-specific comparative anchored tagged sequences (CATS;Lyons et al. 1997). Black arrowheads mark the BACs from which the respective BAC and STS were derived. The length of the lines do not reflect the actual insert size of the corresponding BACs. The BAC contig is aligned with the orthologous human HSA8q24.3 genomic “golden path” sequence (Lander et al. 2001) represented according to the Ensembl Human Genome Server (http://www.ensembl.org/): individual sequence contigs are shown in alternating dark and light blue; a horizontal line indicates a gap in the sequence assembly; genetic markers are indicated in green under the contig map; and green, red, and black boxes represent “curated,” “predicted known,” and “predicted novel” genes, respectively.
DGAT1 Maps to the BULGE13-BULGE09 Interval and Is a Strong Positional Candidate for the QTL
A gene encoding a protein with acylCoA:diacylglycerol acyltransferase (DGAT1 - EC 2.3.1.20) activity was recently identified (Cases et al. 1998) and shown to completely inhibit lactation when knocked out in the mouse (Smith et al. 2000). This gene was initially mapped to HSA8qter by FISH analysis (Cases et al. 1998), and is now known to be located at position 143.8 Mb on the HSA8q24.3 genomic sequence (Lander et al. 2001) (Fig. 1). We screened the publicly available databases with the published murine and humanDGAT1 cDNA sequences and identified three bovine expressed sequence tags (ESTs) (AW446908, AW446985, AW652329) jointly covering approximately two-thirds of the bovine gene. By aligning the humanDGAT1 genomic sequences with the human and bovine cDNA sequences, we could identify the corresponding intron–exon boundaries and develop PCR primers that would amplify a portion of the bovineDGAT1 gene from genomic DNA. We screened our contig with this STS and clearly showed that the bovine DGAT1 gene was contained in a subset of our BACs, allowing us to accurately position the DGAT1 gene in the BULGE13-BULGE09 interval (Fig.1). These results demonstrated that the map position of DGAT1coincides precisely with the most likely position of the chromosome 14 QTL as determined by linkage and linkage disequilibrium (LD) analyses. Knowing that this QTL primarily affects milk fat content, that 98% of milk lipids are triglycerides, that DGAT1 catalyzes the final step in triglyceride synthesis, and knowing the effect of aDGAT1 knock-out on lactation, we considered this gene to be a very strong positional candidate for the corresponding QTL.
Genomic Organization of the Bovine DGAT1 Gene
We determined the organization of the bovine DGAT1 gene by sequence analysis of one of our DGAT1-containing BACs. We designed primers based on the available bovine, murine, and human cDNA sequences which we used either for direct sequencing of the BAC clone or to generate PCR products which were then cycle-sequenced. We merged all available sequences using the software programPhred/Phrap (Ewing et al. 1998; Ewing and Green 1998;Gordon et al. 1998) to yield a consensus sequence that has been submitted to Genbank under accession number AY065621. We performed RT-PCR and 5′ and 3′ RACE experiments on mRNA isolated from bovine mammary gland, and we cycle-sequenced the obtained PCR products.
By comparing the genomic and cDNA sequences, we showed that the bovineDGAT1 gene spans 8.6 Kb and comprises 17 exons measuring 121.8 bp on average (range: 42–436 bp). Whereas the first two introns are respectively 3.6- and 1.9 Kb-long, the remaining 14 introns are only 92.4 bp-long on average (range: 70–215 bp). All introns conform to the GT-AG rule and are strictly conserved between human and bovine. The bovine DGAT1 gene is transcribed in an mRNA comprising 245 bp of 5'UTR sequence, 1470 bp coding for a protein of 489 amino acids, and 275 bp of 3'UTR sequence including a canonical AATAAA polyadenylation signal (Fig. 2). The human and bovine DGAT1 nucleotide (coding) and protein sequences are respectively 89.5% and 92.5% identical.
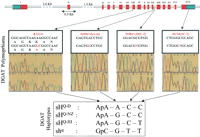
Genomic organization, polymorphisms, and haplotypes found in the bovineDGAT1 gene. Leader and trailer sequences are shown in green, coding sequences in red, and intronic sequences in gray. The positions of the four identified polymorphisms are marked by yellow lines on the gene, and detailed in the underlying boxes including the corresponding sequence traces. The four DGAT1 haplotypes which were found in the Dutch and New Zealand Holstein-Friesian population are shown and referred to as “sHQ-D ”, “sHQ-NZ ”, and “sHQ-III ” for the fat-increasing haplotypes and “shq “ for the fat-decreasing haplotype.
The Predicted “Q” and “q” QTL Alleles Differ by a Nonconservative Lysine to Alanine Amino Acid Substitution in the DGAT1 Gene
In previous linkage studies, we identified 13 sires that were predicted to be heterozygous “Qq” for the chromosome 14 QTL (Coppieters et al. 1998; Riquet et al. 1999). These sires were indeed shown to carry a “Q” allele that markedly increases milk fat content when transmitted to offspring, compared to the alternate “q” allele. We then showed that these “Q” alleles are in strong LD with two specific microsatellite haplotypes referred to as μHQ-D andμHQ-NZ as they occur respectively in the Dutch and New-Zealand Holstein-Friesian populations (Riquet et al. 1999; Farnir et al. 2002). The “q” QTL alleles, in contrast, were found in both populations to be associated with multiple microsatellite haplotypes, which were jointly referred to as μhq .
Assuming that DGAT1 is indeed the QTL, we predicted that the identified “Q” and “q” QTL alleles would correspond to functionally distinct DGAT1 alleles, that is, they would differ at one or more mutations, causing these alleles to be functionally different. To test this hypothesis, we sequenced theDGAT1 gene from (1) two Dutch “Qq” sires withμHQ-D /μhq genotype as well as two of their μHQ-D /μHQ-D offspring, two of theirμhq /μhq offspring, and one μHQ-D /μhq offspring, and (2) one New Zealand “Qq” sire withμHQ-NZ /μhq genotype and one of its μHQ-NZ /μHQ-NZ offspring.
We designed primer pairs that allowed for the amplification from genomic DNA of (1) the coding portion of exon I, (2) exon II, and (3) the chromosome segment spanning exons III to XVII. We amplified the corresponding PCR products from genomic DNA of the selected individuals, cycle-sequenced these and examined the resulting traces with the software program Polyphred (Nickerson et al. 1997).
This analysis revealed four polymorphisms in the DGAT1 gene (Fig. 2): (1) an ApA to GpC dinucleotide substitution in exon VIII, causing a K to A amino acid substitution (K232A), (2) an A to Gsubstitution in intron 12, eight base pairs downstream of exon XII [Nt984 + 8(A-G)], (iii) a C toT substitution in intron 12, 26 bp downstream of exon XII [Nt984 + 26(C-T)], and (4) a C toT transition in the 3′UTR region [Nt1501(C-T))].
These four polymorphisms were shown to assort into three distinctSNP haplotypes referred to as sHQ-D ,sHQ-NZ , and shq because in the sequenced samples they coincided with microsatellitehaplotypes μHQ-D ,μHQ-NZ , andμhq , respectively. The base pair compositions of these three SNP haplotypes are shown in Figure 2.
Because the sHQ-NZ and shq marker haplotypes share the G residue at the DGAT1 Nt984 + 8(A-G) site, the causality of this polymorphism in the determinism of the QTL could be excluded. For the three remaining polymorphic sites, however, the DGAT1haplotypes associated with marker haplotypes sHQ-D and sHQ-NZ proved identical to each other while different from the shq DGAT1 haplotype. Any of these three polymorphisms could therefore be responsible for the observed QTL effect. The Nt984 + 26(C-T) andNt1501(C-T) polymorphisms are a priori more likely to be neutral with respect to DGAT1 activity because of their respective location in an intron and the 3'UTR. A direct effect of theK232A mutation on DGAT1 activity, however, seems very plausible. Indeed, the corresponding mutation causes the nonconservative substitution of a positively charged lysine residue with a neutral, hydrophobic alanine residue. With the exception ofCercopithecus aethiops, where it is nevertheless replaced by a positively charged arginine, the corresponding lysine residue is conserved among all examined mammals (i.e., human, mouse, rat, pig, sheep, bison), demonstrating its functional importance (Fig.3). The evolutionary conservation of this lysine residue also demonstrates that the K residue characterizing the sHQ-D andsHQ-NZ marker haplotypes (associated with an increase in milk fat content) is more than likely the ancestral state and that it is the A residue characterizing theshq haplotypes that corresponds to a more recently evolved state.

Multiple sequence alignment of a portion of the DGAT1 protein of Bos taurus, Bison bison, Ovis aries,Sus scrofa, Homo sapiens, Cercopithecus aethiops, Mus musculus domesticus, and Rattus norvegicus showing the evolutionary conservation of the lysine mutated in the bovine K232A polymorphism.
The K232A Mutation Is Associated with Major Effects on Milk Yield and Composition
We developed an oligonucleotide ligation assay (OLA) as described (Karim et al. 2000) that allows for efficient genotyping of the fourDGAT1 polymorphisms simultaneously. This OLA-test was used to genotype a previously described (Coppieters et al. 1998) “granddaughter design” (i.e., series of 84 paternal half-brother pedigrees) comprising 1,818 Dutch Holstein-Friesian sires as well as a “daughter” design (i.e., series of 51 paternal half-sister pedigrees) comprising 529 New Zealand Holstein-Friesian cows. We determined the marker linkage phase for each individual as described (Farnir et al. 2000).
Figure 4 summarizes the frequency distribution of DGAT1 haplotypes encountered in the Dutch and New Zealand populations. We identified four distinct SNPhaplotypes. Three of these correspond to the sHQ-D ,sHQ-NZ , and shq that were previously identified by sequencing, and jointly account for 99% and 98% of the chromosomes in the Dutch and New Zealand populations, respectively. A fourth minor haplotype was found accounting for the remaining 1% and 2% of the chromosomes. As this haplotype codes for aK residue at position 232, it was assumed to correspond to a fat-increasing “Q” allele and was therefore referred to assHQ-III (Fig. 2). The observation that theK residue is found on three distinct DGAT1 haplotypes whereas the A residue is found on a unique DGAT1haplotype is in agreement with K being the more ancient state.
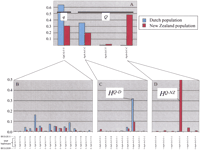
(A) Frequency distribution of observed DGAT1 SNP haplotypes in the Dutch (blue) and New Zealand (red) Holstein-Friesian dairy cattle populations. The correspondence with the previously defined “Q” and “q” QTL alleles (Farnir et al. 2002) is shown. (B–D) Frequency distribution of the combined microsatellite (BULGE09-BULGE11) andSNP DGAT1 haplotypes. The previously definedHQ-D and HQ-NZ haplotypes (Farnir et al. 2002) are shown.
The sHQ-D and sHQ-NZ SNP haplotypes (coding for a K residue at position 232) appear to be in strong LD with the flanking microsatellite markersBULGE09 and BULGE11, as they are in essence associated with unique microsatellite haplotypes corresponding respectively to the previously definedμHQ-D andμHQ-NZ haplotypes (Fig. 4C,D). In sharp contrast, the shq haplotype (coding for anA residue at position 232) is nearly evenly distributed across more than 10 distinct microsatellite haplotypes (Fig. 4B).
These observations are in excellent agreement with the results of the combined linkage and LD analysis (Farnir et al. 2002). These studies indeed predicted that (1) in the Dutch population, the vast majority (estimates ranging from 81% to 92%) of “Q” allele (= K) would reside on theμHQ-D microsatellite haplotype, (2) in the New Zealand population, a large fraction (estimates ranging from 36% to 51%) of “Q” alleles would reside on haplotype μHQ-NZ (we now see that the remainder correspond mainly to theμHQ-D microsatellite haplotype), and (3) in both populations, the “q” alleles (= A) would correspond to multiple marker haplotypes, corresponding to hq .
Figure 5 illustrates the gain in LD signal that could be obtained in the Dutch Holstein-Friesian granddaughter design when adding the DGAT1 polymorphisms to the previously available markers for proximal BTA14q and performing a joint linkage and LD multipoint analysis (Farnir et al. 2002) using the sires “daughter yield deviations” (DYD; see Methods) for milk fat percentage as phenotype. It can be seen that the lod score (see Methods) attributable to LD essentially doubles (from 3.7 to 7.8), and maximizes exactly at the position of the DGAT1 gene. This result strongly supports the causal involvement of theDGAT1 gene in the QTL effect. The corresponding ML estimates of the “Q” to “q” allele substitution effect (α/2) (as defined in Falconer and Mackay 1996), residual standard deviation (ς), population frequency of the “Q” allele (fQ ), number of generations to coalescence (g), and heterogeneity parameter (ρ) (see Methods) were 0.11% (α/2), 0.06% (ς), 0.20 (fQ ), 5 (g), and 0.84 (ρ), respectively.
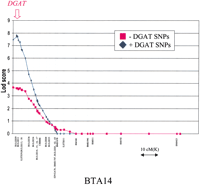
Lod score due to LD when including (blue) or excluding (red) the fourDGAT1 polymorphisms in a combined linkage and LD multipoint maximum likelihood mapping method (Farnir et al. 2002). The lod score corresponds to the log10 of the ratio between the likelihood of the data assuming linkage and LD between the markers and the QTL and the likelihood of the data assuming linkage in the absence of LD. The positions of the microsatellites and SNP markers used in the analysis are shown on the x-axis, and the position of theDGAT1 SNPs is marked by a red arrow at the top of the figure.
Using the same Dutch Holstein-Friesian population, we then examined the additive effect of the DGAT1 K232A polymorphism on milk yield and composition. The sons' DYDs for milk yield (kg), protein yield (kg), fat yield (kg), protein percentage, and fat percentage were analyzed using a mixed model including (1) a regression on the number of K alleles in the genotype (0, 1, or 2), and (2) a random polygenic component estimated using an individual animal model and accounting for all known pedigree relationships. Table1 reports the obtained results. It can be seen that the K232A mutation has an extremely significant effect on the DYDs of the five dairy traits analyzed. The proportion of the DYD variance explained by this polymorphism in this population ranges from 8% (protein yield) to 51% (fat percentage), corresponding to between 10% (protein yield) and 64% (fat percentage) of the genetic variance (= QTL + polygenic). Note that the proportion of the variance explained by the full model (1-r2 error) is of the order of 70% for the yield DYDs and 80% for the percentage DYDs, which is in agreement with their known reliabilities. An interesting feature of this QTL effect is that the “q” to “Q” substitution increases fat yield while decreasing milk and protein yield, despite the overall positive correlation characterizing the three yield traits.
Effect of the DGAT1 K232A Mutation on Sires' Daughter Yield Deviations (DYDs) for Milk Yield and Composition
The two previous analyses examined the effect of the DGAT1polymorphism on estimated breeding values. By definition, this phenotype will only account for the additive component of theDGAT1 effect, and justifies the use of a regression on the number of K alleles in the mixed model. To evaluate the dominance relationship between the DGAT1 alleles, we analyzed the effect of the K232A genotype on the lactation values (first-yield deviations) of the cows composing the New Zealand daughter design. This was achieved by using a mixed model including (1) a fixed effect corresponding to the K232A genotype, and (2) a random polygenic component accounting for all known pedigree relationships (“animal model”). Very significant effects of K232Agenotype on all examined yield and composition traits were found in this population as well (Table 2), accounting for between 1% (protein yield) and 31% (fat percentage) of the variance of lactation values. The observed dominance deviations,d, corresponding to the difference between the genotypic value of the KA genotype and the midpoint between the AAand KK genotypic values (Falconer and Mackay 1996) are shown in Table 2. Genotypic values of the heterozygous genotype are systematically in between alternate homozygotes. None of thed-values proved to be significantly different from zero, indicating an absence of dominance. Average K to AQTL allele substitution effects, α (Falconer and Mackay 1996), were computed from the estimates of a- and d-values, as well as the population frequencies of the K and Aalleles (Table 2). The predicted substitution effects are generally in agreement with those computed from the granddaughter design (estimates of α/2): the K allele increases fat yield, fat percentage, and protein percentage, while decreasing milk and protein yields. For the yield traits, the absolute values of α estimated from the granddaughter are larger when compared to the daughter design. The exact reasons for this are being explored. It could be due to the fact that the sire population in the granddaughter design is not representative of the cow population in general, or to intrinsic differences between the Dutch and New Zealand populations and /or environment. The estimates of α for the percentage traits cannot be directly compared as these are computed from the yield traits using different conversion formulas in the two countries.
Effect of the DGAT1 K232A Mutation on Cows' Lactation Values for Milk Yield and Composition
DISCUSSION
We herein report one of the first successful positional cloning efforts of a QTL in an outbred species, including human. This success was undoubtedly enabled by two factors that facilitated fine-mapping of this QTL: the magnitude of its effect and the fact that it was attributable to a single mutation in one gene. In addition, the identified mutation proved to be a more easily interpretable missense mutation (rather than for instance a regulatory promoter mutation) in a very strongly supported candidate gene. Forthcoming QTL cloning experiments are likely to be more complicated, because all of these conditions will in general not apply. Our present results, however, demonstrate the feasibility of positional cloning as an approach to identify QTL and should encourage further efforts along these lines.
Several lines of evidence strongly support the fact that K232Ais indeed the causal mutation or “quantitative trait nucleotide” (QTN; Mackay 2001):
(1) DGAT1 has very strong candidacy given its known role in fat metabolism and knockout effect (Cases et al. 1998; Smith et al. 2000).
(2) The evolutionary conservation of the affected lysine residue among mammals indicates the functional importance of a positively charged, hydrophilic residue at that position. Its substitution by a neutral, hydrophobic alanine residue can therefore safely be predicted to alter the functionality of the enzyme.
(3) Including the K232A mutation in the combined linkage and LD analysis has a dramatic effect on the lod score that maximizes exactly at the DGAT1 position (Fig. 5)
(4) The allele substitution effect, α, estimated by association studies in the granddaughter design (Table 1) fits the previous estimates obtained by linkage analysis (Coppieters et al. 1998) in this same population.
(5) The frequency distribution of the DGAT1(microsatellite + SNP) haplotypes corroborates the predictions of the combined linkage and LD analysis (Fig. 4; Farnir et al. 2002).
(6) The same K232A mutation was unexpectedly found to be associated with two distinct haplotypes (μHQ-D andμHQ-NZ ) predicted to carry fat-increasing QTL alleles. It is noteworthy in this regard that sequencing more than 100 individuals from a broad range of different breeds didn't uncover a single other DGAT1 amino acid substitution (R. Spelman, in prep.).
Despite this multiple and mutually reinforcing evidence, we do not know at present how the K232A mutation causes the observed effect. Experiments are now being conducted to examine the influence of theK232A mutation on DGAT1 enzymatic activity as well as to generate transgenic mice harboring the two allelic variants using gene-targeting methods.
Our results provide interesting insights into the population genetics of the analyzed dairy cattle populations. The most obvious interpretation of the previously reported QTL fine-mapping experiments exploiting LD (Farnir et al. 2002) suggested two different “Q” alleles resulting from independent neo-mutations that occurred respectively in the Dutch and New Zealand dairy cattle populations. The long-range LD observed around these two “Q” alleles was considered evidence in favor of their relative youth compared to the “q” alleles. The results presented here, however, provide strong evidence that the Kresidue characterizing the “Q” alleles in fact represents the ancestral state, and that the A residue corresponding to the “q” alleles corresponds to the younger acquired state. However, the absence of strong LD between flanking microsatellites and this “novel” A allele indicates that the corresponding neo-mutation is likely to be quite old. This hypothesis is also corroborated by the presence of this allele in numerous distant cattle populations (R. Spelman, in prep.). The long-range LD observed for the “Q” alleles in the Dutch and New Zealand populations probably testifies for recent, independent selective sweeps. This would fit with the change in selection criteria that occurred in the 1950s, when the amount of total fat rather than total milk produced became the predominant breeding objective. Haplotypes carrying the K residue and consequently increasing fat yield could have then rapidly spread throughout the population, assisted by the extensive use of artificial insemination that was generalized in dairy cattle at about the same time. Since then, breeding objectives have continued to evolve, now targeting both fat and protein yield. It is interesting to note that the K232A DGAT1 polymorphism is essentially neutral with respect to present day selection indexes in the Netherlands (INET) and New Zealand (Breeding Worth)(data not shown). This may explain why both the K and A alleles are still segregating at intermediate frequency in these populations.
Having identified the causal mutation will greatly facilitate and reduce the cost of marker-assisted selection for this QTL. Although at present both DGAT1 alleles have very similar economic values in the Dutch and New Zealand economic context, this is not the case in some other parts of the world and is susceptible to change with time. The DGAT1 gene also becomes a prime target for manipulation by transgenic or other routes to modify the milk composition to satisfy consumer demand.
METHODS
Pedigree Material and Phenotypes
The pedigree material used for the association studies comprised a “granddaughter” design (Weller et al. 1990) counting 1818 Holstein-Friesian bulls sampled in the Netherlands, as well as a “daughter” design (Weller et al. 1990) counting 529 Holstein-Friesian cows sampled in New Zealand. The phenotypes of the sires were “daughter yield deviations“ (DYDs) which were obtained directly from CR-Delta (Arnhem, The Netherlands). The DYD of a sire corresponds to the average of the lactation performances of his daughters. More specifically, DYDs correspond to unregressed weighted averages of the daughters' lactation performances adjusted for systematic environmental effects and breeding values of the daughters' dams and expressed as deviations from the population mean (Van Raden and Wiggans 1991). The phenotypes of the cows were “lactation values” (first lactation yield deviations [YD], that is, lactation performances expressed as deviations from the population mean, adjusted for management group, permanent environmental effects, and herd-sire interaction effects [Van Raden and Wiggans 1991]) obtained directly from Livestock Improvement Corp. (Hamilton, New Zealand).
Combined Linkage and Linkage Disequilibrium Analysis
The maximum likelihood procedure for combined linkage and linkage disequilibrium analysis is described in detail in Farnir et al. (2002). In brief, it is an extension to quantitative traits of an approach developed by Terwilliger (1995) for discrete traits and is specifically adapted for large half-sib pedigrees which are common in livestock populations. It
assumes the segregation of a biallelic QTL (allele “Q” and “q”) in the population of interest with allele substitution effect α (defined as in Falconer & Mackay 1996). It also assumes that a fraction ρ (heterogeneity parameter) of the “Q” alleles derives from a “Q” allele that appeared “g” generations ago in the population by migration or mutation on a founder haplotype defined by specific
alleles (denoted 1) at M linked marker loci. As a consequence, QTL and marker loci are expected to be in LD, and the “Q-1” haplotypes are expected
to occur at an excess frequency of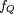 where fQ
is the population frequency of theQ QTL allele, f
1 is the population frequency of the “1” marker allele, f
0 the population frequency of all other marker alleles combined, and θ the distance between the marker under consideration
and the QTL.
where fQ
is the population frequency of theQ QTL allele, f
1 is the population frequency of the “1” marker allele, f
0 the population frequency of all other marker alleles combined, and θ the distance between the marker under consideration
and the QTL.
Using this model and following Terwilliger (1995), one can compute an approximation of the likelihood of the pedigree data, L, as:
 where ∏
where ∏
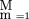 is the product over all M markers composing the chromosome map,
is the product over all M markers composing the chromosome map,
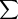
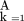 is the sum over all A alleles of marker m, fk
is the population frequency of allele k, ∏
is the sum over all A alleles of marker m, fk
is the population frequency of allele k, ∏
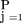 is the product over the P half-sib pedigrees composing the pedigree data,
is the product over the P half-sib pedigrees composing the pedigree data,
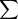
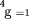 is the sum over the four possible QTL genotypes (i.e.,QQ, Qq, qQ, and qq),P(QGg
‖MGj
) is the probability that sire j has QTL genotype g given its genotype for marker m and assuming gametic association betweenQ and k, ∏
is the sum over the four possible QTL genotypes (i.e.,QQ, Qq, qQ, and qq),P(QGg
‖MGj
) is the probability that sire j has QTL genotype g given its genotype for marker m and assuming gametic association betweenQ and k, ∏
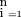 is the product over the n half-sibs composing pedigreej, P(QGg
‖MGi
) is the probability that half-sib i has QTL genotypeg given MGi
(i.e., its own marker genotype, the phase-known marker genotype of its sire, and the marker genotype of the gamete inherited
from its dam), andP(Phi
‖QGg
) is the probability for half-sib i to have phenotypePhi
given its QTL genotype QGg
, which is computed from the normal density function with appropriate mean (see Farnir et al. 2002) and residual variance ς2.
is the product over the n half-sibs composing pedigreej, P(QGg
‖MGi
) is the probability that half-sib i has QTL genotypeg given MGi
(i.e., its own marker genotype, the phase-known marker genotype of its sire, and the marker genotype of the gamete inherited
from its dam), andP(Phi
‖QGg
) is the probability for half-sib i to have phenotypePhi
given its QTL genotype QGg
, which is computed from the normal density function with appropriate mean (see Farnir et al. 2002) and residual variance ς2.
Using optimization routines such as GEMINI (Lalouel 1983), one can maximize the likelihood of the pedigree data, L, with respect to the unknown parameters α, ς2, ρ,fQ , and g, thereby extracting information from both linkage and LD. We refer to this hypothesis as HL+LD. Alternatively, one can fix the value of g at ∞, thereby ignoring all LD information: HL. In addition, one can compute the likelihood of the data under the null hypothesis H0 of no QTL at the corresponding map position by setting α at zero. The significance of the alternative hypotheses can be evaluated by generating different likelihood ratio statistics: HL+LD/H0 tests the combined linkage + LD signal, HL/H0 tests the linkage signal, and HL+LD/HL tests the LD signal (see Farnir et al. 2002 for further details).
Association Studies
The association study in the granddaughter design was performed using the following model: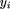 where yi
is the DYD of son i, μ is the overall population mean, β is a fixed regression coefficient estimating the A to K allele substitution effect,xi
is an indicator variable corresponding to the number of K alleles in the K232A genotype,ai
is a random polygenic component accounting for all known pedigree relationships (“animal model”; [Lynch and Walsh 1997], including ungenotyped individuals whose phenotypes were ignored), and ei
is a random residual. The error variance was assumed to be identical for all sons.
where yi
is the DYD of son i, μ is the overall population mean, β is a fixed regression coefficient estimating the A to K allele substitution effect,xi
is an indicator variable corresponding to the number of K alleles in the K232A genotype,ai
is a random polygenic component accounting for all known pedigree relationships (“animal model”; [Lynch and Walsh 1997], including ungenotyped individuals whose phenotypes were ignored), and ei
is a random residual. The error variance was assumed to be identical for all sons.
The association study in the daughter design was performed using the model: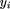 where yi
is the lactation value of cow i,gi
is a fixed effect corresponding to theDGAT1 genotype (KK, KA, or AA),ai
is a random polygenic component accounting for all known pedigree relationships (“animal model”; [Lynch and Walsh 1997], including ungenotyped individuals whose phenotypes were ignored), and ei
is a random residual. In both instances, maximum likelihood solutions for β,gi,ai, ei
, ς2
a, and ς2
ewere obtained using the MTDFREML program (Boldman et al. 1995).
where yi
is the lactation value of cow i,gi
is a fixed effect corresponding to theDGAT1 genotype (KK, KA, or AA),ai
is a random polygenic component accounting for all known pedigree relationships (“animal model”; [Lynch and Walsh 1997], including ungenotyped individuals whose phenotypes were ignored), and ei
is a random residual. In both instances, maximum likelihood solutions for β,gi,ai, ei
, ς2
a, and ς2
ewere obtained using the MTDFREML program (Boldman et al. 1995).
Acknowledgments
This work was funded by research grants from Vialactia Biosciences (Auckland, New Zealand), Cr-Delta, Livestock Improvement Corp., the Vlaamse Rundvee Vereniging, the Belgian Ministry of Agriculture, and the European Union. We are grateful to James Womack for providing us with DNA from the radiation hybrid panel.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 Corresponding author.
-
E-MAIL michel.georges{at}ulg.ac.be; FAX 32-4-366-4122.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.224202.
-
- Received November 16, 2001.
- Accepted December 4, 2001.
- Cold Spring Harbor Laboratory Press








