
Mosaic Organization of Orthologous Sequences in Grass Genomes
Abstract
Although comparative genetic mapping studies show extensive genome conservation among grasses, recent data provide many exceptions to gene collinearity at the DNA sequence level. Rice, sorghum, and maize are closely related grass species, once sharing a common ancestor. Because they diverged at different times during evolution, they provide an excellent model to investigate sequence divergence. We isolated, sequenced, and compared orthologous regions from two rice subspecies, sorghum, and maize to investigate the nature of their sequence differences. This study represents the most extensive sequence comparison among grasses, including the largest contiguous genomic sequences from sorghum (425 kb) and maize (435 kb) to date. Our results reveal a mosaic organization of the orthologous regions, with conserved sequences interspersed with nonconserved sequences. Gene amplification, gene movement, and retrotransposition account for the majority of the nonconserved sequences. Our analysis also shows that gene amplification is frequently linked with gene movement. Analyzing an additional 2.9 Mb of genomic sequence from rice not only corroborates our observations, but also suggests that a significant portion of grass genomes may consist of paralogous sequences derived from gene amplification. We propose that sequence divergence started from hotspots along chromosomes and expanded by accumulating small-scale genomic changes during evolution.
[GenBank Accession Numbers: Rice (Oryza sativa L. ssp. japonica)php200725 region: AF119222; rice (Oryza sativa L. ssp. indica) php200725 region: AF128457; sorghum (Sorghum bicolor) php200725 region: AF114171,AF527807, AF727808, AF527809; maize (Zea mays)php200725 region: AF090447, AF528565; rice chromosome 10 region (2.9 Mb): AC073391, AC087549, AC027657, AC087547, AC027658,AC087546, AC027659, AC087550, AC025905, AC087545, AF229187, AC027660,AC087544, AC027661, AC027662, AC087543, AC073392, AC087542, AC025906,AC073393, AC025907.]
All grasses belong to a single family,Gramineae, that contains ∼10,000 species, and originated ∼65 million years ago (mya; Kellogg 2000).Gramineae family members include important cereal crops such as rice, corn (maize), and wheat. Despite the vast diversity of their chromosome numbers and the DNA content of their genomes (Arumunganathan and Earle 1991), members of the Gramineae family show extensive genome conservation based on comparative genetic mapping using common DNA markers (Bennetzen and Freeling 1993; Gale and Devos 1998; Bennetzen 2000; Devos and Gale 2000; Keller and Feuillet 2000). Therefore, grasses have been considered a single genetic system (Bennetzen and Freeling 1993). With the discovery of massive retrotransposition in some large grass genomes, it was theorized that nested retrotransposition was the root cause of genome expansion, and that gene islands between seas of repetitive DNA were largely conserved (SanMiguel et al. 1996). However, recent data of genomic comparison at the DNA sequence level among grass species have shown many exceptions to the conservation of gene islands as well, resulting in the disruption of gene collinearity between closely related species (Bennetzen 2000; Devos and Gale 2000; Keller and Feuillet 2000). Exceptions to gene collinearity include microrearrangement or small-scale genomic changes, such as gene insertion, deletion, duplication, or inversion (Bancroft 2000). In this study, we investigate additional causes of genome expansion besides retrotransposition, and how small-scale changes occurred during evolution.
We recently reported the characterization of a region of maize Chromosome 4S. This region contains DNA markers conserved among maize (2700 Mb; million base pairs), sorghum (770 Mb), and rice (430 Mb;Arumunganathan and Earle 1991). In maize this region contains a large storage-protein gene family, the 22-kD zein gene family, with 23 members (Song et al. 2001). Interestingly, 22 members are tandemly arrayed within 168 kb, whereas one copy is located 20 cM closer to the centromere of maize Chromosome 4S. Sorghum also has a storage protein, 22-kD kafirin, homologous to the maize 22-kD zein (DeRose et al. 1989). Rice does not have a corresponding storage protein (Kim and Okita 1988). Phylogenetic analysis of the 22-kD zein gene family indicates that most of the copies arose from gene amplification after maize and sorghum diverged (Song et al. 2001). It would be interesting to see how such a large gene cluster could contribute to genomic expansion in this region, and how active gene amplification in the past could have affected the flanking orthologous sequences. Therefore, we isolated and sequenced the orthologous regions corresponding to the maize 22-kD zein cluster region from two other closely related species: rice and sorghum. In rice, the orthologous regions from two different subspecies, Oryza sativa L. ssp. japonicaand Oryza sativa L. ssp. indica, were characterized. These two subspecies separated <1 mya (Bennetzen 2000); sorghum and maize separated from each other ∼16.5 mya (Gaut and Doebley 1997); and rice separated from the ancestor of sorghum and maize ∼50 mya (Kellogg 2000). The sequences from japonica andindica could indicate how sequence divergence initiated, whereas those sequences from sorghum and maize could indicate how sequence divergence accumulated during a long evolutionary period. By comparing these orthologous regions, we gained insight on how genome structure evolved.
RESULTS
Isolation of Orthologous Regions
We previously sequenced a 346-kb maize genomic region that contained the 22-kD α-zein storage protein gene family (Song et al. 2001). This region is linked to the maize RFLP marker php200725, a gene of unknown function that is conserved among maize, rice, and sorghum (data not shown). Using this marker, two rice BAC (bacterial artificial chromosome) clones were selected from BAC libraries of the rice subspecies japonica and indica. Although these rice BAC clones have a rather small insert size (77 kb and 70 kb, respectively), we have determined that they comprise a region larger than the entire 346-kb maize region, exemplifying the expansion of the large maize genome versus the small rice genome. To maximize alignments between rice and maize, the maize contig was extended by selecting another BAC clone from the same maize BAC library overlapping with its 3′ end. This yielded a total of 435 kb from maize, of which 380 kb was suitable for alignment with the rice inserts. Using sequence information from rice and maize, four BAC clones were selected from sorghum BAC libraries. These clones yielded a contig of 425 kb. Only 220 kb was suitable for alignment with sequences from the other two species.
Sequence Comparison of Orthologous Regions
These orthologous regions around the php200725 marker from rice, sorghum, and maize were sequenced and aligned (Fig.1). Using rice as a reference, four CRs (conserved sequence regions) are found across the three species (CR1–CR4, Fig. 1). The conserved markerphp200725 is part of CR2. These conserved regions contain either single genes (CR1 and CR2, Table 1) or gene blocks (CR3 and CR4, Table 1). CR4 in maize was further divided by a retrotransposition between two genes. These conserved regions are separated by nonconserved sequences of various sizes.
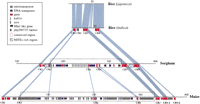
Sequence comparison of orthologous regions from rice, sorghum, and maize. Sequences of genomic regions from rice (subspeciesjaponica and indica), sorghum, and maize have been vertically arranged with a size ruler (in kilobases) for each sequence. Only orthologous regions (CR1–CR4) are highlighted. Light-blue areas indicate the homologous regions. The boxed sections between CR2 and CR3 in sorghum and maize represent an insertion relative to rice. Pink boxes along the rulers indicate conserved sequence regions across species (CR1–CR4, CRa, CRb). In maize, CR4 was divided into CR4-1 and CR4-2, reflecting an insertion relative to sorghum and rice. In sorghum, CRb was split into two pieces because of an insertion relative to maize. Green bars indicate RFLP marker php200725 and its orthologs. The second green bar in the sorghum region marks the duplication of php200725. Small triangles indicate amplified storage-protein gene copies and their orientation (22-kD zeinin maize and 22-kD kafirin in sorghum, respectively). Violet arrows indicate Mla1-like disease-resistance genes and their orientation. All other genes are indicated by small red bars. Genes without a pink outer box indicate nonconserved genes. Gray bars indicate areas of retrotransposons, and blue bars DNA transposons. Two hatched boxes in sorghum indicate two MITE-rich regions (5′ region, <1 kb/MITE; 3′ region, <2 kb/MITE). All kafirins andzeins showed homology to each other, but only the oldest copies are indicated. A summary of predicted genes and their positions is given in Table 1 for convenience.
Genes in Orthologous Regions
The nonconserved spacers contain a blend of genes, transposable elements, and small repetitive sequences. The spacer between CR1 and CR2 shows a gradual increase in size, consistent with the genome sizes of the three species. This spacer does not contain any genes in rice, two genes and a cluster of MITEs (miniature-inverted-repeat-transposableelements) in sorghum (Bureau and Wessler 1994), and two nonconserved genes and a retrotransposon in maize. The spacer between CR3 and CR4 shows very different characteristics. Its size is not proportional to genome size and varies even between the two rice subspecies. In rice, it differs by small repetitive sequences, such as simple sequence repeats (SSRs), but does not contain any genes. In sorghum, this spacer does not contain any genes, but multiple MITE sequences. In maize, although the size of the spacer is surprisingly shorter than in rice, it contains two genes.
The largest expansion of sorghum (∼155 kb) and maize (∼290 kb) relative to rice occurs between CR2 and CR3 (boxed sections in Fig. 1). In sorghum, the expansion is comprised of three amplified genes, fragments of two DNA transposable elements, and five other genes, but no retrotransposons. The most extensive gene amplification is that of a storage-protein gene, 22-kD α-kafirin, with 11 copies, all with the same orientation, and 10 forming a nearly perfect tandem array with a unit size of 3290 bp. We found that the second amplified gene, a putative Mla1-like disease resistance gene (Halterman et al. 2001), has four copies, but they are not in tandem. The third amplified gene is the php200725 ortholog, with two tandem copies.
The corresponding expansion in maize is attributed to a combination of gene amplification and retrotransposition. Only the storage-protein gene that is orthologous to sorghum was amplified, resulting in 22 tandem copies of the 22-kD α-zein gene. This tandem array ofzein genes is interrupted by transposable elements and other genes spread over 168 kb. The entire expansion consists of several DNA transposable elements (blue bars in Fig. 1) and 110 kb of retrotransposons (gray bars), as well as two other genes, one in the middle and the other downstream of the zein cluster, which are conserved between maize and sorghum (CRa and CRb, Fig. 1).
Within the orthologous regions (CR1–CR4), rice has six genes; sorghum has 15 different genes, of which three have been amplified, resulting in a total of 29 genes; and maize has 13 different genes, of which one has been amplified, resulting in a total of 34 genes. Although gene amplification has caused a large sequence expansion, it is remarkable that the amplified copies in maize and sorghum are confined and do not disrupt the order and orientation of nonamplified conserved genes, such as CRa and CRb (Fig. 1). As a result, gene amplification increased local gene density of the respective genome (in CR1–CR4, rice, 1 gene/9.5 kb; sorghum, 1 gene/7.4 kb; maize, 1 gene/11 kb).
Analysis of Paralogous Sequences
A major finding of this study is the presence of different genes in nonconserved sequences of the sorghum and maize php200725regions (Table 1). Although these genes are missing from the ricephp200725 region, based on sequence and hybridization data, in many cases copies of these genes are present in the rice genome, but mapped to nonorthologous chromosomal locations (Table2). One exception is the 22-kD storage-protein gene, which does not have a homologous sequence in the rice genome. We also found at least four genes that are not only in a nonorthologous position, but also amplified at the same time. A single putative receptor kinase in the sorghum region (gene 10, Tables 1 and2) has 10 clustered homologs in a single BAC clone on rice Chromosome 4. Also in the sorghum region, a single putative glucosyl transferase gene (gene 19, Tables 1 and 2) has four clustered homologs on rice Chromosome 1. These clustered gene copies are apparently derived from gene amplification. The Mla1-like disease resistance gene that is amplified in sorghum in the php200725 region has a single homologous copy on rice Chromosome 1. CRa, which encodes a putative disease response gene and is orthologous between maize and sorghum, is positioned on the other arm of rice Chromosome 11, 65 cM away from the rice php200725 region. The rice CRa is also amplified in contrast to maize and sorghum (see Methods). These data strongly suggest that gene amplification and gene movement are functionally associated with each other.
Rice Homologous Genes of Nonconserved Genes Fromphp200725 Regions
Additional conserved sequences could be found within extra sequences 3′ to CR4 from rice and sorghum (data not shown), including a predicted gene consisting of conserved MATH/TRAF and BTB/POZ domains (Albagli et al. 1995; Uren and Vaux 1996). In addition to the orthologous position in the php200725 region of rice on Chromosome 11, a large number of paralogous sequences of this gene were found at the central portion of rice Chromosome 10. We sequenced ∼2.9 Mb surrounding these gene copies. Subsequent sequence analysis revealed a total of 48 copies of this gene, with 36 gene copies clustered within a 330-kb region, 9 copies clustered within another 85-kb region, and 3 other copies scattered elsewhere. Further analysis of the same region provided several other examples of gene amplification, including 27 copies of a glycine-rich protein within 230 kb (Table3). Of a total of 360 predicted genes within this region, ∼26% were the result of gene amplification. Given a gene density of 8 kb/gene within the 3-Mb region, one would predict a total of 49,000 genes for the rice genome. Discounting the amplified gene copies, this number would be only 36,000. Therefore, it appears that gene amplification contributes to a large proportion of genome size.
Gene Amplification Within 2.9-Mb Region on Rice Chromosome 10
DISCUSSION
Contiguous Genomic Sequences From Large Plant Genomes
BAC libraries from three grass species, maize, sorghum, and rice, have been used to clone orthologous sequences based on conserved gene sequences. With a small genome such as rice, a single BAC clone was sufficient to comprise all aligned markers. However, with larger genomes like sorghum and maize, contiguous and overlapping BAC clones had to be isolated. The absence of STC (sequence tagged connector) and FPC (finger printing contig) databases (see Methods) for the BAC libraries from maize and sorghum has slowed the effort to obtain contiguous sequence information. The sequencing of 2.9 Mb of the central portion of rice Chromosome 10, on the other hand, has been greatly facilitated by the rice STC and FPC resources (Mao et al. 2000). Once these resources become available for maize and sorghum, additional comparison analysis among these three species will be simplified. For now, the two contiguous sequences from maize and sorghum described here provide us with the greatest amount of contiguous-sequence information of both genomes.
Mosaic Organization of Orthologous Sequences
In this study we observed a mosaic organization of evolutionarily conserved (orthologous) sequences across the php200725 regions among rice, maize, and sorghum. The distinct feature of such an organization is that conserved sequences were interspersed with nonconserved sequences. This mosaic structure is apparent only between sufficiently diverged species. For example, between the two subspecies of rice, sequence divergence is not sufficient to uncover such a mosaic structure, whereas the mosaic structure is quite apparent in the other pairwise or triple comparisons. Besides sequence divergence, large regions are also required to visualize a mosaic structure. The sequence should be long enough to cover several conserved islands separated by nonconserved seas. This is particularly challenging when dealing with a large, complex genome like maize for two main reasons. First, it is common for conserved gene islands to be separated by a long stretch of nonconserved sequences. Secondly, many repetitive sequences usually contain nested retrotransposons, further complicating the analysis. In this study, the nonconserved regions in between conserved sequences could be as long as 155 kb, as in the sorghum region, or 290 kb as in the maize region.
Between sufficiently diverged species, the conserved regions usually only contain genes, whereas the nonconserved regions contain a mixture of genes, transposable elements, and other diverged sequences. However, it is surprising to see so many genes present in nonconserved regions in this study. These genes must have originated from different positions in the genome. This indicates that gene movement must have been quite frequent.
Gene Amplification and Gene Movement
How can genes move between different genomic positions? This study suggests a possible mechanism of gene movement that is associated with gene amplification. Besides examples provided in this study, gene movement associated with gene amplification has also been documented in other studies. For example, the floury-2 locus in maize is one copy of the amplified 22-kD zein gene translocated 20 cM away from other copies (Song et al. 2001). The adh1 locus in maize and sorghum is a translocated copy from one of the two duplicated genes (Tikhonov et al. 1999; Tarchini et al. 2000). Therefore, one can envision a mechanism by which gene amplification generates free gene copies that could be inserted elsewhere in the genome by illegitimate recombination. Such a mechanism is reminiscent of plant DNA transformation by direct DNA uptake, or the particle bombardment method in which free DNA gets inserted by illegitimate recombination (Krens et al. 1982; Klein et al. 1988). On the other hand, gene copies translocated to nonorthologous positions are also prone to amplification, like the Mla1-like gene in the sorghum region, or the extreme amplification of the gene containing MATH/TRAF and BTB/POZ domains at the nonorthologous 2.9-Mb region in rice. The association of gene amplification with gene translocation could quickly generate divergent sequences in certain regions of the genome, and it might also underlie the rapid reorganization of resistance gene homologs in grass genomes (Leister et al. 1998).
Formation of Mosaic Organization by Hotspots Expansion
In this study, we were able to analyze sequences with different degrees of divergence. The sequences from the two rice subspecies diverged separately for <1 mya (Bennetzen 2000). The conserved sequence regions are also comprised of genes, but they extend into intergenic regions, leaving rather narrow nonconserved regions (Fig. 1, light-blue areas). These regions consist of a mixture of short repeat sequences and mobile elements. However, these elements appear to be defective elements rather than the result of homologous recombination of LTRs. Solo LTRs could indicate a reversal of genome expansion, which, however, does not appear to be the case in the ricephp200725 region, because defective elements could transpose without further truncations. Interestingly, the position of these narrow nonconserved regions aligns with those in sorghum and maize, where more significant changes occurred. Sorghum and maize provide sequences that diverged 16.5 mya (Gaut and Doebley 1997). We found three more genes not present in rice, but conserved between sorghum and maize. Almost all other changes occurred after maize and sorghum separated. The three extra conserved genes are the oldest copy of the 22-kD storage protein (22-kD zein and kafirinwere amplified after sorghum and maize separated), CRa and CRb (Fig.1). During this 16.5-million-year period, extensive gene movement and amplifications occurred. In addition, whereas massive retrotransposition occurred in maize, sorghum had extensive MITEs insertion. It is remarkable that gene amplification, retrotransposition, and MITEs insertions are all confined within a restricted space, promoting the formation of clustered gene families, nested transposons, and MITE-rich regions. Conserved sequence regions were further diminished at the same time, and only gene sequences or the exons of genes were conserved. Furthermore, rice and sorghum or rice and maize sequences diverged nearly 50 mya. Except for the changes to the three conserved genes between CR2 and CR3, other changes were either already saturated or difficult to detect on top of the changes that occurred within 16.5 million years.
Therefore, we believe that when orthologous sequences were just starting to diverge, small nonconserved regions formed in intergenic regions. These nonconserved regions formed hotspots for further sequence divergence. Orthologous sequences further diverged by expanding from these hotspots to form larger nonconserved regions. Such expansion occurred by accumulating small-scale changes, such as gene movement, gene amplification, and transposition (Bancroft 2000). Because all the changes were confined within or between the hotspots regions, we can predict that gene amplification could form gene family clusters (The Arabidopsis Genome Initiative 2000; Song et al. 2001); retrotranspositions could form nested retrotransposon regions (SanMiguel et al. 1996); and MITE transpositions form MITE-rich regions (Jang and Wessler 2001), and so on. Genes from one hotspot might move to another hotspot owing to their active status, translocating new genes into a different nonconserved region. The genes newly moved into a new hotspot might continue to undergo further small-scale changes, such as gene amplification. In addition, conserved regions and nonconserved regions could also change their state. Hotspots could turn cold by switching a nonconserved region into a conserved region. As an example, CRa and CRb within sorghum and maize regions, as compared with rice, were once nonconserved sequences. Even before sorghum and maize separated, CRa and CRb turned into conserved sequences. Reciprocally, conserved regions also could switch to nonconserved regions, for example, the 22-kD zein andkafirin genes, whose oldest copies were conserved sequences between maize and sorghum. After maize and sorghum separated, both of them turned into hotspots for gene amplification, and turned conserved sequences into nonconserved sequences.
Differential divergence of these nonconserved hotspots along chromosomes is then manifested during speciation, and a mosaic of conserved segments interspersed with nonconserved segments becomes apparent in the comparison of orthologous regions of different species.
Do Grass Genomes Have a Faster Evolutionary Clock?
Mammalian genomes, which diverged much earlier (100 mya) than the grass genomes described here (50 mya; Burt et al. 1999; Kellogg 2000), also contain rapidly changing nonorthologous sequences (Chiaromonte et al. 2001), although not to the same degree as in this case. In grasses, even between much more closely related species such as sorghum and maize, the sequence divergence could be very drastic. It appears that grass genomes may have evolved much more rapidly than one would have predicted by only comparing coding sequence substitution rates (Gaut et al. 1996) or chromosome structural mutation rates (Paterson et al. 1996). Because polyploidization and segmental chromosomal duplication occur on top of gene amplification in many higher plants (Vision et al. 2000; Gaut 2001), the number of their paralogous sequences exceeds those of other eukaryotic organisms sequenced so far, including the human genome (Messing 2001). Such a fast evolutionary clock might also explain the great success of speciation of certain large families, such as Gramineae.
METHODS
BAC Clone Isolation and Sequencing
Maize RFLP marker php200725, which is conserved among maize, rice, and sorghum, was used to isolate two rice BAC clones from BAC libraries of two rice subspecies japonica (Oryza sativa japonica cv. Lemont) and indica (Oryza sativa indica cv. Teqing), available from Clemson University Genome Institute (CUGI). Clone 1H19 from the japonica Lemont library and clone 16F19 from the indica Teqing library were sequenced.
Previously we sequenced a 346-kb maize region (Song et al. 2001). To maximize alignments between rice and maize, an additional BAC clone (BAC #072) from the same maize BAC library (Zea mays cv BSSS53; Song et al. 2001) was selected, which extended the maize 346-kb contig from the 3′-end.
Using sequence information from rice and maize, BAC clones containing orthologous sequences were selected from two sorghum BAC libraries. BAC clones SB25M18 and SB40L16 came from Sorghum bicolor cv BTx623, digested with HindIII and BamHI, respectively, available from the Texas A&M BAC Center. BAC clones SB126P21 and SB234M12 came from the Sorghum bicolor cv BTx623HindIII library, available from CUGI.
A minimum-tiling path based on end-sequence information and using sequence-tagged connectors (STCs), was used to select neighboring BAC clones (Mao et al. 2000). A total of 22 clones yielded a complete contiguous sequence of 2,883,802 bp that spans 12 cM on rice Chromosome 10, spanning between markers S11069 at position 29.8 cM and C1286 at position 41.8 cM of the Oryza sative japonica cv. Nipponbare map from the Rice Genome Research Program (RGP) in Japan (Rice Genome Research Program in Japan, 2001;http://rgp.dna.affrc.go.jp/).
All BAC clones were sequenced by the shotgun DNA sequencing method (Messing et al. 1981) using sequence mates of pUC-based templates (Vieira and Messing 1982) as described before (Song et al. 2001).
Sequence Analysis
We performed homology searching using BLAST2 (http://www.ncbi.nlm.nih.gov/blast/ bl2seq/bl2.html; Tatusova and Madden 1999). Gene prediction was based on both GeneScan (http://genes.mit.edu/Genscan.html) and FGENESH++ (http://www.softberry.com./berry.phtml?topic=gfind). Protein sequences generated from gene prediction programs were used to search the protein database (Altschul et al. 1997). Expressed sequence tag (EST) database searches were used to improve the prediction of expressed genes. Transfer RNA (tRNA) was predicted using the tRNA-scan-SE program (http://www.genetics.wustl.edu/eddy/tRNAscan-SE/). RepeatMasker (http://repeatmasker.genome.washington.edu/cgi-bin/RepeatMasker) was used to screen interspersed repeats and low-complexity DNA sequences.
Rice BAC Clone Fingerprint Contig (FPC) Analysis
Rice BAC filters (Oryza sative japonica cv. Nipponbare,HindIII section) from the International Rice Genome Sequencing Project (IRGSP) were used to screen positive clones containing the putative disease-resistance response gene (CRa in Fig. 1). These clones have been assembled in Fingerprinting Contigs (FPCs) that have been anchored to the genetic map (http://www.genome.clemson.edu/projects/rice/fpc/). Positives clones are within FPC #206 on rice Chromosome 11, between markersR682 (30.5 cM) and RZ316 (32.1 cM), immediately adjacent to the rice Adh homologous region. Thephp200725 ortholog falls within FPC #256, also on Chromosome 11 but on the other side of the centromere, at position 97.4 cM. In addition, Southern blot analysis of these BAC clones detected multiple copies of this gene (data not shown), indicating that this gene became amplified in rice.
WEB SITE REFERENCES
http://genes.mit.edu/GENSCAN.html; the new Genscan Web server at MIT.
http://repeatmasker.genome.washington.edu/cgi-bin/RepeatMasker; RepeatMasker Web server.
http://rgp.dna.affrc.go.jp/; Rice Genome Research Programe.
http://www.genetics.wustl.edu/eddy/tRNAscan-SE; tRNAscan-SE Search server.
http://www.genome.clemson.edu/projects/rice/fpc; Rice FPC map.
http://www.ncbi.nlm.nih.gov/blast/bl2seq/bl2.html; NCBI BLAST2 Web server.
http://www.softberry.com./berry.phtml?topic=gfind; Softberry Gene Finding Web page, FGENESH++.
Acknowledgments
We thank S. Kavchok and S. Young for technical assistance; J. Bennetzen of Purdue University, M. Vaudin, and B. Barbarzuk of Monsanto for critical reading of the manuscript. This work has been supported by DOE grant #DE-FG05-95ER20194, USDA-NRI grant #98-35300-6165, and NSF grant #9975618 to J.M.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵1 Present address: LPG-ATC, NCI Advanced Technology Center, 8717 Grovemont Circle, Gaithersburg, MD 20877, USA.
-
↵2 Corresponding author.
-
E-MAIL messing{at}mbcl.rutgers.edu; FAX (732) 445-0072.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.268302. Article published online before print in September 2002.
-
- Received March 11, 2002.
- Accepted July 25, 2002.
- Cold Spring Harbor Laboratory Press








